

Our novel algorithm for delimiting protein structural domains provides insights into protein folding, function, and evolution.
Keywords: Protein structure, protein domains, structure partitioning, structural bioinformatics
Abstract
Ambiguity is the quality of being open to several interpretations. For an image, it arises when the contained elements can be delimited in two or more distinct ways, which may cause confusion. We postulate that it also applies to the analysis of protein three-dimensional structure, which consists in dividing the molecule into subunits called domains. Because different definitions of what constitutes a domain can be used to partition a given structure, the same protein may have different but equally valid domain annotations. However, knowledge and experience generally displace our ability to accept more than one way to decompose the structure of an object—in this case, a protein. This human bias in structure analysis is particularly harmful because it leads to ignoring potential avenues of research. We present an automated method capable of producing multiple alternative decompositions of protein structure (web server and source code available at www.dsimb.inserm.fr/sword/). Our innovative algorithm assigns structural domains through the hierarchical merging of protein units, which are evolutionarily preserved substructures that describe protein architecture at an intermediate level, between domain and secondary structure. To validate the use of these protein units for decomposing protein structures into domains, we set up an extensive benchmark made of expert annotations of structural domains and including state-of-the-art domain parsing algorithms. The relevance of our “multipartitioning” approach is shown through numerous examples of applications covering protein function, evolution, folding, and structure prediction. Finally, we introduce a measure for the structural ambiguity of protein molecules.
INTRODUCTION
Analysis is the process of separating a whole into its constituent parts to gain a better understanding of it. Applied to the three-dimensional (3D) structure of proteins, it often consists in dividing a macromolecule into simpler yet informative subunits, called domains, which can be studied independently. Thus, investigating protein function, folding, or evolution often starts by delineating structural domains. This strategy also helps overcome challenges associated with structural studies of full-length proteins by molecular dynamics or de novo predictions. In addition, the classifications of protein structural domains are at the basis of every protein structure prediction method relying on fold recognition.
The idea of dividing protein structure into domains was introduced more than four decades ago by Wetlaufer (1), who defined protein domains as structurally compact and separate regions of the macromolecule. After this geometrical definition, many manual and automated methods for assigning structural domains have been based on additional criteria, such as folding autonomy, function, thermodynamic stability, or domain motions (2). As a result, many proteins are annotated differently from one domain database to another, depending on the methods and criteria used for structure partitioning (3). Paradoxically, although protein structure partitioning is a multiple-criteria problem—which, by its definition, can often accept more than one solution—different domain decompositions of the same protein are still considered to be mutually exclusive, rather than compatible or complementary. This issue inherent to human perception has been previously raised (4, 5) and continues to be a challenge (6), because it biases the analysis of protein molecules and restricts the number of avenues to explore, by not allowing more than one way to decompose their 3D structure. A domain partitioning based on a particular criterion, for example, geometry, may be useful for studying certain properties of the protein, such as function or dynamics, while being irrelevant regarding other characteristics, such as evolution or folding. This is well illustrated by the actin structure, which is divided into either two functional and evolutionary domains in the Structural Classification of Proteins (SCOP) (7) and Evolutionary Classification of Protein Domains (ECOD) (8) databases, or four domains, based on secondary structure elements, in the CATH (Class, Architecture, Topology, Homology) database (Fig. 1A) (9). Moreover, the delineation into two domains made by the authors of the structure (10), who used spatial separation of the domains as a criterion, differs from the function-based partitioning in terms of boundaries.
Fig. 1. Analogy between recognition of structural domains and image interpretation.
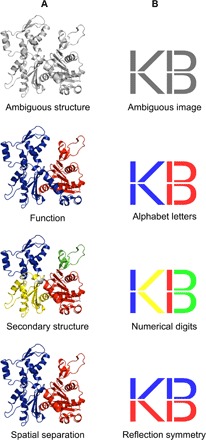
(A) Three equally valid assignments of structural domains for the actin protein (PDB: 1ATNA), each resulting from different partitioning criteria. (B) Ambiguous image that can be interpreted as either the letters “KB,” the mathematical inequality “1 < 13,” or the letters “VD” with their mirror image.
Here, to get around the human tendency to reject potentially crucial options when studying proteins, we propose an automated approach for structure partitioning, which considers the fact that protein structures may be ambiguous and have different but equally valid domain delineations in the same way that an ambiguous image has equally valid interpretations (Fig. 1B). This concept is also analogous to the syntactic ambiguity, a situation where a sentence may be interpreted in several ways because of its ambiguous structure. Thus, unlike other methods developed to date that provide single partitioning solutions, our algorithm—named SWORD (Swift and Optimized Recognition of Domains)—is aimed at cutting protein structures into multiple alternative domain decompositions. It operates through the hierarchical clustering of protein units (PUs), which are structural descriptors of intermediate size, between secondary structures and domains (11). These evolutionarily preserved substructures (12), into which the input protein is initially decomposed, characterize protein architecture in a more elementary way than domains while being large enough to contain relevant structural information. Here, we first validate the use of PUs to delineate structural domains, taking annotations from the CATH, SCOP, and ECOD databases as reference and comparing our results with those obtained with state-of-the-art algorithms. Then, we show the power of our multipartitioning approach for cases of protein structure prediction and studies of folding, function, and evolution. Finally, we show how we made SWORD able to detect complex cases of partitioning through the development of an original measure of the ambiguity in protein structures.
RESULTS AND DISCUSSION
Quantitative validation of the method
The ability of SWORD to find single partitioning solutions in agreement with structural domains assignments made by human experts was evaluated and compared with three reference algorithms: Protein Domain Parser (PDP), DomainParser, and DDomain (Fig. 2A). Considering the four benchmarks as a whole, SWORD performs slightly better than the other methods, ranking first for three annotation data sets and third on the Islam90 set (n = 90). Although SWORD is equaled by PDP for the Jones set (n = 55) and DomainParser for the Broad-consensus set (n = 329), our method is unmatched for the largest benchmark, that is the Consensus set (n = 3523), for which it identified 87.7% of the manual annotations. Regarding incorrect domain assignments, the four automatic methods have similar propensities to over- or undercut protein structures. Finally, all these accuracies are hardly inferior to those calculated without the 85% boundary overlap criterion (fig. S1), which confirms that the main difficulty of protein structure partitioning lies more in finding the correct number of domains than in delimiting accurate boundaries (4).
Fig. 2. Benchmark results for the SWORD algorithm.
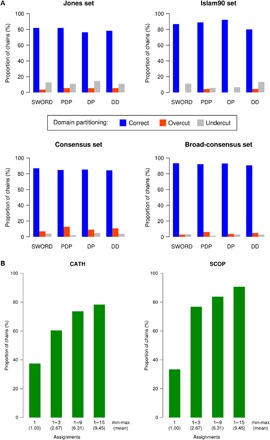
(A) Partitioning accuracies of SWORD, PDP, DomainParser (DP), and DDomain (DD) calculated for the four data sets of structural domain annotations (values are given in fig. S1). (B) Agreement between SWORD and either CATH or SCOP annotations, depending on the number of assignments provided, for the structures of the Dissensus data set (values are given in table S1).
For a given protein structure, our method can propose multiple alternative decompositions. Considering more than one assignment necessarily increases SWORD’s ability to find a domain arrangement that corresponds to expert annotations. Thus, the rate of agreement with data set annotations reached 94.6% for the Jones set, 95.6% for the Islam set, 96.9% for the Consensus set, and 97.6% for the Broad-consensus set, with averages of 5.3, 3.7, 4.3, and 3.9 alternative delineations, respectively (see table S1). Moreover, by computing multiple domain assignments, SWORD can solve difficult cases of protein structure partitioning. This has been evaluated by using the Dissensus set (n = 1025), which contains domain annotations that differ between CATH and SCOP databases. When considering only the optimal partitioning for each protein structure of the Dissensus set, SWORD found the correct assignment for 37.5 and 33.4% of CATH and SCOP annotations, respectively (Fig. 2B). However, these proportions of correct assignments markedly increased to 60.4 and 76.8% of CATH and SCOP annotations, respectively, when taking into account up to three decompositions (2.67 on average) provided by our multipartitioning method. The same benchmark has been conducted on the Strong-dissensus data set (n = 98) made of discrepancies between CATH, SCOP, and ECOD (fig. S2). By providing an average of 7.03 decompositions, SWORD manages to find about half of CATH and ECOD annotations (48.57 and 52.38%, respectively) and two-thirds of SCOP annotations (67.62%). These lower, although still remarkable, performances primarily reflect the higher difficulty of being in simultaneous agreement with three diverging methods. Moreover, because most of the ECOD annotations are based on SCOP, one can expect that these partitioning cases, for which ECOD and SCOP disagree (in addition to differing from CATH), are particularly complex.
Besides validating our PU-based algorithm, these results also highlight the theoretical limit that automated methods have reached regarding their ability to converge toward domain annotations manually made by the authors. This limit is due to the exclusive use of stereochemical information by algorithms, whereas human experts can additionally take into account experimental data from molecular biology and biochemistry. Although SWORD may be more accurate than the other algorithms on our benchmark, the little improvement it brings about leads us to believe that further development of the currently used “monopartitioning” approach is now of limited interest.
Dealing with ambiguity: Applications
The multiple view of protein architecture that we propose here can be advantageously applied to the numerous fields of molecular biology involving domain assignment, such as structure prediction or studies about protein folding, function, and evolution. This can be illustrated first with the set of structures used by Wetlaufer (1) when he introduced the concept of protein domains. For these proteins, neither the CATH nor the SCOP databases contain domain assignments similar to those he made four decades ago, although these remain valid today. The same goes for the recent ECOD database, probably because it mainly relies on domain assignments from SCOP. On the other hand, our partitioning algorithm finds all these expert annotations of domains through the alternative structural decompositions it computes for each protein (Fig. 3, A to E). Thus, without the results produced by SWORD, any scientists interested in these proteins risk missing important avenues of research.
Fig. 3. Practical cases requiring alternative domain decompositions.
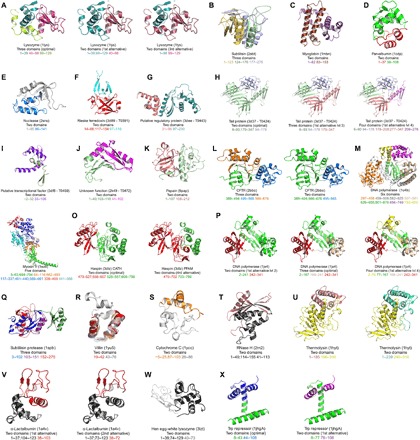
(A to X) Examples illustrating the usefulness of SWORD structural partitioning (for details, please refer to the main text).
More specifically, SWORD multipartitioning finds applications in protein structure prediction. In this field, it is well established that fold recognition methods perform better if the template library includes, along with full protein chains, protein structures partitioned into domains (13) because the global-local algorithm, typically used in fold recognition methods, cannot efficiently assess sequence-fold compatibilities over short portions of protein structures (14). Therefore, extending the fold library with alternative domain decompositions for each template structure can improve the search for compatible folds and, consequently, the quality of protein structure predictions. This can be verified by using target structures from the eighth edition of the Critical Assessment of Structure Prediction (CASP8) competition, for which the modeling difficulty is lowered when treating the structural domains separately rather than the whole protein chain (15), the challenge being reduced to the relative positioning of the individually modeled domains. For these target proteins, unlike annotations from other methods, SWORD partitioning finds the number of domains that most facilitates structure prediction (Fig. 3, F to J). Thus, we can speculate that a fold library derived from SWORD domain assignments would likely have helped the prediction of these protein structures by containing template domains more relevant for fold recognition than those from other databases.
In detail, the Rieske ferredoxin (target T0391) is annotated as a one-domain protein by PDP, CATH, and Pfam (no data in SCOP), although this target is an obvious two–evolutionary domain protein, constituted of a rubredoxin-like domain (residues 55 to 117) inserted into a six-strand β barrel (residues 14 to 54 and 118 to 154), as shown by N. Grishin in his analysis of the CASP8 results (15) and as can be found in his ECOD database. This two-domain partitioning corresponds to what SWORD proposes as the best alternative decomposition (Fig. 3F). Because the modeling difficulty goes from “hard” to “medium,” depending on whether the target is treated as a one- or a two-domain protein, respectively, it can be concluded that predicting the structure would have been easier if it were based on SWORD, which can successfully identify the two domains, rather than on CATH or Pfam domain assignments. Also worth mentioning is the particular case of the Mu-like prophage tail protein gpP from Neisseria meningitidis (target T0424), in which structural modeling is favored when considering the protein as a whole (that is, one-domain assignment) because of the existence of close homologs. For this protein, the best decomposition identified by SWORD is similar to that of SCOP and divides the structure into two domains (Fig. 3H). Our algorithm also finds the same three-domain organization as CATH and the four-domain assignment made by N. Grishin (15)—and therefore annotated as a manual assignment in ECOD. Although SWORD does not find the domain assignment that is most favorable for predicting this target structure, the multiplicity of partitioning solutions it provides remarkably reflects the complex evolutionary history of this protein. The comparison between the two- and three-domain decompositions shows that the two β-strand domains result from a duplication event, which was followed by the insertion of a third domain. Then, by comparing the three- and four-domain assignments, we can deduce that one of the duplicated β-strand domains has undergone another insertion event of a 68-residue domain.
Our partitioning algorithm is also helpful in understanding the molecular mechanisms underlying protein functions. For example, although the papain protease [Protein Data Bank (PDB): 9pap] is annotated as a one-domain enzyme in CATH, SCOP, and ECOD databases, the best alternative decomposition provided by SWORD successfully identifies the two structural domains that form the cleft in which the active site is located (Fig. 3K) (16). This also goes for the cystic fibrosis transmembrane conductance regulator (CFTR) (PDB: 2bbo), for which the most thorough partitioning computed by SWORD corresponds to the three functional subdomains identified by the authors of the structure (Fig. 3L, left) (17), whereas CATH and ECOD assign a unique structural domain. Another example is the structure of the high-fidelity DNA polymerase I (PDB: 1u4bA), for which only SWORD properly isolates the catalytic domain through a decomposition into six domains (Fig. 3M), our method being relevant regarding the molecular mechanisms of this enzyme. Finally, the complex structure of the myosin V molecular motor (PDB: 1oe9A), which SWORD optimally partitions into the five subcomponents delimited by a study of allosteric motions, is worth mentioning (Fig. 3N) (18), whereas other methods fail at identifying these dynamic/functional domains. In this case, the function of the protein is related to its structure and internal dynamics. These structural motions locally modify the geometry of the protein so that the resulting domain assignment can vary depending on the conformational state of the protein, hence the importance of providing multiple possibilities of protein structure partitioning. By doing so, SWORD can delimit protein domains that are compatible with dynamic experimental data while still providing alternative decompositions that agree with those based on structural and functional criteria.
The above examples of the DNA polymerase I and CASP8 targets are actually cases where SWORD identifies structural domains that correspond to mobile evolutionary units. These domain decompositions agree with the manual annotations from the MultiDom database (19), in which structural domains are assigned mainly on the basis of evolutionary information. This is also true for the optimal decomposition that SWORD provides for the CFTR structure, which fits with the two evolutionary domains annotated in Pfam 2bbo (Fig. 3L, right). Our method can also compute alternative partitioning solutions that have the same number of domains but different boundaries. Because of this capacity, SWORD can identify the two evolutionary domains of the kinase haspin (PDB: 3dlz), in agreement with the different but equally plausible annotations of Pfam and CATH/MultiDom (Fig. 3O). Finally, the advantage of using SWORD is also well illustrated by the structural partitioning of DNA polymerase IV (PDB: 1jx4). Two structural domains have been assigned in SCOP, and this protein was annotated in CATH 3.4 as containing three domains, whereas both the version 3.5 and the current version 4.0 identify four functional domains, as does ECOD. Instead of considering these three assignments as mutually exclusive, all should be retained because these three-, four-, and two-domain assignments are actually valid in terms of evolution, function, and geometry, respectively. This is what SWORD does by providing all these decompositions of the structure (Fig. 3P). Thus, we can see that the use of the evolutionarily preserved PU substructures to delimit protein domains can make SWORD assignments consistent with both geometrical and evolutionary definitions of domains.
The intermediate size and compactness of PUs, their content in regular secondary structure, and their conservation throughout evolution suggest an important role of these substructures in protein folding. Thus, it is certainly no coincidence that SWORD succeeds in demarcating the folding nucleus of the subtilisin protease (PDB: 1spb) (20), whereas other methods do not distinguish any domain from this protein structure (Fig. 3Q, folding nucleus in purple). This is also the case with the partitioning of the villin headpiece structure (PDB: 1yu5), for which the sole alternative decomposition provided by SWORD precisely delimits the ultrafast folding subdomain of this protein (21), whereas other methods do not isolate any domain (Fig. 3R, folding subdomain in red). Similarly, the best alternative assignment provided by SWORD for cytochrome c (PDB: 1ycc), or for RNase H (PDB: 2rn2), isolates a subdomain that corresponds to a stable autonomous folding region (Fig. 3, S, in orange, and T, in black) (22, 23), whereas CATH, SCOP, ECOD, and Pfam consider it as a one-domain protein. For thermolysin (PDB: 1hyt), SCOP and ECOD identify only one domain, whereas CATH and Pfam assign two functional and evolutionary domains, respectively, as does SWORD. However, our method proposes two different boundaries that are both relevant regarding protein folding experiments. One decomposition isolates an autonomous folding unit (Fig. 3U, in salmon) (24), whereas its alternative delimits a domain that has been shown to be able to fold partially (Fig. 3U, in green cyan) (25). Another example is α-lactalbumin (PDB: 1a4v), which is annotated as a one-domain protein in CATH, SCOP, ECOD, and Pfam, whereas our algorithm isolates an α-helical domain in its best alternative assignment (Fig. 3V, left, in black) and a β-strand domain (in red, residues 38 to 103) that can independently fold while keeping its ability to bind calcium (26). A shorter delineation of this latter β-strand domain (residues 38 to 72) that can still fold partially as a molten globule (27) is also identified by SWORD in its second best alternative assignment (Fig. 3V, right). A homolog of α-lactalbumin is the hen egg-white lysozyme (PDB: 3lzt), for which the same main folding domains are isolated by SWORD (Fig. 3W): the α-helical domain (residues 1 to 39 and 74 to 129), which forms early during the folding of the lysozyme, and the β-strand folding domain (residues 40 to 73) (28, 29). As for α-lactalbumin, CATH, SCOP, ECOD, and Pfam annotate this lysozyme as a one-domain protein. Finally, the same situation is once again observed with the Trp repressor (PDB: 1jhgA), which is considered by CATH, SCOP, ECOD, and Pfam as made of one functional or evolutionary domain. Although the best partitioning solution provided by SWORD for this structure is also a one-domain assignment, our algorithm produces two alternative decompositions (Fig. 3X) identifying two domains (in blue and magenta), which have been shown to fold, or partially fold, in an independent manner (30, 31).
Measure and source of structural ambiguity
Given the above results showing the success of our method at finding multiple domain decompositions, one can expect that the number and quality of the alternative partitioning solutions produced by SWORD may provide a relevant measure of ambiguity in protein structures. This is why we have developed an ambiguity index (A-index; see Materials and Methods) and compared its average value and distribution for the Consensus, Dissensus, and Strong-dissensus data sets. Protein size is an obvious source of ambiguity because a larger structure naturally means more possible decompositions. Therefore, to avoid this size-related bias, the A-index comparisons have been (i) based on structures annotated in the SCOP database as having two domains (Fig. 4A) and (ii) performed according to different categories of chain lengths (Fig. 4B). When comparing the A-index means, we can see that the A-index is significantly higher for proteins of the Dissensus and Strong-dissensus data sets than for those of the Consensus data set (Fig. 4A). Moreover, proteins of the Strong-dissensus data set are significantly more ambiguous than those of the Dissensus data set. When comparing the A-index distributions, we can also observe that the A-indexes are significantly higher in the Dissensus than in the Consensus data set for each size category (Fig. 4B). For both the Dissensus and Consensus data sets, we can see on the bar plots that the A-index gradually increases with the chain length, which was expected: the longer the protein chain, the more complex the structure can be. These differences in the mean and distribution of the A-index show that it is a pertinent measure of structural ambiguity as we define it, that is, a quantifiable property that is positively related to the number of valid domain decompositions of the protein structure.
Fig. 4. Assessement of the structural ambiguity measure.
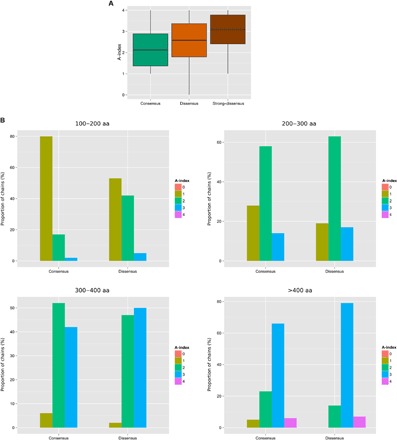
(A) Box plots of the A-indexes calculated by SWORD for the two-domain protein structures (SCOP boundaries) of the Consensus, Dissensus, and Strong-dissensus data sets (statistics in table S3). (B) A-index distributions of the structures from the Consensus and Dissensus data sets for four categories of chain lengths from 100 to 200 amino acids to >400 amino acids (statistics in table S4).
Despite the statistical significance of these results, one can observe that a fraction of the structures from the Dissensus set show a lower A-index than those from the Consensus set (Fig. 4A). Although seemingly contradictory, the large majority of these cases from the Dissensus set actually correspond to one-domain assignments from either CATH or SCOP. When considering the structures from the Dissensus set (n = 1025) that have an A-index of 0 or 1 (that is, 136 structures detected as unambiguous by SWORD), we observe that 93.4% (127 of 136) of them are annotated in CATH or SCOP as being made of only one domain. These disagreements on annotations that involve one-domain assignments are special cases of discrepancies because a “decomposition into one domain” could actually correspond to an absence of analysis (except for small proteins). Therefore, these structures that have a relatively low A-index, although belonging to the Dissensus set, may turn out to be “false discrepancies” if additional data confirm their organization into more than one domain. In the Strong-consensus data set (n = 98), only two structures have an A-index of ≤1, and both are annotated as one-domain proteins in SCOP. Thus, all these results show the efficiency of SWORD at identifying unambiguous protein structures.
Reciprocally, a fraction of the structures from the Consensus set show a higher A-index than those from the Dissensus set (Fig. 4A). The fact that a protein is similarly annotated in CATH, SCOP, and ECOD is actually not incompatible with having an ambiguous structure. For example, when the decomposition into domains only relies on the 3D structure itself (either because there are no other data available or because the method focuses on structural features to delimit domains), the use of the sole geometric criterion is likely to lead different algorithms or experts to the same annotation. However, when functional, evolutionary, folding, or dynamic information is used, databases would rather tend to disagree, by selecting only one out of several valid decompositions, according to the type of information they favor. Thus, the protein structures of the Consensus set that have a high A-index should fall into two categories: (i) those for which the lack of data has made all databases converge toward the same domain assignment or (ii) those for which several valid possibilities of partitioning have been found throughout their different studies, but only one has been arbitrarily conserved in CATH, SCOP, and ECOD.
Although it is difficult to identify structures that belong to the first category, examples of proteins with alternative annotations to those of CATH, SCOP, and ECOD can be easily found among the most ambiguous structures of the Consensus set (n = 3523): 34 proteins with an A-index of 4 (table S2). For these protein structures, the ambiguity (that is, high A-index) finds its source in the potential number (actually >1) of equally valid domain assignments. Thus, although the elongation factor Tu from Thermus thermophilus (EF-Tu; PDB: 2c78A) is consensually decomposed into three domains, based on spatial separation, it can be alternatively annotated as a two-domain structure (Fig. 5A) when using either the secondary structure content or the domain motion, which is consequent to the hydrolysis of GTP, as a criterion (see http://pdb101.rcsb.org/motm/81). A second example is the structure of the interferon-γ–induced guanylate-binding protein 1 (GBP1; PDB: 1f5nA), which is a two-domain protein in CATH, SCOP, and ECOD, but could be decomposed into three or four domains (Fig. 5B), when considering its conformational change induced by the binding of a 4-azapodophyllotoxin derivative (32). A third example is the clathrin heavy chain proximal leg segment from Bos taurus (PDB: 1b89A), which is a large protein chain annotated as a one-domain protein in the Consensus, whereas it is made of three clathrin domains according to Pfam (Fig. 5C). The opposite case can be illustrated by the rabbit skeletal muscle calsequestrin (PDB: 1a8yA), which is decomposed into three thioredoxin-like domains by CATH, SCOP, and ECOD, whereas Pfam identifies the whole chain as one calsequestrin domain (Fig. 5D). For the 30 other structures of table S2, multiple decompositions can also be found when using evolutionary, functional, or folding criteria.
Fig. 5. Revealing the structural ambiguity with the A-index.
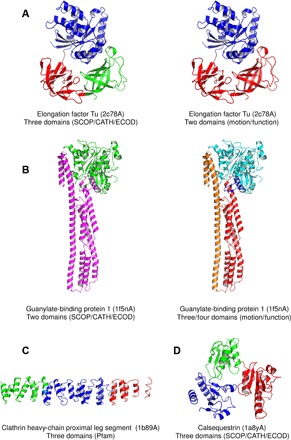
(A to D) Examples of domain assignments for protein structures from the Consensus set that are detected as ambiguous by SWORD.
The examples presented in this article show that ambiguous cases of domain assignment occur when the formation of domains in a protein structure has been driven by different and diverging forces. Thus, the evolutionary domains of the Mu-like prophage tail protein gpP (A-index = 3; Fig. 3H) could not be identified by a method that focuses on the protein function. Similarly, the folding domains of the thermolysin structure (A-index = 3; Fig. 3U) could not be identified if functional and evolutionary criteria are used. Finally, the high ambiguity measured by SWORD for calsequestrin (A-index = 4; Fig. 5D) may result from the fact that its structure is partly made of random coil and has its secondary structure content modified upon the binding of Ca2+ (33), which would blur or multiply the possibilities of partitioning. These structural particularities and the numerous domain decompositions found by SWORD must be related to the multifunctional nature of calsequestrin, which can bind Ca2+ and K+, polymerize, and directly regulate other protein activities via protein-protein interactions (34) and has a reported kinase activity (35).
Conclusions and perspectives
Here, we show that our conceptually new multipartitioning approach can tackle the analytical bias caused by the ambiguity of protein structure and can therefore be helpful for any field of molecular biology involving protein domain assignment. We did not mention the DHcL (Domain Hierarchy and closed Loops) method (36, 37), although it can compute alternative partitionings because of its low performance reported in an independent benchmarking (38). We did not also mention the DOMIRE web server (39) because its ability to produce multiple structural decompositions is more a consequence of using multiple algorithms rather than its purpose.
Finally, we have used our partitioning algorithm as a basis for developing an original measure of ambiguity in protein structures. We have shown that our A-index is sensitive to ambiguous cases of structural domain assignment. Some structures show a high A-index despite having the same annotation in CATH, SCOP, and ECOD. These cases may fall into the category of proteins that have alternative, yet undiscovered, biological functions. Thus, future work will investigate how measuring the architectural complexity of protein structures can be used to detect functionally complex (that is, multifunctional) proteins.
MATERIALS AND METHODS
Benchmark data sets
Jones set
The well-known Jones domain data set (40) contains 55 protein chains, for which domain assignments had been reported in the literature by the authors of the structures. This data set is widely used as a benchmark for domain assignment methods.
Islam90 set
The Islam2363 domain data set (41) contains 2363 manual protein domain assignments and has previously served as a benchmark (5). Here, we used Islam90, a subset of 90 annotations, with a maximum sequence identity of 30% (to avoid the bias of overrepresented protein families) and excluding theoretical models.
CATH and SCOP set
The CATH and SCOP (CS) domain data set contains 4660 proteins that share ≤30% sequence identity and are annotated in both SCOP 1.75 and CATH 3.4 protein domain databases. Here, for a given protein structure, we considered domain annotations as similar when having an equal number of identified domains and ≥85% overlap between domain boundaries (9). Thus, we derived two benchmark data sets from the CS set: a Consensus set of 3635 proteins and a Dissensus set of 1025 proteins, for which database annotations (CATH and SCOP) were similar and different, respectively. The number of proteins in the Consensus set was further reduced to 3523 by selecting the annotations that are similar in CATH, SCOP, and ECOD. Finally, we derived from the CS set a third data set, named the Strong-dissensus set, which contains 98 proteins, for which annotations from CATH, SCOP, and ECOD are all different.
Broad-consensus set
The Broad-consensus data set contains the 333 proteins of the CS set, for which CATH, SCOP, and Islam annotations were similar. Our decision model was optimized using this set of structural domain annotations. The number of proteins in the Broad-consensus set was further reduced to 329 by selecting the annotations that are similar in CATH, SCOP, Islam, and ECOD.
Domain assignment using PUs
Since the first automated method for structural domain assignment (which was published only 1 year after Wetlaufer’s definition of protein domains and was based on Cα-Cα distance maps) (42), a wide variety of algorithms have been developed, continuously improving the quality of automatic annotations. Different partitioning strategies have been used. Thus, algorithmic methods can delimit domains by iteratively segmenting the entire protein structure (“top-down” strategy) and/or by defining and clustering smaller substructures (“bottom-up” strategy). Hence, domain assignment is achieved using different representations of the protein structure, such as maps, graphs (43–45), or Gaussian network models (46). Although maximizing the ratio of intradomain contacts over domain-domain interface is the most popular approach (47–50), other criteria have also been used successfully, such as energy (51) or secondary structure (52).
The SWORD method is a top-down/bottom-up approach in which PUs are generated using Protein Peeling (53) and then gradually merged while testing several possible 30-residue-long domain delineations. Thus, each PU merging event defines a domain partitioning level, which is evaluated using two criteria: the separation (σ) and the compactness (κ), inspired by the PDP (47) and PUU [Parser for protein Unfolding Units (48)] methods, respectively. The separation criterion σi,j measures the independence between two PUs, i and j, and can be written as follows
where pi,j is the contact probability (11) between PUs i and j (pi,j is a real number between 0 and 1; see the Supplementary Materials), Si and Sj are the amino acid lengths of PUs i and j, α = 0.43 (47), and pi+j is the contact probability of the whole domain formed by merging the two PUs. Thus, a high value of σi,j indicates a high number of contacts between PUs i and j, meaning that these PUs are good candidates for being merged into one protein domain; otherwise, a low σi,j implies two independent PUs that should remain separated between two protein domains. The merging of PUs i and j is also evaluated using the κi,j compactness criterion, which measures the contact density of the resulting protein domain, and can be written as follows
where pa,b is the contact probability between residues a and b of the resulting protein domain, and Si and Sj are the amino acid lengths of PUs i and j. Thus, a high value of κi,j indicates a high compactness of the protein domain, meaning a favorable merging event of PUs i and j. Finally, the choice of using PUs as building blocks to reconstruct protein domains is justified by their potential content in evolutionary information. This assumption is supported by a recent study, which demonstrates that alternative splicing events tend to spare PUs while modifying the overall protein structure (12). These substructures are also relevant regarding the protein folding because they have been successfully used to identify early folded elements (11). The relevance of PUs was also confirmed by their very recent use in the design of small HIV-1 antigens (54).
Parameter optimization
For each protein structure of the training set, after each level of PU merging, the different possibilities of domain delineations are sorted by their compactness, the best delineation being the one with the highest κ compactness value. Then, when matching with the domain delineation reported in the literature, each of these best domain delineations, as well as the corresponding undercut delineation (that is, the domain delineation from the previous level), is labeled “correct”; the corresponding delineation of the next level (overcut) is labeled “incorrect.” Thus, following this bimodal classification, quasi-optimal values of σ and κ for separating correct and incorrect delineations were determined using a grid search algorithm. Stable values of the model parameters σ and κ were obtained with 10-fold cross-validation. An illustration of the model is given in fig. S3.
Alternative delineations and measure of ambiguity
At the end of the structure partitioning process, several domain decompositions may fall in the acceptance region because of their σ and κ values, enabling SWORD to provide several domain assignments for a single query structure. In addition, SWORD provides decompositions that are outside the acceptance region but close to the model’s threshold. The best domain assignment is selected among the accepted decompositions of the highest level (that is, those with the highest number of domains) as the one with the highest domain compactness. A qualitative assessment is also provided for each assignment by calculating f(di), where di is the Euclidean distance between the decomposition i and the threshold of the acceptance region and f is a step function used to sort the distances di into five classes. The f function takes five values, from “*” to “*****,” defined on five intervals delimited by the values {−0.15, −0.05, 0.05, 0.15} (rejected decompositions have negative distances). This implies that a quality of “***” corresponds to a decomposition that is close to the model’s threshold, either inside or outside the acceptance region. Finally, we have developed a measure of structural ambiguity, called the A-index, which is similar to the Hirsch index (h-index) used in scientometrics (55), except that it is based on the decomposition quality described above, instead of article citations. Thus, a protein structure with an A-index of 3 has at least three different decompositions, each with a quality of *** or more. Therefore, the A-index is an integer ranging from 0 to 5 (0 being the value attributed to proteins that do not have any partitioning, that is, one-domain assignment). On the web server [where the different decompositions can be visualized with the PV molecular viewer (56)], the structural ambiguity is represented by n “+” symbols, where n is the A-index.
Performance evaluation
The accuracy of SWORD in identifying protein domains was evaluated using the benchmark data sets defined above and compared with that of three widely used methods: PDP (47), DomainParser (43), and DDomain (50). We used DDomain trained on the “AUTHORS” data set of annotations [see the study by Zhou et al. (50)] because it gave the best results on our benchmark. Nevertheless, results for the CATH- and SCOP-optimized versions are also provided in the Supplementary Materials. For the evaluation of all methods, domain decompositions were considered similar when boundary overlap was ≥85% (9). In addition, the accuracy in finding the correct number of domains regardless of the boundary overlap was also evaluated. The notions of accuracy and correctness must be considered in light of the fact that CATH, SCOP, and ECOD annotations do not represent the absolute truth. Here, the purpose was to see whether SWORD decompositions agreed with domain annotations made by human experts. Finally, to determine which algorithm is the most accurate, the distributions of delineations (“correct” and “incorrect”) were compared using the Wilcoxon signed-rank test, with an α error of 0.05. Finally, the capacity of SWORD to identify alternative domain delineations was evaluated using (i) the Dissensus benchmark data set of CATH and SCOP annotation discrepancies and (ii) the Strong-dissensus set of CATH, SCOP, and ECOD discrepancies.
Supplementary Material
Acknowledgments
We thank the anonymous reviewers for their helpful comments. Funding: This work was supported by grants from the Ministry of Research (France), the University Paris Diderot, Sorbonne Paris Cité (France), the National Institute for Blood Transfusion (INTS, France), the National Institute for Health and Medical Research (INSERM, France), and the Laboratory of Excellence (LabEx) GR-Ex. The LabEx GR-Ex, reference ANR-11-LABX-0051, is funded by the program “Investissements d'avenir” of the French National Research Agency, reference ANR-11-IDEX-0005-02. Author contributions: G.P. and R.C. implemented the method. G.P., Y.G., and J.-C.G. analyzed the results. G.P. and J.-C.G. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors and are available at www.dsimb.inserm.fr/sword/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1600552/DC1
fig. S1. Monopartitioning accuracies of SWORD, PDP, DomainParser, and DDomain.
fig. S2. Rate of agreement between SWORD and CATH, SCOP, or ECOD annotations, depending on the number of assignments provided, for the structures of the Strong-dissensus data set.
fig. S3. Representation of the domain assignment model.
fig. S4. Domain assignments of the 1A8YA protein structure, as displayed by SWORD.
table S1. Rate of agreement between SWORD and annotations from the five data sets of structural domains, depending on the number of assignments provided.
table S2. The 34 most ambiguous protein structures of the Consensus set.
table S3. The P values of the Mann-Whitney-Wilcoxon tests comparing the A-index means of the Consensus, Dissensus, and Strong-dissensus sets.
table S4. The P values of the Mann-Whitney-Wilcoxon and Pearson’s χ2 tests comparing the A-index distributions of the Consensus and Dissensus sets.
equation S1. The contact probability between two PUs.
REFERENCES AND NOTES
- 1.Wetlaufer D. B., Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. U.S.A. 70, 697–701 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janin J., Wodak S. J., Structural domains in proteins and their role in the dynamics of protein function. Prog. Biophys. Mol. Biol. 42, 21–78 (1983). [DOI] [PubMed] [Google Scholar]
- 3.Csaba G., Birzele F., Zimmer R., Systematic comparison of SCOP and CATH: A new gold standard for protein structure analysis. BMC Struct. Biol. 9, 23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.S. Veretnik, J. Gu, S. Wodak, Identifying structural domains in proteins, in Structural Bioinformatics, J. Gu, P. E. Bourne, Eds. (Wiley-Blackwell, ed. 2, 2009), pp. 485–513. [Google Scholar]
- 5.Holland T. A., Veretnik S., Shindyalov I. N., Bourne P. E., Partitioning protein structures into domains: Why is it so difficult? J. Mol. Biol. 361, 562–590 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Kelley L. A., Sternberg M. J. E., Partial protein domains: Evolutionary insights and bioinformatics challenges. Genome Biol. 16, 100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murzin A. G., Brenner S. E., Hubbard T., Chothia C., SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Cheng H., Schaeffer R. D., Liao Y., Kinch L. N., Pei J., Shi S., Kim B.-H., Grishin N. V., ECOD: An evolutionary classification of protein domains. PLOS Comput. Biol. 10, e1003926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orengo C. A., Michie A. D., Jones S., Jones D. T., Swindells M. B., Thornton J. M., CATH—A hierarchic classification of protein domain structures. Structure 5, 1093–1109 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Kabsch W., Mannherz H. G., Suck D., Pai E. F., Holmes K. C., Atomic structure of the actin: DNase I complex. Nature 347, 37–44 (1990). [DOI] [PubMed] [Google Scholar]
- 11.Gelly J.-C., de Brevern A. G., Hazout S., ‘Protein Peeling’: An approach for splitting a 3D protein structure into compact fragments. Bioinformatics 22, 129–133 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Gelly J.-C., Lin H.-Y., de Brevern A. G., Chuang T.-J., Chen F.-C., Selective constraint on human pre-mRNA splicing by protein structural properties. Genome Biol. Evol. 4, 966–975 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D. T. Jones, C. Hadley, Threading methods for protein structure prediction, in Bioinformatics, Sequence, Structure and Databanks, D. Higgins, W. Taylor, Eds. (Oxford Univ. Press, 2000), pp. 1–13. [Google Scholar]
- 14.Fischer D., Elofsson A., Rice D., Eisenberg D., Assessing the performance of fold recognition methods by means of a comprehensive benchmark. Pac. Symp. Biocomput. 1996, 300–318 (1996). [PubMed] [Google Scholar]
- 15.Shi S., Pei J., Sadreyev R. I., Kinch L. N., Majumdar I., Tong J., Cheng H., Kim B.-H., Grishin N. V., Analysis of CASP8 targets, predictions and assessment methods. Database 2009, bap003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamphuis I. G., Kalk K. H., Swarte M. B. A., Drenth J., Structure of papain refined at 1.65 Å resolution. J. Mol. Biol. 179, 233–256 (1984). [DOI] [PubMed] [Google Scholar]
- 17.Lewis H. A., Wang C., Zhao X., Hamuro Y., Conners K., Kearins M. C., Lu F., Sauder J. M., Molnar K. S., Coales S. J., Maloney P. C., Guggino W. B., Wetmore D. R., Weber P. C., Hunt J. F., Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 396, 406–430 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Cecchini M., Houdusse A., Karplus M., Allosteric communication in myosin V: From small conformational changes to large directed movements. PLOS Comput. Biol. 4, e1000129 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majumdar I., Kinch L. N., Grishin N. V., A database of domain definitions for proteins with complex interdomain geometry. PLOS ONE 4, e5084 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fersht A. R., Nucleation mechanisms in protein folding. Curr. Opin. Struct. Biol. 7, 3–9 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Chiu T. K., Kubelka J., Herbst-Irmer R., Eaton W. A., Hofrichter J., Davies D. R., High-resolution x-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc. Natl. Acad. Sci. U.S.A. 102, 7517–7522 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chamberlain A. K., Fischer K. F., Reardon D., Handel T. M., Marqusee S., Folding of an isolated ribonuclease H core fragment. Protein Sci. 8, 2251–2257 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu L. C., Laub P. B., Elove G. A., Carey J., Roder H., A noncovalent peptide complex as a model for an early folding intermediate of cytochrome c. Biochemistry 32, 10271–10276 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Rico M., Jimenez M. A., Gonzalez C., De Filippis V., Fontana A., NMR solution structure of the C-terminal fragment 255–316 of thermolysin: A dimer formed by subunits having the native structure. Biochemistry 33, 14834–14847 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Conejero-Lara F., González C., Jiménez M. A., Padmanabhan S., Mateo P. L., Rico M., NMR solution structure of the 205–316 C-terminal fragment of thermolysin. An example of dimerization coupled to partial unfolding. Biochemistry 36, 11975–11983 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Hendrix T. M., Griko Y., Privalov P., Energetics of structural domains in α-lactalbumin. Protein Sci. 5, 923–931 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng Z. Y., Kim P. S., A protein dissection study of a molten globule. Biochemistry 33, 2136–2141 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Miranker A., Radford S. E., Karplus M., Dobson C. M., Demonstration by NMR of folding domains in lysozyme. Nature 349, 633–636 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Radford S. E., Dobson C. M., Evans P. A., The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature 358, 302–307 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Wallqvist A., Lavoie T. A., Chanatry J. A., Covell D. G., Carey J., Cooperative folding units of Escherichia coli tryptophan repressor. Biophys. J. 77, 1619–1626 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tasayco M. L., Carey J., Ordered self-assembly of polypeptide fragments to form nativelike dimeric trp repressor. Science 255, 594–597 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Andreoli M., Persico M., Kumar A., Orteca N., Kumar V., Pepe A., Mahalingam S., Alegria A. E., Petrella L., Sevciunaite L., Camperchioli A., Mariani M., Di Dato A., Novellino E., Scambia G., Malhotra S. V., Ferlini C., Fattorusso C., Identification of the first inhibitor of the GBP1:PIM1 interaction. Implications for the development of a new class of anticancer agents against paclitaxel resistant cancer cells. J. Med. Chem. 57, 7916–7932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slupsky J. R., Ohnishi M., Carpenter M. R., Reithmeier R. A. F., Characterization of cardiac calsequestrin. Biochemistry 26, 6539–6544 (1987). [DOI] [PubMed] [Google Scholar]
- 34.Valle G., Galla D., Nori A., Priori S. G., Gyorke S., de Filippis V., Volpe P., Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem. J. 413, 291–303 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Beard N. A., Laver D. R., Dulhunty A. F., Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 85, 33–69 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Berezovsky I. N., Discrete structure of van der Waals domains in globular proteins. Protein Eng. 16, 161–167 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Koczyk G., Berezovsky I. N., Domain Hierarchy and closed Loops (DHcL): A server for exploring hierarchy of protein domain structure. Nucleic Acids Res. 36, W239–W245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alden K., Veretnik S., Bourne P. E., dConsensus: A tool for displaying domain assignments by multiple structure-based algorithms and for construction of a consensus assignment. BMC Bioinformatics 11, 310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samson F., Shrager R., Tai C.-H., Sam V., Lee B., Munson P. J., Gibrat J.-F., Garnier J., DOMIRE: A web server for identifying structural domains and their neighbors in proteins. Bioinformatics 28, 1040–1041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones S., Stewart M., Michie A., Swindells M. B., Orengo C., Thornton J. M., Domain assignment for protein structures using a consensus approach: Characterization and analysis. Protein Sci. 7, 233–242 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Islam S. A., Luo J., Sternberg M. J. E., Identification and analysis of domains in proteins. Protein Eng. 8, 513–526 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Rossman M. G., Liljas A., Recognition of structural domains in globular proteins. J. Mol. Biol. 85, 177–181 (1974). [DOI] [PubMed] [Google Scholar]
- 43.Guo J.-t., Xu D., Kim D., Xu Y., Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res. 31, 944–952 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor T. J., Vaisman I. I., Graph theoretic properties of networks formed by the Delaunay tessellation of protein structures. Phys. Rev. E 73, 041925 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Wernisch L., Hunting M., Wodak S. J., Identification of structural domains in proteins by a graph heuristic. Proteins 35, 338–352 (1999). [PubMed] [Google Scholar]
- 46.Kundu S., Sorensen D. C., Phillips G. N. Jr, Automatic domain decomposition of proteins by a Gaussian Network Model. Proteins 57, 725–733 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Alexandrov N., Shindyalov I., PDP: Protein domain parser. Bioinformatics 19, 429–430 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Holm L., Sander C., Parser for protein folding units. Proteins 19, 256–268 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Siddiqui A. S., Barton G. J., Continuous and discontinuous domains: An algorithm for the automatic generation of reliable protein domain definitions. Protein Sci. 4, 872–884 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou H., Xue B., Zhou Y., DDOMAIN: Dividing structures into domains using a normalized domain–domain interaction profile. Protein Sci. 16, 947–955 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porter L. L., Rose G. D., A thermodynamic definition of protein domains. Proc. Natl. Acad. Sci. U.S.A. 109, 9420–9425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sowdhamini R., Blundell T. L., An automatic method involving cluster analysis of secondary structures for the identification of domains in proteins. Protein Sci. 4, 506–520 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gelly J.-C., de Brevern A. G., Protein Peeling 3D: New tools for analyzing protein structures. Bioinformatics 27, 132–133 (2011). [DOI] [PubMed] [Google Scholar]
- 54.D. Verma, J. Lai, E. Brown, J. Suarez, M. Ackerman, C. Bailey-Kellogg, Designing small HIV-1 antigens by protein peeling and rewiring techniques, Proceedings of the 3DSIG Structural Bioinformatics and Computational Biophysics, Orlando, FL, 8 to 9 July 2016. [Google Scholar]
- 55.Hirsch J. E., An index to quantify an individual’s scientific research output. Proc. Natl. Acad. Sci. U.S.A. 102, 16569–16572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.M. Biasini, PV—WebGL-based protein viewer (Zenodo, 2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1600552/DC1
fig. S1. Monopartitioning accuracies of SWORD, PDP, DomainParser, and DDomain.
fig. S2. Rate of agreement between SWORD and CATH, SCOP, or ECOD annotations, depending on the number of assignments provided, for the structures of the Strong-dissensus data set.
fig. S3. Representation of the domain assignment model.
fig. S4. Domain assignments of the 1A8YA protein structure, as displayed by SWORD.
table S1. Rate of agreement between SWORD and annotations from the five data sets of structural domains, depending on the number of assignments provided.
table S2. The 34 most ambiguous protein structures of the Consensus set.
table S3. The P values of the Mann-Whitney-Wilcoxon tests comparing the A-index means of the Consensus, Dissensus, and Strong-dissensus sets.
table S4. The P values of the Mann-Whitney-Wilcoxon and Pearson’s χ2 tests comparing the A-index distributions of the Consensus and Dissensus sets.
equation S1. The contact probability between two PUs.