

SUMMARY
Most ribosomal proteins (RP) are regarded as essential, static components that only contribute to ribosome biogenesis and protein synthesis. However, emerging evidence suggests that RNA-binding RP are dynamic and can influence cellular processes by performing “extraribosomal”, regulatory functions involving binding to select, critical target mRNAs. We report here that the RP, Rpl22, and its highly homologous paralog, Rpl22-Like1 (Rpl22l1 or Like1), play critical, extraribosomal roles in embryogenesis. Indeed, they antagonistically control morphogenesis through developmentally-regulated localization to the nucleus where they modulate splicing of the pre-mRNA encoding smad2, an essential transcriptional effector of Nodal/TGF-β signaling. During gastrulation, Rpl22 binds to intronic sequences of smad2 pre-mRNA and induces exon 9 skipping in cooperation with hnRNP-A1. This action is opposed by its paralog, Like1, which promotes exon 9 inclusion in the mature transcript. The nuclear roles of these RP in controlling morphogenesis represent a fundamentally different and paradigm-shifting mode of action for RP.
Keywords: Ribosomal protein, paralog, morphogenesis, extraribosomal function, Rpl22, Rpl22l1, pre-mRNA splicing, Smad2, hnRNP-A1
Graphical Abstract

eTOC
Zhang et al., reveal that ribosomal proteins can perform regulatory functions of fundamental importance while separated from the ribosome. They demonstrate that ribosomal proteins Rpl22 and Rpl22-Like1 (Like1) are retained in the nucleus during gastrulation where they control morphogenesis by modulating the splicing of Smad2 pre-mRNA in cooperation with HNRNPA1.
INTRODUCTION
Ribosomal proteins (RP) are basic components of the ribosome that are generally thought to contribute to the assembly of the ribosome and its ability to synthesize proteins. Inactivation of RP has been linked to clinical syndromes collectively known as ‘ribosomopathies’(Narla and Ebert, 2010). Ribosomopathies exhibit not only impaired erythropoiesis and increased risk for development of leukemia, but also other abnormalities in a variety of organ systems (Narla and Ebert, 2010). These developmental anomalies include short stature, craniofacial defects, thumb malformation, urogenital abnormalities and heart defects, which strongly suggests that RP might play a role in embryogenesis (Narla and Ebert, 2010). A recent example of this is the finding that haploinsufficiency of RPSA causes human asplenia, suggesting that RP inactivation can cause distinct and tissue-restricted developmental abnormalities (Bolze et al., 2013). Mutations of RP have been shown in many cases to impair ribosome biogenesis, which activates p53 by attenuating the function of the p53 ubiquitin ligase, MDM2 (Zhang and Lu, 2009). However, emerging evidence has begun to reveal that certain RP may also perform specialized regulatory roles in biological processes through p53-independent ‘‘extraribosomal functions’’, acting to regulate context-dependent translation or transcription, and doing so from outside of the ribosome (Warner and McIntosh, 2009; Xue and Barna, 2012). For example, extraribosomal Rpl26 and Rpl13a can modulate the translation of selected target RNAs by binding to their 5′ and 3′ untranslated regions, respectively (Mukhopadhyay et al., 2008; Takagi et al., 2005). Likewise, Rps3 and Rps14 have been shown to interact with DNA-binding complexes and regulate gene-specific transcription (Wan et al., 2007; Zhou et al., 2013).
We previously established that neither of the highly homologous RP paralogs, Rpl22 or Rpl22-Like1 (Rpl22l1 or Like1), is required for general protein synthesis; however, these paralogs do play distinct, antagonistic, regulatory roles in blood cell development (Anderson et al., 2007; Rao et al., 2012; Zhang et al., 2013). Indeed, the ability of Like1 to promote the emergence of embryonic hematopoietic stem cells (HSC) is dependent upon its ability to directly bind and facilitate the translation of mRNA encoding the essential transcription factor, smad1. In contrast, Rpl22 acts in direct opposition to repress smad1 translation (Zhang et al., 2013). These findings revealed that the RNA-binding RP, Rpl22 and Like1, played an important regulatory role in hematopoiesis, from outside of the ribosome (Zhang et al., 2013). Our previous analysis in zebrafish also revealed that Rpl22 and Like1 are abundantly expressed throughout early development from the 2-cell stage to 18 hours post fertilization (hpf) (Zhang et al., 2013), raising the possibility that they might also act to regulate early development, as has been found for other molecular effectors exhibiting similar expression patterns (Langdon and Mullins, 2011; Schier, 2007).
Here, we employed antisense morpholinos (MO) targeting the ATG translational start codons of Rpl22 and Like1 to attenuate the translation of both maternal and zygotic mRNA encoding these proteins to repress their expression and assess the impact on development of zebrafish embryos. We showed that Like1 knockdown disrupted the convergence & extension (C&E) phase during gastrulation and, consequently, elaboration of the normal body plan. The molecular basis for disruption of C&E is that splicing of pre-mRNA encoding smad2, an essential mediator of Nodal/TGF-β signaling, was impaired, revealing nuclear functions for Rpl22 and Like1 in regulating pre-mRNA splicing. Indeed, Like1-knockdown resulted in skipping of exon 9 of smad2, which blocked protein expression. The skipping of exon 9 was caused by Rpl22, as both the mis-splicing of smad2 and the defect in C&E observed in Like1 morphants were rescued by simultaneous knocking down Rpl22. The ability of Rpl22 and Like1 to control smad2 pre-mRNA splicing coincided with a developmentally controlled retention of these proteins in the nucleus and was associated with direct binding of Rpl22 to a consensus motif in smad2 intron 8, immediately preceding skipped exon 9. Using RNA-seq we determined that numerous other pre-mRNAs were mispliced in Like1 morphants and these targets shared the features of having consensus Rpl22/Like1 binding sites in the intron preceding the skipped exon and potential binding sites for splicing modulator hnRNA-A1. hnRNP-A1 function is required for the ability of Rpl22 to disrupt smad2 splicing, as exon 9 inclusion is restored upon knockdown of hnRNP-A1 in Like1 morphants. Together, these data reveal that the RP, Rpl22 and Like1 antagonistically control gastrulation through a nuclear role in regulating mRNA splicing, in zebrafish as well as in mammals.
RESULTS
Rpl22 and Like1 play distinct and antagonistic roles during gastrulation
We previously demonstrated that neither of the highly homologous RP paralogs, Rpl22 or Like1, is required for general protein synthesis; however, these paralogs do play distinct, antagonistic, regulatory roles in hematopoiesis that are mediated by binding to and controlling the translation of smad1 mRNA (Anderson et al., 2007; Rao et al., 2012; Shen et al., 2012; Zhang et al., 2013). Moreover, we showed that the mRNAs encoding Rpl22 and Like1 were abundantly expressed in zebrafish embryos beginning at the 2-cell stage (Zhang et al., 2013). To determine if Rpl22 and Like1 regulate morphogenesis, we employed antisense MO that target the ATG start codons and repress the translation of both maternal and newly synthesized zygotic Rpl22 and Like1 mRNA (Zhang et al., 2013) (Figure S1A,B), enabling analysis of the role of these proteins in early development. Like1 morpholino-treated embryos (Like1 morphants) exhibited defects in anterior-posterior extension during late gastrulation (Figure 1A), as indicated by the reduced angle between the anterior and posterior ends (Figure 1B). Importantly, whereas knockdown of Rpl22 did not disrupt extension (Figure 1A,B), simultaneous knockdown of Rpl22 and Like1 (double morphants; D-MOs) completely corrected the extension defect observed upon knockdown of Like1 alone (Figure 1A, right panel; Figure 1B). This indicates that Like1 plays a critical role in promoting C&E during gastrulation and that its ability to promote C&E is antagonized by its paralog, Rpl22. Co-injecting Like-MO and mCherry-CAAX (membrane-targeted mCherry) mRNA revealed that knockdown of Like1 caused a broadening of the notochord in 10 hpf Like1 morphants (Figure 1C), indicating that the convergence of notochord precursors was also impaired by Like1 knockdown. The shortening and widening of mesoderm (ntl, gsc), endoderm (sox32/sox17), paraxial mesoderm (papc), adaxial (myod1) and brain/neural tissue (six3, pax2, krox20 and hgg1/dlx3b) in Like1 morphants was confirmed by whole mount in situ hybridization (WISH) using probes for markers of those tissues (Figure 1D–E; Figure S1C–J). To gain insight into the molecular basis for the disruption of C&E during gastrulation, we assessed whether signaling pathways previously determined to be critical for early morphogenesis were disrupted. We found that signaling through the Bmp/pSmad5, canonical and noncanonical Wnt, PI3-Kinase/Akt, and Stat3/Liv1/e-cadherin pathways controlling migration of developing tissues were unaffected by Like1 knockdown (Figure S2K–P) (Heisenberg and Solnica-Krezel, 2008; Kimelman and Griffin, 2000; Solnica-Krezel, 2005). However, the expression of lefty1 was essentially eliminated in Like1 morphants at 16hpf (Figure 1F). Because lefty1 is a direct target of Nodal/Smad2 signaling, we assessed whether Smad2 signaling was altered in Like1 morphants (Smith et al., 2011; Stemple, 2000; Thisse et al., 2000). Indeed, both Smad2 phosphorylation and expression were reduced between 10 and 16 hpf (Figure 1G,H). Smad2 signaling plays an essential role in regulating gastrulation and later morphogenesis (Heyer et al., 1999; Nomura and Li, 1998; Waldrip et al., 1998). Interestingly, maternal Smad2/Nodal signaling and expression of the downstream target squint (sqt) were intact at 4.7hpf, prior to the onset of gastrulation (Figure 1I, Figure S1Q), indicating that the ability of Like1 to control Smad2 expression was developmentally regulated.
Figure 1. Opposing roles of RP paralogs, Rpl22 and Like1, in regulating gastrulation.

(A,B) One-cell stage zebrafish embryos were injected with translational-blocking Like1-A-MO (2ng), L22-A-MO (6ng) or both (D-MOs), following which effects on gastrulation were assessed. The images of embryos represent lateral views at 10hpf. The red and black lines indicate the anterior and posterior ends of the body axis, respectively. The angle (θ), which defines the degree of extension, was measured between the red arrow and dashed black line, and is represented graphically as the mean ± standard deviation (S.D.). Control (black); Like1 MO (red); Rpl22 MO (yellow); and double-morphants (D-MO; green). Triplicate samples were quantified and the mean ± S.D. is depicted graphically. **, p < 0.01. (C) Imaging of notochord in Like1 morphants (10hpf) co-injected with 100pg mCherry-CAAX mRNA at the 1-cell stage. The dorsal view, anterior is at the top. The lateral notochord boundaries are indicated by the dotted yellow lines, and the width of notochord was marked by white lines. (D,D’) Expression patterns of ntl in 10hpf Like1 morphants. Red Arrows mark changes in distribution in the images representing lateral (D) and dorsal (D’) views. (E,F) sox32 and lefty1 expression in Like1 morphants. Red Arrows indicated changes in expression or distibution. (E) 75%-epiboly stage, dorsal view. (F) lateral view, 16hpf. (G-I) Phospho-Smad2 and total-Smad2 were assessed in 10hpf (G), 16hpf (H), and 4.7hpf (I) Like1 morphants by immunoblotting. All results are representative of at least 3 experiments performed. See also Figure S1.
Smad2 splicing is regulated by Rpl22 and Like1 during gastrulation
To determine how Like1 knockdown repressed the expression of zygotic Smad2, we tested whether smad2 mRNA levels were altered. Interestingly, while total smad2 mRNA levels were unchanged, most of the smad2 mRNA was significantly smaller in size in 10hpf Like1 morphants (Figure 2A). The alteration of smad2 mRNA size was not observed until 6hpf, after the switch from maternal to zygotic transcription (Kane and Kimmel, 1993) (Figure S2A). Sequence analysis revealed that the basis for the reduction in smad2 mRNA size was that it lacked exon 9, such that exon 8 was directly spliced to exon 10 (Figure 2B,C). While knockdown of Rpl22 alone did not affect smad2 splicing, simultaneous knockdown of Rpl22 with Like1 significantly reduced the smad2 mis-splicing observed in Like1 morphants, suggesting antagonistic regulation by these RP paralogs (Figure 2A, right lane). Isoform-specific qPCR confirmed that normal smad2 splicing was increased from ~35% in Like1 morphants to ~70% in morphants where Rpl22 was knocked down together with Like1 (i.e., in double-morphants; D-MOs; Figure 2D).
Figure 2. The gastrulation defects in Like1 morphants result from skipping of smad2 exon 9.

(A,B) RT-PCR analysis of smad2 mRNA in Like1, Rpl22 and double morphants (D-MOs). smad2 mRNA was evaluated by RT-PCR using primers (black arrowheads) amplifying the sequences between exons 5 to 10. (C) Like1 knockdown causes skipping of smad2 exon 9. Sequence analysis of the smaller smad2 mRNA species caused by Like1 knockdown (red arrow in panel A). (D) Quantitative RT-PCR analysis of the relative expression of intact smad2 mRNA (indicated by left panel, blue arrow). The blue and red lines identify the position of real-time primers employed to detect intact smad2 mRNA. Triplicate samples were quantified and the mean ± S.D. is depicted graphically. p-values are indicated. All results are representative of at least 3 experiments performed. See also Figure S2.
The C&E defects in Like1 morphants result from Smad2 mis-splicing
To assess whether smad2 pre-mRNA mis-splicing was responsible for the C&E defects, we sought to specifically replicate the mis-splicing of smad2 using MO targeting the smad2 intron8- exon9 boundary (S2-i8e9-MO; Figure 3A). We verified that the S2-i8e9-MO replicates the mis-splicing of smad2, and reduces Smad2 protein levels (Figure 3B and Figure S2B,C). Moreover, the S2-i8e9-MO also phenocopied the morphological defects and impaired lefty1 expression observed in Like1 morphants (Figure 3C–E). The mis-spliced smad2 transcript appeared to be disrupting development by reducing Smad2 expression rather than functioning as a dominant negative. Indeed, ectopic expression of the mis-spliced smad2 transcript failed to block Smad2-dependent signaling events induced by a constitutively activated Alk4 mutant (Tar*) (Figure S3A, B). This is not unexpected because, despite the retention of the translational reading frame, we failed to detect a truncated protein produce from the exon9-skipped smad2 mRNA, either endogenously or upon ectopic expression of the mutant cRNA (Figure S3C,D). The inability to produce a stable, truncated protein has also been reported for a murine Smad2 splice variant lacking exons 9 and 10, in which the translational reading frame was preserved (Liu et al., 2004). Having found that the mis-splicing of smad2 was sufficient to phenocopy the C&E defects observed in Like1 morphants, we next asked if restoring Smad2 signaling could rescue those defects. Indeed, ectopic expression of constitutively active Smad2 (Ca-Smad2) did alleviate the C&E defects in Like1 morphants (morphology and ntl/myod1 distribution; Figure 3F–H) (Dick et al., 2000). Taken together, these results suggest that Rpl22 and Like1 antagonistically regulate C&E by controlling the splicing of smad2, a critical regulator of gastrulation.
Figure 3. The C&E defects in Like1 morphants can be rescued by re-establishing smad2 signaling.
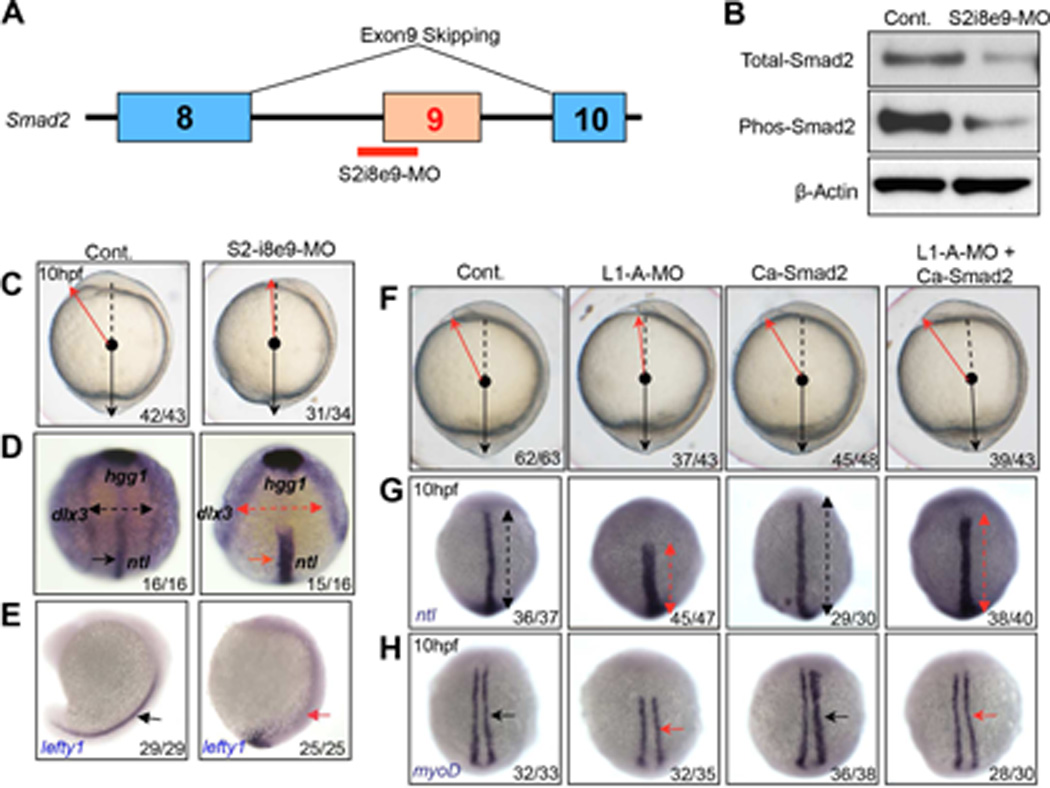
(A) Schematic of the morpholino employed to induce exon9 skipping (S2-i8e9-MO). (B) Immunoblotting of detergent extracts reveals a reduction in total and phospho-Smad2 protein expression in the S2-i8e9-morphants. (C–E) S2-i8e9-MO induction of smad2 mis-splicing phenocopied the C&E defects caused by Like1 knockdown, as indicated by altered morphology (C, red arrow) and alterations in ntl/hgg1/dlx3b and lefty1 expression and distribution, as measured by in situ hybridization (D, 10hpf, red arrows, anterior dorsal view; E, 16hpf, lateral view). (F–H) mRNA encoding constitutively activated smad2 (Ca-Smad2, 20pg) was utilized for injection alone or co-injected with Like1-A-MO. Embryo morphology (F) as well as the abnormal distribution of ntl (G, red arrows) and myod1 (H, red arrows) at 10hpf, can be rescued by ectopic expression of Ca-Smad2. All embryos are dorsal view with the anterior on top at 10hpf. All results are representative of at least 3 experiments performed. See also Figure S3.
The subcelluar localization of Rpl22 and Like1 in zebrafish embryos is developmentally regulated
For Rpl22 and Like1 to directly regulate Smad2 pre-mRNA splicing during gastrulation, Rpl22 and Like1 would have to be located in the nucleus. Indeed, immunofluorescence analysis of ectopically expressed HA-tagged RP revealed that Like1 and Rpl22 were primarily located in the nucleus at 6 and 10hpf (Figure 4A and Figure S4A). The nuclear localization of Like1 and Rpl22 appears to be developmentally regulated, as they relocalized to the cytoplasm at 24hpf, after gastrulation and morphogenesis are complete (Figure 4B). Importantly, at 24hpf, Like1 knockdown no longer caused smad2 mis-splicing (Figure 4C). The nuclear retention of Rpl22 and Like1 requires both their NLS motifs and their ability to bind RNA (Figure S4B–E). These results indicate that Rpl22 and Like1 regulate smad2 pre-mRNA splicing in a developmentally controlled manner, and their ability to do so is tightly linked to nuclear localization during gastrulation.
Figure 4. The regulation of smad2 pre-mRNA splicing during gastrulation by Rpl22 and Like1 is associated with their retention in the nucleus.

(A,B) Subcellular location of epitope-tagged Rpl22 and Like1 at 10hpf (A) and 24hpf (B). 100pg of mRNA encoding HA-zRpl22 (HA/L22) and HA-zLike1 (HA/L1) was co-injected with mRNA encoding mCherry-CAAX into 1-cell stage embryos and visualized by HA antibody immunostaining. mCherry-CAAX marked the cell membrane and DAPI marked the nucleus. Red scale bar =10 µm. (C) RT-PCR detection of smad2 mis-splicing. Following Like1 MO injection, smad2 mis-splicing was assessed by RT-PCR in 10hpf and 24hpf Like1 morphants. All results are representative of at least 3 experiments performed. See also Figure S4.
Like1-deficiency disrupts Smad2 splicing in mouse embryos
To determine if these RP also regulate Smad2 splicing during mammalian gastrulation, we generated mice in which the Rpl22l1 gene was ablated (Figure 5A–C). Intercrossing Rpl22l1+/− mice revealed that Like1-deficiency was embryonic lethal, as no Rpl22l1−/− mice were observed among 54 offspring. Moreover, Rpl22l1−/− embryos were absent embryonically by 12 days post coitus (dpc) and displayed aberrant morphology at 9.5 dpc (Figure 5D). When the status of Smad2 pre-mRNA splicing was assessed at 6.5 dpc, we observed mis-splicing of the Smad2 pre-mRNA (Figure 5E and F); however, unlike the mis-splicing observed in zebrafish, both exons 7 and 8 of murine Smad2 were skipped in the Rpl22l1−/− embryos (Figure 5G). Together, these data indicate that the regulation of Smad2 splicing during gastrulation is not only observed in zebrafish, but is also conserved in mammals.
Figure 5. Effect of Like1-deficiency on Smad2 splicing during murine gastrulation.

(A) Molecular strategy for targeted deletion of Rpl22l1. A Loxp-FRT-neo resistance-FRT cassette was inserted 3’ to the first exon of Rpl22l1 and a second LoxP site was inserted 3’ to the third exon. F1 heterozygous offspring were bred to Mox2-cre mice to delete the 2nd and 3rd exons of Rpl22l1, disrupting expression at the genomic locus. (B,C) Strategy to genotype Rpl22l1+/L or Rpl22l1+/− mice. To genotype mice with LoxP sites flanking exons 2 and 3 of Rpl22l1, primers C and D were used to amplify a 223 bp or 263 bp product for wildtype and Rpl22L1-LoxP, respectively. Deletion of Rpl22l1 is genotyped with primers B and D, which amplify a 607 bp product for the WT allele and A and D, which amplify a 420 bp product for the mutant allele after cre recombination. (D) Representative Rpl22l1−/− embryo compared to Rpl22l1+/+ littermate control at 9.5 dpc. (E–G) Effect of Like1-deficiency on splicing of Smad2 pre-mRNA during murine gastrulation. Embryos derived from timed matings of Rpl22l1+/− mice were isolated at 6.5 dpc (mid gastrulation), genotyped as above, and analyzed by RT-PCR using the indicated primers to identify alterations in Smad2 splicing (E,F). Sequencing of the mis-spliced Smad2 species found in Rpl22l1−/− embryos revealed that it represented a species in which exon 6 was fused directly to exon 9, eliminating exons 7 and 8 (G). All results are representative of at least 3 experiments performed.
Like1 knockdown disrupts the splicing of numerous pre-mRNA targets
To gain insight into how Rpl22 and Like1 might antagonistically regulate smad2 splicing, we performed RNA-Seq to identify the set of pre-mRNAs whose splicing they regulate. RNA-Seq analysis of Like1 morphants revealed more than 300 mis-spliced targets, including smad2 (Figure 6A). Pathway analysis revealed that the mis-spliced targets were enriched for those involved in DNA-replication, morphogenesis, and regulation of BMP signaling (Figure 6B). Most of the mis-splicing events represented exon skipping, with a few instances of alternative exon usage (Figure 6C). Moreover, MaxEntScan analysis of the mis-spliced targets revealed that the skipped exons (Figure 6D; skipped, “S”) exhibited weaker 5´ splice donor and 3´ splice acceptor sites than the included exons in the same genes (Figure 6D; not skipped, “NS”) (Lu et al., 2013). Hence, Like1 knockdown preferentially induced the skipping of exons with weak splice sites, suggesting that additional splicing factors were necessary for recognition of these exons by the spliceosome (Lopez, 1998). It is well established that trans-acting splicing factors such as hnRNP A/B family members are able to preferentially bind to exonic splicing silencers (ESS) and antagonize the binding of SR proteins to exonic splicing enhancers (ESE), thereby leading to exon skipping (Wang and Burge, 2008; Zhu et al., 2001). The observation that most of the mis-splicing events in Like1 morphants were exon-skips, raised the possibility that the exonic elements involved might be bound by trans-acting factors that could influence their function and cause exon skipping (Wang et al., 2004). To test this hypothesis, we validated a set of mis-spliced target genes by RT-PCR (Figure S5) and interrogated the sequences surrounding the skipped exons to identify common features. The FAS-ESS and RESCUE-ESE algorithms were used to predict potential ESS or ESE (Wang et al., 2004). Indeed, we discovered that G-rich (GGGG or GGG) motifs were enriched in skipped exons (Figure S6A). Furthermore, the consensus Rpl22/like1 stem-loop binding motif was found in introns immediately preceding the skipped exons (Figure 6E), suggesting that Rpl22/Like1 can directly bind to target pre-mRNAs and regulate their splicing. Importantly, these features were also found in smad2 pre-mRNA (Figure S6B). Thus, these data support a model where Rpl22 and Like1 antagonistically regulate gastrulation by directly binding to pre-mRNA targets, including smad2, and promoting their mis-splicing in conjunction with a trans-acting factor(s) that recognizes a G-rich motif (Figure 6F).
Figure 6. Common features of mRNA targets mis-spliced in Like1 morphants.

(A) smad2 exon9 skipping detected by RNA-Seq. Alignment of RNA-seq reads to the genome reveals exclusion of smad2 exon9 in 10hpf Like1 morphants (blue box). (B) Gene ontology analysis of transcripts affected by Like1 knockdown from RNA-seq analysis. Significant gene ontology terms (p < 0.05) are depicted as a bar graph with the p values represented as –log10 on the X-axis. (C) Schematic illustrating the type of alternative splicing identified by RNA-Seq in 10hpf Like1 morphants. (D) Calculation of the strength of 5´ and 3´ splice sites of skipped (S) versus non-skipped (NS) exons in pre-mRNAs targeted in Like1 morphants. (E) Schematic of the consensus hairpin bound by Rpl22/Like1. (F) Schematic model of the common features of pre-mRNA targets affected by Like1 knockdown. Targets contained both a consensus Rpl22/Like1 binding motif in the preceding intron and a G-rich motif in the skipped exon. RNA-Seq analysis was performed on at least 3 independent biological replicates per condition. See also Figures S5 and S6
The ability of Rpl22 to disrupt smad2 pre-mRNA splicing in Like1 morphants is dependent upon hnRNP-A1
To test this model, we used in vivo RNA:Protein cross-linking immunoprecipitation (CLIP) analysis to determine if Rpl22 and Like1 can bind their consensus motif in smad2 pre-mRNA. Both Rpl22 and Like1, but not their m88 RNA-binding mutant forms, were able to bind to intron 8 of smad2 pre-mRNA, which immediately precedes the skipped exon 9 (Figure 7A,B), suggesting that the regulation of smad2 splicing by Rpl22 and Like1 is direct. We next sought to identify the trans-acting factor(s) that bind the G-rich motif present in the pre-mRNAs mis-spliced in Like1 morphants. hnRNP-A1 (A1) has been reported to recognize a motif similar to the G-rich sequence motif observed in the targets mis-spliced in Like1 morphants, and zebrafish A1 is expressed and localized in the nucleus during gastrulation (Figure S7A)(Gabut et al., 2008). Moreover, A1 is a well-established splicing modulator that can bind ESS and inhibit exon inclusion (Zhu et al., 2001). To determine if A1 was contributing to the ability of Rpl22 and Like1 to modulate smad2 splicing, we overexpressed A1 mRNA and found that A1, but not other hnRNP such as hnRNP-H and I (Y.Z., data not shown), induced smad2 exon9-skipping (Figure 7C, red arrow). Moreover, knocking down A1 (validated in Figure S7B) in Like1 morphants significantly reduced the skipping of smad2 exon 9 in Like1 morphants (Figure 7D, red arrow). Finally, knockdown of both Rpl22 and A1 completely suppressed the smad2 mis-splicing normally observed in Like1 morphants (Figure 7D, red arrow), suggesting that Rpl22 and A1 collaborate in promoting smad2 mis-splicing in the absence of Like1. hnRNPs have been reported to physically and specifically interact with selected RPs (Kristensen et al., 2012). To determine if the ability of Rpl22 and A1 to promote smad2 mis-splicing involved their physical association, we performed co-precipitation analysis. We found that A1 co-precipitated with Rpl22, but not Like1, in detergent extracts of 10hpf zebrafish embryos, demonstrating a physical interaction between Rpl22 and A1 (Figure 7E). Importantly, the Rpl22-A1 association was not mediated by an RNA bridge as it was not disrupted by RNAse treatment (Figure 7E).
Figure 7.

Factors through which Rpl22 and Like1 regulate smad2 alternative splicing. (A) Position of the real-time primers (red arrows) flanking the consensus Rpl22/Like1 binding site in intron 8 of smad2 pre-mRNA. (B) RNA-CLIP analysis of Rpl22/Like1 binding to smad2 pre-mRNA. Embryos injected with mRNA encoding HA tagged Rpl22, Like1, or their RNA-binding mutants (m88) were harvested at 10hpf. After light crosslinking, detergent nuclear extracts were immunoprecipitated using anti-HA antibody, and the co-precipitated RNA quantified by RT-PCR. Triplicate measurements are depicted graphically as mean ± S.D. p-values are indicated. (C) Overexpression of hnRNP-A1 (A1) mRNA induces smad2 mis-splicing. Embryos were injected with differing amounts of A1 mRNA, following which the effect on smad2 mis-splicing was determined by RT-PCR at 10hpf. The relative ratio of exon 9 skipped mRNA to intact smad2 mRNA was quantified in triplicate and depicted graphically as the mean ± S.D. (D) Genetic interaction of Rpl22 and A1. The ratio of exon9 skipped to intact smad2 was quantified in Like1 morphants in which A1 and/or Rpl22 was knocked down. The mean ± S.D of triplicate measurements was depicted graphically as in (C). (E) Physical association between Rpl22, Like1 and A1. Anti-HA immunoprecipitation (IP) and anti-Flag immunoblots (IB) were performed on detergent extracts of embryos injected with 100pg of mRNA encoding Flag-A1 and either HA-Rpl22 or HA-Like1, either before or after treatment with RNAse. All results are representative of at least 3 experiments performed. See also Figure S7.
DISCUSSION
Mutations in RP have long been linked to developmental anomalies manifested among the group of inherited syndromes collectively known as ribosomopathies. While the notion that these anomalies result from generalized perturbations of ribosome biogenesis or function is increasingly viewed as too simplistic, little insight has been gained into the molecular basis for their genesis. We report here nuclear functions for Rpl22 and Like1, which when disrupted, perturb embryonic patterning by impairing C&E during gastrulation. Indeed, Rpl22 and Like1 antagonistically control C&E through their developmentally-regulated retention in the nucleus and their ability to influence the splicing of pre-mRNAs of key regulators of gastrulation, principally smad2. The outcome of these splicing events is determined by the antagonistic balance of Rpl22 and Like1 and entails cooperation with the splicing modulator hnRNP-A1. These findings reveal a fundamentally different way to view the function of RP, in that they are capable of not only existing independent of intact ribosomes but are capable, in that form, of exerting a profound influence on critical events during development in zebrafish as well as in mammals. These findings also raise a number of crucial questions relating to how Rpl22 and Like1 are retained in the nucleus, the molecular basis by which they regulate the splicing of pre-mRNA targets, and how Rpl22 and Like1 are able to exert opposing effects on splicing.
Because the control of splicing by Rpl22 and Like1 is linked to their retention in the nuclear, the effects on splicing are clearly mediated in an extraribosomal fashion; however, the basis for the developmentally-controlled retention of Rpl22 and Like1 in the nucleus remains a critical, unanswered question. There are two likely explanations. First, Rpl22 and Like1 may assemble into the ribosome in the nucleolus, traffic to the cytosol and then separate from the ribosome in the cytosol, following which they traffic back to the nucleus. The separation of Rpl22 and Like1 from the ribosome could be induced by post-translational modifications. This has been observed for Rpl13a, which is displaced from the ribosome by interferon-mediated phosphorylation. Upon release, Rpl13a is able to bind to cytosolic mRNA species and silence their translation as part of the GAIT complex (Mukhopadhyay et al., 2008). Motif prediction analysis suggests that both Rpl22 and Like1 possess numerous consensus phosphorylation sites; however, it remains unclear if any are actually utilized in vivo. Rpl22 has been reported to be modified by SUMO in Drosophila, which is required to localize Rpl22e to the nucleoplasm (Kearse et al., 2013).
Nevertheless, because Drosophila Rpl22 contains a large, unique N-terminal extension, it remains unclear whether the SUMOylation of Rpl22 or Like1 plays a role in influencing their subcellular localization in other species. Rpl22 and Like1 might also be induced to dissociate from the ribosome through protein:protein interactions. We have shown that Rpl22 associates hnRNA-A1, a protein whose localization is reported to change dynamically during embryonic development (Vautier et al., 2001). Specifically, when zygotic transcription is activated, hnRNP-A1 accumulates in the nucleus (Vautier et al., 2001). The developmental changes in hnRNP-A1 localization, along with its genetic and physical interaction with Rpl22 raise the possibility that hnRNP-A1 may play a role in retention of Rpl22 in the nucleus during gastrulation. Nevertheless, hnRNP-A1 does not display the same association with Like1, suggesting that Like1 retention would be mediated by association with a different factor(s). Another mode of controlling the localization of Rpl22 and Like1 might entail direct trafficking to the nucleus after synthesis in the cytosol, through a process that does not involve their assembly onto the ribosome. We have shown that Rpl22 and Like1 localization in the nucleus requires both their NLS and RNA-binding motifs. Because the retention of Rpl22 and Like1 in the nucleus coincides with the switch to zygotic transcription during zebrafish embryogenesis, Rpl22 and Like1 retention in the nucleus is likely to be due, at least in part, to association with the large number of nascent transcripts being made in the nucleus during gastrulation. This is consistent with a previous report indicating that RP can associate with nascent RNAs in budding yeast (Schroder and Moore, 2005). Given the intimate connection between transcription and splicing, this represents a plausible explanation for developmental control of the retention of Rpl22 and Like1 in the nucleus and their involvement in regulating splicing.
The critical pre-mRNA target through which Rpl22 and Like1 modulation of splicing controls gastrulation is Smad2, a transcription factor whose function is essential for this process. Indeed, Like1 knockdown induces the skipping of smad2 exon 9, which preserves the translational reading frame of the splice variant, but nevertheless renders in the loss of Smad2 protein. This results either from repression of translation, or more likely, from instability of the truncated protein product. The inability to generate a stable protein product appears to be linked to the particular exonic sequences lost (i.e., exon 9), since a Smad2 variant lacking exon3 is capable of supporting production of a truncated Smad2 protein product (Dunn et al., 2005). Accordingly, the protein domain encoded by exon9 is likely to play a critical role in maintaining the structural integrity of Smad2. Interestingly, while restoration of Smad2 expression is able to alleviate the C&E defect observed in Like1-morphants, restoration of Smad2 expression is not sufficient to rescue development beyond gastrulation, between 16hpf and 3dpf (data not shown). Thus, is likely because the other targets that are mis-spliced upon Like1-knockdown are playing a role at more distal stages of development. The role of these mis-spliced targets in later morphogenesis processes, such as heart development and left-right patterning, is currently under investigation.
While it is clear that Rpl22 and Like1 regulate the splicing of numerous pre-mRNA targets in addition to smad2, it remains unclear how they do so. Rpl22 and Like1 could regulate splicing by directly binding to their pre-mRNA targets. Consistent with this possibility, we found that Rpl22 and Like1 can bind smad2 pre-mRNA. Moreover, there are consensus Rpl22/Like1 bindign motifs in the intron immediately preceding the skipped exon in all of validated, mis-spliced targets. Collectively, these data strongly suggest that Rpl22 and Like1, both RNA-binding proteins, are regulating splicing through direct binding to pre-mRNA targets (Zhang et al., 2013). Alternatively, it should be noted that the zebrafish and human U2-snRNA, on which the U2-snRNP is assembled, also contains a consensus Rpl22/Like1 binding motif. Accordingly, it is also possible that Rpl22 and Like1 could influence splicing indirectly, through effects on the U2-snRNP, which plays a central role in RNA-splicing (Matera and Wang, 2014). Nevertheless, even if altered U2-snRNP function were contributing to mis-splicing, because consensus Rpl22 and Like1 binding sites were found in all validated targets, it remains likely that direct binding of Rpl22 and Like1 to targets plays an important role.
One of the most interesting aspects of the regulation of development by the paralogs, Rpl22 and Like1, is the basis for their antagonistic functions. It was formerly thought that RP paralogs in lower organisms served largely redundant roles, but emerging evidence hints at functional specialization (Komili et al., 2007). Consistent with this notion, a few RP paralogs have been conserved in vertebrates, where they exhibit some tissue restriction in their expression patterns, but their functions remain largely unexplored. We have previously reported that the antagonistic functions of Rpl22 and Like1 regulate the emergence of embryonic hematopoietic stem cells (Zhang et al., 2013). We now find that Rpl22/Like1 antagonism extends to the control of pre-mRNA splicing during gastrulation. Because Rpl22 and Like1 are more than 70% identical at the amino acid level, this raises the question of how proteins as highly-homologous as these are able to perform antagonistic functions. The RNA-binding cores of Rpl22 and Like1 are nearly identical, and the helices that contact RNA are entirely identical and conserved from human to zebrafish; however, the amino (N) and carboxy (C) termini are more divergent (Zhang et al., 2013). Accordingly, we hypothesize that Rpl22 and Like1 are able to bind to a largely overlapping set of RNA targets, but have opposing effects on those targets, most likely because of the influence of their divergent termini. The termini might alter the way the RNA-binding domains function. Alternatively, the termini of Rpl22 and Like1 might recruit distinct trans-acting factors that serve as the effectors of antagonism. The preferential association of hnRNP-A1 with Rpl22 and not Like1 certainly supports this notion, but the basis for the selective interaction of hnRNP-A1 with Rpl22 remains to be determined. Nevertheless, the simplest interpretation of our data is that Like1 binding to the intronic sequences of a pre-mRNA target interferes with the ability of Rpl22 to recruit hnRNP-A1, thereby preventing it from disrupting normal splicing. It is unclear whether Like1 alone is capable of opposing the disruptive effects of Rpl22 and hnRNP-A1, or if it requires assistance from a positive regulator of splicing. Interestingly, the recurrent G-rich element observed in the mis-spliced targets of Like1 morphants can be bound either by splicing repressors or activators (Expert-Bezancon et al., 2004). Indeed, SR Proteins ASF/SF2 and SC35 can compete with hnRNP-A1 to bind to same Grich element. hnRNP-A1 binding has been reported to antagonize the action of SR proteins and cause mis-plicing of beta-tropomyosin exon6 (Expert-Bezancon et al., 2004). Accordingly, it is possible that Like1 may require the physical or genetic interaction with a factor(s) that facilitates normal splicing (e.g., SR proteins) in order to overcome the splice disruption induced by hnRNP-A1 and Rpl22. Our finding that some paralogs are not functionally redundant, but can instead perform biologically important antagonistic functions is unusual but not without precedent. Indeed a recent report revealed that paralogs Upf3a and Upf3b perform antagonistic functions in regulating nonsense mediated decay (Shum et al., 2016). Thus, the use of antagonistic paralogs as molecular rheostats to fine-tune biological processes appears to be an emerging theme.
Altogether our findings reveal that Rpl22 and Like1 are not functionally redundant during early development, but instead act as an antagonistic regulatory node where the balance of these antagonistic activities is crucial. This balance appears to be set not only by their expression level but also by control of their localization in the nucleus. Beyond their role in causing developmental abnormalities in ribosomopathies, inactivation of RP has also been associated with increased cancer risk both in inherited ribosomopathy syndromes and when RP inactivation occurs somatically. This raises the possibility that alterations in pre-mRNA splicing caused by Rpl22/Like1 imbalances might also occur in, and contribute to, transformation. We’ve shown that Rpl22 is a tumor suppressor as inactivation of Rpl22 promotes lymphoma formation (Rao et al., 2012). In contrast to Rpl22, the Like1 locus (RPL22L1) is frequently amplified in many types of human cancers (data not shown). Moreover, mutations in RNA splicing factors (e.g. U2AF1, SF3B1 and SFPQ) have been recently implicated in the pathogenesis of human MDS and acute myelogenous leukemia (Dolnik et al., 2012; Yoshida et al., 2011). Thus, imbalances in Rpl22 and Like1 expression might disrupt splicing in these diseases when splice factors are not mutated. Understanding how Rpl22 and Like1 interact with the splicing machinery to control smad2 splicing, may reveal insights not only into how mutations in RP can cause physical defects but also how they alter cancer risk in humans.
EXPERIMENTAL PROCEDURES
Zebrafish
Zebrafish were bred and maintained at 28.5 °C under standard aquaculture conditions in the Fox Chase Association for Assessment and Accreditation of Laboratory Animal Care (AALAAC) accredited Zebrafish Facility under the auspices of an Institutional Animal Care and Use Committee (IACUC) protocol. Embryos were staged as described previously (Kimmel et al., 1995).
Morpholino antisense oligonucleotides (MO)
MOs (GeneTools) were designed against the ATG translational start sites of the indicated targets as previously reported (Zhang et al., 2013).
Plasmids construction, RNA synthesis and overexpression
Epitope tagged constructs were generated by standard molecular biological approaches, following which mRNA were produced by in vitro transcription, and injected into 1-cell embryos to achieve overexpression.
In situ hybridizations
Whole mount RNA in situ hybridizations were performed as previously described (Thisse and Thisse, 2008). The stained embryos were mounted in 3% methylcellulose and photographed from the Nikon SMZ1500 stereomicroscope.
Antibodies, Western blotting and Immunofluorescent staining
Immunoblotting and immunofluorescent analysis of zebrafish embryos was performed as described (Zhang et al., 2013).
Generation of Rpl22l1−/− mice
Rpl22l1−/− mice were produced using the Targeting Vector 4595 D8 in order to insert LoxP sites in the introns between exons 1 and 2 and between exons 3 and 4. Following electroporation into R1 ES cells, the ES cells were injected into C57BL/6 blastocysts. To ubiquitously disrupt Rpl22l1 expression, Rpl22l1Loxp/+ mice were mated to Mox2-cre mice to generate Rpl22l1Loxp/− mice, which were subsequently bred to C57BL/6 mice to create the Rpl22l1+/− mice employed in timed matings. Maintenance and analysis of these mice were performed under the auspices of an IACUC approved protocol.
RT-PCR and Quantitative Real time PCR
Total RNA was isolated from embryos using Trizol (Life technologies) and glycogen (Ambion), following which RNA was reverse transcribed and subjected to RT-PCR or quantitative real-time PCR as indicated using SYBR-Green detection.
RNA-Seq Analysis
RNA-Seq analysis was performed on RNA extracted from zebrafish embryos at the end of gastrulation (10hpf). Briefly, the mRNA-Seq library was prepared from the total RNA using poly(A) selection (Truseq™ RNA Sample Preparation Kit V2, Illumina). RNA concentration was quantitated using a NanoDrop and RNA integrity was measured using a BioAnalyzer chip (Agilent), followed by 50–100bp paired end sequencing on a HiSeq2000 according to manufacturer protocols (Illumina). RNA-Seq sequence-read data for control and Like1 morphants was deposited into the NCBI Short Read Archive (SRA) database (SRP093436). mRNA-seq reads for both control and Like1 morphants were mapped to the latest zebrafish genome assembly (Zv9) using TopHat (version 2) alignment algorithm (Trapnell et al., 2009). For detection of splicing changes in Like1 morphants, the MATS algorithm was implemented using the aligned BAM files (Shen et al., 2012). Each splicing change was visualized using the IGV program (Integrative Genomics Viewer). Enrichment analysis for Gene Ontology (GO) terms was assessed using the GOstats program (Falcon and Gentleman, 2007). To evaluate the splicing strength for splice sites, we employed the MaxEntScan algorithm based on the maximum entropy model using candidate genes from Like1 morphant RNA-Seq data (Yeo and Burge, 2004). The significance of differences in the strength of 5’ and 3’ splice sites was evaluated using the paired Wilcoxon signed rank test.
Co-immunoprecipitation (CoIP) of Rpl22 and Like1 with hnRNP-A1
Co-immunoprecipitation of Rpl22 and Like1 with hnRNP-A1 was performed on detergent extracts of zebrafish embryos using minor modifications to a well-established protocol (Little and Mullins, 2009).
Embryonic RNA-Protein Crosslinking and Immunoprecipitation (E-CLIP)
To develop the E-CLIP method for Rpl22/Like1 interaction with smad2 mRNA in zebrafish embryos, we modified published protocols (Lu et al., 2013; Niranjanakumari et al., 2002; Ule et al., 2003). Briefly, mRNAs encoding HA-tagged Rpl22 and Like1 were injected into 1-cell embryos. Embryos were collected at 10hpf, cross-linked by adding 37% formaldehyde (Sigma) to 1.85% vol/vol final concentration, and lysed in hypotonic lysis buffer (10mM Tris-HCl pH 7.5, 10mM NaCl, 0.5% NP40, containing protease inhibitors and SuperRNase inhibitor). Isolated nuclei were lysed by sonication (Bioruptor sonication system), following which RNA was partially trimmed by treatment with RNase T1 (Ambion) for 5min at 37°C as described (Lu et al., 2013). The nuclear extract was clarified by centrifugation at 22,000g for 30 min, pre-cleared with Protein A Magnetic Beads (NEB), and immunoprecipitated with the indicated antibodies following which the immunoisolated RNA was quantified as above.
Statistical Analysis
A two-tailed Student’s t test was used in the experiments shown in Figure 1B, Figure 2D, and Figure 7B. Statistical significance was accepted when p< 0.05. All experiments are repeated at least three times.
Supplementary Material
HIGHLIGHTS.
Ribosomal proteins Rpl22 and Like1 antagonistically control morphogenesis
During gastrulation, Rpl22 and Like1 act away from the ribosome in the nucleus
Rpl22 and Like1 control gastrulation by regulating the splicing of Smad2 pre-mRNA
Control of gastrulation by Rpl22 and Like1 involves cooperation with HNRNP-A1
Acknowledgments
This work was supported by NIH grants AI110985 (D.L.W.), P30CA006927 (D.L.W.), an appropriation from the Commonwealth of Pennsylvania (D.L.W.), and the Bishop Fund (D.L.W.). Y.Z. was supported in part by grants from the MD Anderson Cancer Center Leukemia SPORE CA100632, When Every One Survives foundation, William J. Avery postdoctoral fellowship and the institutional postdoctoral training grant, T32 CA009035. We are grateful to the Wiest laboratory and Dr. Pengfei Xu for helpful suggestions and Drs. Siddharth Balachandran, Dietmar Kappes, and Charles Query for critical evaluation of the manuscript. We also thank Ms. Robin G. Kunkel, M.S., for illustrations. These studies were supported by the following core facilities at Fox Chase: Genomic, DNA Sequencing, Imaging and Laboratory Animal/Zebrafish.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Y.Z. designed and performed all the experiments, interpreted data and wrote the manuscript. S.P. and S.M. contributed to the RNA-Seq analysis. D.K., M.W, and J.R. helped with experiments. P.A., Q.F, and D.M. produced the Rpl22l1−/− mice. M.O. analyzed the development of Rpl22l1−/− mice and B.K. oversaw those analyses. D.L.W. oversaw the design and research, interpreted all the data and wrote the manuscript.
REFERENCE
- Anderson SJ, Lauritsen JP, Hartman MG, Foushee AM, Lefebvre JM, Shinton SA, Gerhardt B, Hardy RR, Oravecz T, Wiest DL. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759–772. doi: 10.1016/j.immuni.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, Abhyankar A, Itan Y, Patin E, Brebner S, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340:976–978. doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick A, Mayr T, Bauer H, Meier A, Hammerschmidt M. Cloning and characterization of zebrafish smad2, smad3 and smad4. Gene. 2000;246:69–80. doi: 10.1016/s0378-1119(00)00056-1. [DOI] [PubMed] [Google Scholar]
- Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, Fries T, Kronke J, Kuhn MW, Paschka P, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–e92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- Dunn NR, Koonce CH, Anderson DC, Islam A, Bikoff EK, Robertson EJ. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes & development. 2005;19:152–163. doi: 10.1101/gad.1243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expert-Bezancon A, Sureau A, Durosay P, Salesse R, Groeneveld H, Lecaer JP, Marie J. hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of beta-tropomyosin exon 6B. The Journal of biological chemistry. 2004;279:38249–38259. doi: 10.1074/jbc.M405377200. [DOI] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Gabut M, Chaudhry S, Blencowe BJ. SnapShot: The splicing regulatory machinery. Cell. 2008;133:192, e191. doi: 10.1016/j.cell.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Heisenberg CP, Solnica-Krezel L. Back and forth between cell fate specification and movement during vertebrate gastrulation. Current opinion in genetics & development. 2008;18:311–316. doi: 10.1016/j.gde.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12595–12600. doi: 10.1073/pnas.96.22.12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane DA, Kimmel CB. The zebrafish midblastula transition. Development. 1993;119:447–456. doi: 10.1242/dev.119.2.447. [DOI] [PubMed] [Google Scholar]
- Kearse MG, Ireland JA, Prem SM, Chen AS, Ware VC. RpL22e, but not RpL22e-like-PA, is SUMOylated and localizes to the nucleoplasm of Drosophila meiotic spermatocytes. Nucleus. 2013;4:241–258. doi: 10.4161/nucl.25261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D, Griffin KJ. Vertebrate mesendoderm induction and patterning. Current opinion in genetics & development. 2000;10:350–356. doi: 10.1016/s0959-437x(00)00095-2. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Komili S, Farny NG, Roth FP, Silver PA. Functional specificity among ribosomal proteins regulates gene expression. Cell. 2007;131:557–571. doi: 10.1016/j.cell.2007.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, Foster LJ. A high-throughput approach for measuring temporal changes in the interactome. Nature methods. 2012;9:907–909. doi: 10.1038/nmeth.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon YG, Mullins MC. Maternal and zygotic control of zebrafish dorsoventral axial patterning. Annual review of genetics. 2011;45:357–377. doi: 10.1146/annurev-genet-110410-132517. [DOI] [PubMed] [Google Scholar]
- Little SC, Mullins MC. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nature cell biology. 2009;11:637–643. doi: 10.1038/ncb1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Festing MH, Hester M, Thompson JC, Weinstein M. Generation of novel conditional and hypomorphic alleles of the Smad2 gene. Genesis. 2004;40:118–123. doi: 10.1002/gene.20072. [DOI] [PubMed] [Google Scholar]
- Lopez AJ. Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annual review of genetics. 1998;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- Lu X, Goke J, Sachs F, Jacques PE, Liang H, Feng B, Bourque G, Bubulya PA, Ng HH. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nature cell biology. 2013;15:1141–1152. doi: 10.1038/ncb2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang Z. A day in the life of the spliceosome. Nature reviews Molecular cell biology. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Molecular cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods. 2002;26:182–190. doi: 10.1016/S1046-2023(02)00021-X. [DOI] [PubMed] [Google Scholar]
- Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- Rao S, Lee SY, Gutierrez A, Perrigoue J, Thapa RJ, Tu Z, Jeffers JR, Rhodes M, Anderson S, Oravecz T, et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012;120:3764–3773. doi: 10.1182/blood-2012-03-415349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science. 2007;316:406–407. doi: 10.1126/science.1140693. [DOI] [PubMed] [Google Scholar]
- Schroder PA, Moore MJ. Association of ribosomal proteins with nascent transcripts in S. cerevisiae. RNA. 2005;11:1521–1529. doi: 10.1261/rna.2134305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic acids research. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH, et al. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell. 2016;165:382–395. doi: 10.1016/j.cell.2016.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KA, Noel E, Thurlings I, Rehmann H, Chocron S, Bakkers J. Bmp and nodal independently regulate lefty1 expression to maintain unilateral nodal activity during left-right axis specification in zebrafish. PLoS genetics. 2011;7:e1002289. doi: 10.1371/journal.pgen.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnica-Krezel L. Conserved patterns of cell movements during vertebrate gastrulation. Current biology : CB. 2005;15:R213–R228. doi: 10.1016/j.cub.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Stemple DL. Vertebrate development: the fast track to nodal signalling. Current biology : CB. 2000;10:R843–R846. doi: 10.1016/s0960-9822(00)00789-2. [DOI] [PubMed] [Google Scholar]
- Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Thisse B, Wright CV, Thisse C. Activin- and Nodal-related factors control antero-posterior patterning of the zebrafish embryo. Nature. 2000;403:425–428. doi: 10.1038/35000200. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature protocols. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Vautier D, Chesne P, Cunha C, Calado A, Renard JP, Carmo-Fonseca M. Transcription-dependent nucleocytoplasmic distribution of hnRNP A1 protein in early mouse embryos. Journal of cell science. 2001;114:1521–1531. doi: 10.1242/jcs.114.8.1521. [DOI] [PubMed] [Google Scholar]
- Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92:797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, Lam LT, Staudt LM, Levens D, et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Molecular cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nature reviews Molecular cell biology. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of computational biology : a journal of computational molecular cell biology. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Duc AC, Rao S, Sun XL, Bilbee AN, Rhodes M, Li Q, Kappes DJ, Rhodes J, Wiest DL. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Developmental cell. 2013;24:411–425. doi: 10.1016/j.devcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Hao Q, Liao JM, Liao P, Lu H. Ribosomal protein S14 negatively regulates c-Myc activity. The Journal of biological chemistry. 2013;288:21793–21801. doi: 10.1074/jbc.M112.445122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Molecular cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.