

To the Editor
Atopic dermatitis (AD) is the most common inflammatory skin disease in the general population. The disease prevalence is associated with age. AD often starts in early childhood, affecting 15-30% of children.1 However, up to 70% of AD children show clearing of the disease or a spontaneous remission around puberty even in patients with filaggrin mutations. AD can persist or start in adulthood. However, the prevalence of AD in adults is only approximately 3%. While multiple factors contribute to AD pathogenesis, skin microorganisms are critical in driving disease development. Shifts in the skin microbiome were observed during disease progression of pediatric AD.2 In adult AD, however, the skin microbiome is not well characterized. Comparisons of the skin microbiome among age groups of AD patients and healthy controls will help delineate age differences in the microbial pathogenesis of this disease.
In this study, we recruited 128 AD patients including 59 young children (age 2-12), 13 teenagers (age 13-17), and 56 adults (age 18-62). A cohort of 68 age-matched non-atopic healthy controls (age 3-59) was also enrolled, which includes 13 young children, 10 teenagers, and 45 adults (Supplementary Table E1). We collected two swab samples from the volar forearm of each AD patient, one from lesional skin and one from adjacent normal-appearing non-lesional skin. One swab sample of the volar forearm was collected from each healthy subject. In total 324 samples were analyzed using 16S rRNA gene sequencing. After data cleaning, on average 32,311 paired-end 16S rRNA sequences were obtained for each sample. Our data provided sufficient sequencing depths at the species level as indicated by the rarefaction curve (Supplementary Figure E1). This large study cohort and the high sequencing coverage enabled us to robustly identify differences in the skin microbiome between age groups in healthy individuals and AD patients.
In the skin microbiome of healthy individuals, we identified 7 prevalent bacterial phyla and 20 genera (Figure 1a). Four of the genera (Propionibacterium, Corynebacterium, Staphylococcus, and Streptococcus) were present in ≥ 90% of the healthy subjects with ≥ 5% relative abundance in at least one age group. Among the 15 species identified (Supplementary Figure E2), Propionibacterium acnes, Staphylococcus epidermidis, and Streptococcus mitis/oralis/pneumoniae/sanguinis were the most prevalent species in healthy subjects, accounting for 98.5% of total Propionibacterium species, 46.6% of total Staphylococcus species, and 32.5% of total Streptococcus species in relative abundance.
Figure 1. The microbiome composition in healthy individuals and patients with AD.

A, The skin microbiome composition of the healthy controls is shown at the phylum level (upper panel) and at the genus level (lower panel). Samples were ordered by subject's age at sampling. B, The representative major skin micro-organisms differ with age in healthy skin. C, The microbiome compositions of AD lesional (upper panel) and nonlesional (lower panel) skin are shown at the genus level. The same color scheme is used as in Fig 1, A. Samples collected from the same patients are shown in the same order in both panels (see details in the extended figure legend in this article's Online Repository at www.jacionline.org).
To determine whether the stages of the human physical development have a significant effect on the skin microbiome,3 we compared the microbiome among the age groups. We found that the healthy skin microbiome was significantly more diverse in young children than in adults (alpha diversity, p = 0.01), and was distinct between the two age groups as indicated by beta diversity (ANOSIM, p = 0.009) (Figure 2a). At the genus level, Streptococcus, Granulicatella, Gemella, Rothia, and Haemophilus were more abundant in young children, while Propionibacterium, Corynebacterium, Staphylococcus, Lactobacillus, Finegoldia, and Anaerococcus were more abundant in adults (Supplementary Table E2). At the species level, Streptococcus salivarius/thermophilus/vestibularis was more abundant in young children (p = 0.045) (Supplementary Table E3), while P. acnes and S. epidermidis were more abundant in adults (p = 0.01 and p < 1E-5, respectively) (Figure 1b). S. aureus was detected in 20.6% of the healthy subjects, but had very low relative abundance (<1%) in all age groups. Increased host sebum production and changes in the skin structure at and after puberty may facilitate the colonization and growth of lipophilic bacteria Propionibacterium and Corynebacterium,4 which replace Streptococcus and become dominant in adulthood. Teenagers are in transition from young children to adults in physical development, and their skin microbiome is in transition as well with a higher similarity to adults than young children,3 reflected in the representative microorganisms (Figure 1b) and the overall microbial community structure (Figure 2a). In subsequent analyses we combined teenagers and adults into one group and compared to young children.
Figure 2. The microbiome is different between young children and adults in both health and AD with age-specific co-occurrence of skin microorganisms.
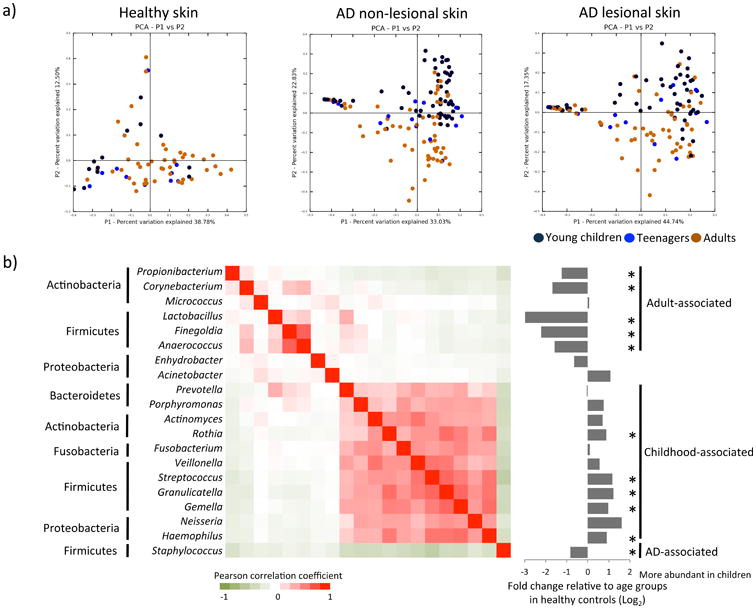
a) A principal coordinate analysis shows significant differences in the microbiome between young children and adults in healthy skin and in AD skin. b) Skin microorganisms were clustered into adult-associated, childhood-associated, and AD-associated groups. Pearson's correlations among the microorganisms are indicated by a gradient from red (co-occurring) to green (co-exclusive), as shown in the heat map. Fold changes of the genera in relative abundance between healthy young children and adults-teenagers are indicated on the right. Genera labeled with asterisks were significantly different in relative abundance between the two age groups of the healthy controls (see details in Supplementary Materials).
Similar to the healthy skin microbiome, in the AD skin microbiome we identified significant differences between young children and adults-teenagers (beta diversity, ANOSIM p < 0.001) (Figure 2a). In AD non-lesional skin, the microbiome diversity was significantly higher in young children than in adults-teenagers (alpha diversity, p = 0.036). The 20 prevalent genera identified in healthy controls were also detected in most of the AD patients (Figure 1c). Eight of the genera were significantly different in relative abundance between young children and adults-teenagers in both lesional and non-lesional skin Supplementary Table E4). The age differences were consistent with those observed in healthy controls.
The microbiome differences in lesional and non-lesional skin were identified previously in AD patients,5 however, it was unclear whether the differences were associated with age. In this study, we found that the microbiome diversity was significantly decreased in lesional skin compared to non-lesional skin in both young children (p < 0.001) and adults-teenagers (p = 0.013). In both age groups, Staphylococcus was significantly more abundant in lesional skin (p ≤ 0.012) and was also more abundant in non-lesional skin compared to healthy skin (p < 0.003), suggesting that non-lesional skin is susceptible to pathogen colonization and is at risk to progress toward diseased state. In contrary, skin commensals Streptococcus and Propionibacterium were observed in lower relative abundance in lesional skin compared to non-lesional skin and in non-lesional skin compared to healthy skin, but their changes were specific to young children and adults-teenagers, respectively.
To better understand the bacterial associations with age in the skin microbiome, we calculated correlations among the 20 prevalent genera based on their relative abundances in AD patients and healthy controls. Three distinct bacterial clusters were identified: adult-associated, childhood-associated, and AD-associated (Figure 2b). Most of the bacterial organisms were clustered in either adult-associated group or in childhood-associated group (Supplementary Table E2 - E5), suggesting that age differences in the skin microbiome may be attributed to shifts in skin microorganisms that are coordinated with each other in abundance during host maturation. AD-associated cluster consisted of only Staphylococcus. We identified multiple species within the major genera. Except for S. aureus, species within each genus were positively correlated in relative abundance (Supplementary Figure E3). S. aureus was inversely correlated with other species, including those from the same genus, suggesting an antagonistic relationship between S. aureus and skin commensals.
We further investigated differences in the gene functions encoded in the genomes of age-specific and AD-associated skin bacteria. We analyzed 46 genomes of 13 major species found in our cohort (Supplementary Methods). 1,910 KEGG orthologous groups (KO genes) were identified, 833 of which were unique to one of the three skin bacterial clusters (Supplementary Figure E4, Table E6). Among the 316 KO genes unique to AD-associated cluster, 63 KO genes were S. aureus-specific, involved in disease-associated pathways including S. aureus infection and bacterial invasion of epithelial cells. Among the 517 KO genes specific to skin commensals, 113 were unique to childhood-associated Streptococcus spp., and 404 were unique to adult-associated P. acnes and Corynebacterium spp.. It has been suggested that Streptococcus can inhibit S. aureus growth by producing hydrogen peroxide,6 while adult-associated commensals Propionibacterium and Corynebacterium harbor genes involved in porphyrin metabolism and can reduce S. aureus infection.7 Additionally, metabolites of adult-associated skin commensals can decrease skin pH and enhance antimicrobial activities, thus suppressing adherence and growth of S. aureus in human keratinocytes.4, 8, 9
In summary, we identified significant differences in the AD skin microbiome between young children and adults-teenagers. Among many other factors that we examined, including host factors, clinical parameters, disease history, and history of concomitant medications (Supplementary Table E7), we found that the microbiome was also correlated with disease severity in AD lesional skin. This is consistent with previous observations that the skin microbiome changes with disease progression.2 While AD pathogenic factors drive the disease development, age-specific skin commensals possess various potentials in defending against pathogens and maintaining skin health at different development stages. Our findings, from a new perspective of the skin microbiome, may partly explain the age differences in AD.
Supplementary Material
The rarefaction curve suggested the sufficient sequencing depths provided in this study. Data are represented as mean ± s.e.m.
In the skin microbiome of the healthy controls, 15 species were present in ≥ 20% of the samples of any age group. Samples were ordered by subject's age.
At the species level, we were able to identify multiple species within the genera of Streptococcus and Staphylococcus. Except for S. aureus, species from the same genus were positively correlated with each other in relative abundance. S. aureus was negatively correlated with other species, including those from the same genus.
The functions encoded in 46 genomes of the representative skin microorganisms were annotated with KO genes. AD-associated (S. aureus and S. epidermidis), childhood-associated (Streptococcus spp.), and adult-associated (P. acnes and Corynebacterium spp.) skin microorganisms have distinct functional profiles. The number of annotated genomes in each species is labeled in brackets after the species name.
Study participants in different age groups.
The taxonomic compositions of the healthy skin microbiome at the genus level
The taxonomic compositions of the healthy skin microbiome at the species level
The taxonomic compositions of the AD skin microbiome at the genus level
The taxonomic compositions of the AD skin microbiome at the species level
The KEGG orthologous groups encoded in skin bacterial species
Correlations between the skin microbiome and host phenotypes
Acknowledgments
This study was funded with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, to support The Atopic Dermatitis Research Network (ADRN) under contract number HHSN272201000020C and HHSN272201000017C. The authors wish to thank Joanne Streib, Gayle Spears, and Caroline Bronchick for their invaluable assistance in the processing and collection of skin swabs as well as recruitment of study subjects into this ADRN protocol. We also thank Keli Artis, Denise Babineau, and Alice Lail from Rho Federal Systems Division Inc. for their help on this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The sequence data from this study have been deposited to NCBI BioProject accession number 268694.
References
- 1.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–94. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 2.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–9. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4:77. doi: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannigan GD, Grice EA. Microbial ecology of the skin in the era of metagenomics and molecular microbiology. Cold Spring Harb Perspect Med. 2013;3:a015362. doi: 10.1101/cshperspect.a015362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flores GE, Seite S, Henley JB, Martin R, Zelenkova H, Aguilar L, et al. Microbiome of affected and unaffected skin of patients with atopic dermatitis before and after emollient treatment. J Drugs Dermatol. 2014;13:1365–72. [PubMed] [Google Scholar]
- 6.Regev-Yochay G, Trzcinski K, Thompson CM, Malley R, Lipsitch M. Interference between Streptococcus pneumoniae and Staphylococcus aureus: In vitro hydrogen peroxide-mediated killing by Streptococcus pneumoniae. J Bacteriol. 2006;188:4996–5001. doi: 10.1128/JB.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orenstein A, Klein D, Kopolovic J, Winkler E, Malik Z, Keller N, et al. The use of porphyrins for eradication of Staphylococcus aureus in burn wound infections. FEMS Immunol Med Microbiol. 1997;19:307–14. doi: 10.1111/j.1574-695X.1997.tb01101.x. [DOI] [PubMed] [Google Scholar]
- 8.Shu M, Wang Y, Yu J, Kuo S, Coda A, Jiang Y, et al. Fermentation of Propionibacterium acnes, a commensal bacterium in the human skin microbiome, as skin probiotics against methicillin-resistant Staphylococcus aureus. PLoS One. 2013;8:e55380. doi: 10.1371/journal.pone.0055380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Dai A, Huang S, Kuo S, Shu M, Tapia CP, et al. Propionic acid and its esterified derivative suppress the growth of methicillin-resistant Staphylococcus aureus USA300. Benef Microbes. 2014;5:161–8. doi: 10.3920/BM2013.0031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The rarefaction curve suggested the sufficient sequencing depths provided in this study. Data are represented as mean ± s.e.m.
In the skin microbiome of the healthy controls, 15 species were present in ≥ 20% of the samples of any age group. Samples were ordered by subject's age.
At the species level, we were able to identify multiple species within the genera of Streptococcus and Staphylococcus. Except for S. aureus, species from the same genus were positively correlated with each other in relative abundance. S. aureus was negatively correlated with other species, including those from the same genus.
The functions encoded in 46 genomes of the representative skin microorganisms were annotated with KO genes. AD-associated (S. aureus and S. epidermidis), childhood-associated (Streptococcus spp.), and adult-associated (P. acnes and Corynebacterium spp.) skin microorganisms have distinct functional profiles. The number of annotated genomes in each species is labeled in brackets after the species name.
Study participants in different age groups.
The taxonomic compositions of the healthy skin microbiome at the genus level
The taxonomic compositions of the healthy skin microbiome at the species level
The taxonomic compositions of the AD skin microbiome at the genus level
The taxonomic compositions of the AD skin microbiome at the species level
The KEGG orthologous groups encoded in skin bacterial species
Correlations between the skin microbiome and host phenotypes