

Abstract
Mussel adhesive moiety, catechol, has been utilized to design a wide variety of biomaterials. However, the biocompatibility and biological responses associated with the byproducts generated during the curing process of catechol has never been characterized. An in situ curable polymer model system, 4-armed polyethylene glycol polymer end-capped with dopamine (PEG-D4), was used to characterize the production of hydrogen peroxide (H2O2) during the oxidative crosslinking of catechol. Although PEG-D4 cured rapidly (under 30 seconds), catechol continues to polymerize over several hours to form a more densely crosslinked network over time. PEG-D4 hydrogels were examined at two different time points; 5 min and 16 hrs after initiation of crosslinking. Catechol in the 5 min-cured PEG-D4 retained the ability to continue to crosslink and generated an order of magnitude higher H2O2 (40 μM) over 6 hrs when compared to 16 hrs-cured samples that ceased to crosslink. H2O2 generated during catechol crosslinking exhibited localized cytotoxicity in culture and upregulated the expression of an antioxidant enzyme, peroxiredoxin 2, in primary dermal and tendon fibroblasts. Subcutaneous implantation study indicated that H2O2 released during oxidative crosslinking of PEG-D4 hydrogel promoted superoxide generation, macrophage recruitment, and M2 macrophage polarization in tissues surrounding the implant. Given the multitude of biological responses associated with H2O2, it is important to monitor and tailor the production of H2O2 generated from catechol-containing biomaterials for a given application.
Keywords: Mussel adhesive protein, oxidative crosslinking, hydrogen peroxide, cytotoxicity, macrophage polarization
Graphical Abstract
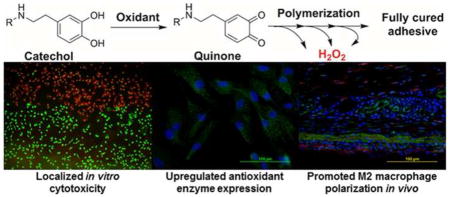
1. Introduction
Marine mussels secrete adhesive proteins that solidify in situ to facilitate the anchoring of these organisms to underwater surfaces (i.e., rocks, boats, other mussels, etc.) [1, 2]. Mussel foot adhesive proteins contain an elevated amount (up to 28 mol%) of a unique catecholic amino acid, 3,4-dihydroxyphenylalanine (DOPA), which imparts these proteins with adhesive characteristics and the ability for crosslink formation [3]. DOPA is a versatile adhesive molecule and its catechol side chain can undergo various reversible and irreversible chemical interactions, making it highly attractive for designing a wide variety of injectable biomaterials and bioadhesives [3–5]. Biomaterials functionalized with DOPA and its derivatives (e.g., dopamine) have demonstrated promising results for suture-less wound closure [6], fetal membrane sealing [7], Achilles tendon repair [8], cell and tissue engineering [9, 10], and localized drug delivery applications [11].
Catechol can be activated by enzymatic (i.e., tyrosinase) or chemical (i.e., periodate, IO4−) oxidants to form the highly reactive quinone, which is a necessary first step in both cohesive (i.e., in situ curing) and interfacial (i.e., adhesion to soft tissues) crosslinking formation [12, 13]. Specifically, the use of IO4−-mediated crosslinking is widely adopted for designing rapidly curable biomaterials [6, 9, 14]. Mussel inspired bioadhesives have exhibited favorable biocompatibility based on in vitro cytotoxicity and in vivo implantation testing [6, 13, 15]. However, these biocompatibility tests were conducted using fully cured adhesives and the potential toxic byproducts generated during the curing process were not examined. To our knowledge, there have been no documented reports that evaluated the biocompatibility of catechol during oxidative crosslinking. For products that polymerize in situ, the International Standard ISO 10993-12 necessitates the assessment of the potential toxicity of the reacting components during their curing process [16]. Therefore, to advance this biomimetic technology for future clinical applications, there is a need to systematically evaluate the biological responses associated with the curing of the catechol moiety.
Our lab recently identified the generation of hydrogen peroxide (H2O2) during autoxidation of catechol as the source of its cytotoxicity in culture [17]. H2O2 is one of the major reactive oxygen species (ROS) released during normal wound healing response and its biological functions is highly dependent on its concentration [18]. At a relatively low concentration (102–103 μM), H2O2 induces vascular endothelial growth factor expression and activates M2 phenotype macrophages differentiation, which promotes angiogenesis and tissue regeneration, respectively [19, 20]. Complete removal of H2O2 from the wound site by antioxidant (i.e., catalase) impairs wound healing [21]. H2O2 also provides a natural defense against bacterial infection [22]. On the other hand, elevated levels of H2O2 (≥ 105 μM) destroys healthy tissues, resulting in the formation of chronic wounds and promote tumor initiation [18, 21]. Besides the biological responses associated with H2O2, H2O2 is a mild oxidant which can oxidize the catechol moiety [23]. H2O2 has also been widely used as an electron acceptor in enzyme (e.g. horseradish peroxidase) mediated crosslinking of phenol-modified polymers and the concentration of H2O2 was correlated to changes in the crosslinking density of the hydrogel network [24, 25]. Trapping the oxidant within a polymer network can potentially affect the mechanical and swelling properties of catechol-containing adhesive. Given the vital roles that H2O2 plays in wound healing and oxidative crosslinking, it is necessary to determine the production of H2O2 during the oxidative crosslinking process of catechol and correlate its release with its biological responses and its effect on the mechanical properties of the catechol-containing adhesive.
Here, we used a model polymer, 4-armed polyethylene glycol end capped with dopamine (PEG-D4, Figure S1), to investigate the production of H2O2 during IO4−-induced crosslinking of catechol. The effect of the H2O2 generation on the crosslinking density of the adhesive, in vitro cytotoxicity, and the upregulation of the antioxidant enzyme, peroxiredoxin (Prx), in primary fibroblasts (rat dermal and tendon fibroblasts) were determined. Finally, PEG-D4 was subcutaneously implanted in rats and the generation of superoxide (O2•−), recruitment of macrophages, and M2 macrophage polarization in the surrounding tissues were evaluated.
2. Materials and methods
2.1 Materials
NaIO4 was purchased from Acros Organics (Fair Lawn, New Jersey). Polytetrafluoroethylene (PTFE) sheet was purchased from McMaster (Chicago, IL). Dulbecco’s Modified Eagle Medium (DMEM; with 4.5 g/L glucose and glutamine, without sodium pyruvate) and trypsin-EDTA (0.05% Trypsin/0.53 mM EDTA in Hank’s balanced salt solution) were obtained from Corning Cellgro (Manassas, VA). Phosphate buffered saline (PBS, BioPerformance certified, pH7.4), bovine liver catalase, dimethyl sulfoxide (DMSO), and PolyFreeze were purchased from Sigma Aldrich (St Louis, MO). H2O2 (30% stock solution) was from Avantor (Center Valley, PA). Pierce Quantitative Peroxide Assay Kit with sorbitol, 18 mm coverslips, fetal bovine serum (FBS), Penicillin-Streptomycin (10 U/mL) and dihydroethidium (DHE) were purchased from Thermo Scientific (Rockford, IL). 4’, 6-Diamidino-2-phenylindole (DAPI) and calcein am/ethidium bromide for cell live/dead stain were purchased from Invitrogen, ThermoFisher Scientific (Rockford, IL). 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide 98% (MTT) was from Alfa Aesar (WardHill, MA). TransWell® Permeable Supports (12 mm insert, 12 well plate, 3.0 μ, polycarbonate membrane) were purchased from Corning Costar (Pittston, PA). Anti-iNOS antibody (ab15323), anti-CD68 (ab125212), anti-Prx 2 antibody (ab109367) and goat anti-rabbit IgG H&L (Alexa Fluor 488, ab150077) were purchased from Abcam (Cambridge, MA). Anti-CD163 antibody (sc-58965) and goat anti-mouse IgG (sc-2781) were purchased from Santa Cruz Biotechnology (Dallas, Texas). Rat dermal fibroblasts and rat tendon fibroblasts were isolated from rat dermal tissue and tendon, respectively, and identified with anti-S100A4 antibody (ab27957) and goat anti-rabbit IgG H&L (Alexa Fluor 488) [17]. PEG-D4 was prepared following a previously published protocol [26] using 4-arm 10k Da Nhydroxysuccinimide ester activated poly (ethylene glycol) purchased from JenKem U.S.A, Inc. (Allen, TX).
2.2 Hydrogel Formation
Equal volume of PEG-D4 (300 mg/mL in pH 7.4 PBS) and NaIO4 (54.5 mM in deionized water) were mixed in a mold consisted of 2 glass plates separated with a spacer (thickness = 0.7, 1.3, or 2.2 mm) to control the thickness of the hydrogel. Hydrogels were allowed to cure for either 5 min or 16 hrs. For the 16 hrs-cured hydrogels, the glass molds with hydrogels were sealed with parafilm and stored in a sealed zip-lock bag at room temperature to prevent liquid evaporation. To determine the effect of PEG-D4 concentration on the release of H2O2, hydrogels (75, 110, and 150 mg/mL PEG-D4) were prepared by mixing equal volume of PEG-D4 (150, 220, and 300 mg/mL) and NaIO4 (the molar ratio of NaIO4: dopamine = 0.5:1) in a glass mold with a 0.7 mm-thick spacer. For in vitro cell culture experiments and subcutaneous implantation in rats, the precursor solutions of PEG-D4 (300 mg/mL in pH 7.4 PBS) and NaIO4 (54.5 mM in deionized water) were separately sterilized using a 0.22 μm filter prior to forming the hydrogel.
2.3 Oscillatory Rheometry Testing
Rheological properties of PEG-D4 hydrogels were characterized using a HR-2 rheometer (TA Instruments, New Castile, DE, USA). A frequency sweep (0.1–100 Hz at 10% strain) experiment was performed to determine the storage (G′) and loss (G″) moduli of PEG-D4. Hydrogel discs (diameter = 10 mm, thickness = 0.7 mm, n = 3) were tested using a parallel plate geometry at a gap distance that is set at 90% of the individual hydrogel thickness, which was measured by a digital caliper before testing.
2.4 H2O2 Measurement
Hydrogels were incubated with 1.5 mL of cell culture medium (DMEM with 10% (v/v) FBS and 0.5% (v/v) Penicillin-Streptomycin; pH = 7.4) for 48 hours at 37°C. H2O2 concentration was measured using the ferrous ion oxidation xylenol orange (FOX) assay by Quantitative Peroxide Assay Kit [17, 27]. 20 μL of the hydrogel extract was mixed with 200 μL of the FOX reagent and incubated at room temperature for 20 min and its absorbance at 590 nm was assessed using a plate reader (SynergyTM HT, BioTek). 20 μL of fresh cell culture medium was added to the hydrogel extract to supplement the loss of volume during sampling at each time point (from 15 min to 48 hrs). To determine the generation of H2O2 from the two precursors of the hydrogel, PEG-D4 and NaIO4 were dissolved in cell culture medium at an equivalent concentration relative to a sample with a diameter of 1 cm and a thickness of 0.7 mm. The H2O2 standard curve was prepared by preparing a serially diluted H2O2 solution (0.5 – 2000 μM of H2O2) from 30% H2O2 stock solution.
2.5 Molecular weight between crosslinks (M̄c)
Hydrogels discs (diameter = 1 cm) were immersed in 1.5 mL of PBS (pH 7.4) for 24 hrs. Swollen hydrogels were dried under vacuum for at least 2 days. The mass of the hydrogel in its relaxed (immediately after preparation; Mr), swollen (after swollen in PBS; Ms) and dried (Md) states were determined to calculate the polymer volume fraction in swollen hydrogel (vs) and relaxed hydrogel (vr) using following equations [28]:
| (1) |
| (2) |
where Vr, Vs, and Vp are the volumes of relaxed, swollen and dried hydrogel, respectively. ρp is the density of PEG (1.123 g/cm3) [29]. The density of water was assumed to be 1 g/cm3. M̄c was calculated using the following equation [30]:
| (3) |
where Mn is the starting molecular weight of PEG-D4, χ is the Flory-Huggins parameter for PEG and water (0.462) [31]. VH2O is the molar volume of water (18.1 mol/cm3).
The equilibrium water content (EWC) was determined based on equation 4:
| (4) |
2.6 Cell Viability
Hydrogels were cured directly onto the outside membrane of a TransWell® insert (Figure S2) so that the cells can be directly exposed to the released H2O2. The hydrogels were formed between the membrane of a TransWell® insert and a sheet of PTFE, and separated by a silicone rubber spacer (thickness = 0.7 mm) with a circular opening (diameter = 1 cm) (Figure S2A). Cell viability was assessed using MTT cytotoxicity assay following the ISO 10993-5 guideline with minor modification [32]. 1.5 mL of L929 mouse fibroblasts were seeded into 12-wells culture plate at a density of 3 × 104 cells/cm2. The cells were incubated for 24 hrs to obtain a confluent monolayer (37 °C, 5% CO2). After which, a TransWell® insert affixed with either a 5 min- or 16 hrs-cured hydrogel was placed into the culture plates and further incubated for another 24 hrs. The samples were removed from the culture plates and the culture medium was replaced by 300 μL of 1 mg/mL MTT solution in PBS. After incubating for 2 hrs at 37°C, the MTT solution was replaced with 600 μL DMSO to dissolve formazan and the absorbance of the DMSO solution was measured at a wavelength of 570 nm (reference 650 nm) using a Synergy HT multimode microplate reader (BioTek, USA). The relative cell viability was calculated using the absorbance values of cells directly exposed to the hydrogel extract divided by those for cells incubated in cell culture medium. To verify that the H2O2 is the source of the cytotoxicity, cell culture medium was also supplemented with 100 U/mL of catalase. The cytotoxicity of PEG-D4 (9.36–150 mg/mL in 10 mM PBS) and NaIO4 (1.71–27.3 mM) precursors were also assessed by directly exposing them to fibroblasts. Samples with relative cell viability less than 70% were considered cytotoxic. Each sample was repeated 3 times independently.
To observe the localized cell response to the hydrogels, after 24 hours incubation with the hydrogels, cells were stained with live (calcine) /dead (ethidium bromide) dyes (1:1000 in PBS) for 15 min at room temperature and imaged using an Olympus BX51 microscope (Melville, NY). Cellular density and live cells ratio were quantified using imageJ.
2.7 Immunofluorescent stain of Prx 2 in rat dermal and tendon fibroblasts
Glass coverslips (diameter = 18 mm) were sterilized by submersing into 70% (v/v) ethanol for 45 min and irradiating with UV light for 1 hour, and placed in the wells of a 12-well cell culture plate. Either rat dermal or tendon fibroblasts were seeded at a density of 104 cells/cm2 with 1.5 mL of cell culture medium and incubated for 24 hrs. TransWell® inserts with 5 min- and 16 hrs-cured hydrogels were placed over the cells for 2 hrs. The TransWells® inserts were removed and the fibroblasts were cultured for another 22 hrs. Coverslips with seeded cells were fixed in formalin and stained with primary anti-Prx 2 antibody, secondary goat anti-rabbit antibody IgG H&L, and DAPI. Fluorescent images were captured using Olympus BX51 microscope and the fluorescent intensity was quantified by ImageJ following the published method with minor modification [33]. An irregular outline was drawn following the edge of the cells so that the area of the cell and fluorescent intensity inside the cell could be measured. The area and the fluorescent intensity surround the cells were also measured, which was considered as the background intensity. The total corrected cellular fluorescent intensity density was measured as the fluorescent intensity per unit area within the cell subtracted from that of the background [33].
2.8 Subcutaneous Implantation
Healthy, weight matched Sprague Dawley rats were provided by Michigan Technological University animal facility. Subcutaneous implantation were performed following the Institutional Animal Care and Use Committee (IACUC) approved protocols. Both 5 min- and 16 hrs-cured hydrogels were randomly and bilaterally implanted into 4 pouches along the dorsal midline of the rats. Hydrogels with surrounding tissue were harvested after 3 and 7 days of implantation, embedded in PolyFreeze, and flash frozen in liquid nitrogen. The frozen tissues with embedded samples were either freshly sectioned for DHE staining or stored at −80°C before sectioning for fluorescent staining.
To detect superoxide in the surrounding tissue, freshly frozen samples were sectioned into 30-μm thick samples and incubated with DHE (10 μM) in PBS for 30 min at 37 °C, while protected from direct exposure to light [21]. To evaluate the macrophage phenotype in the surrounding tissue, samples were cryosectioned into 10-μm thick sections and stained with anti-CD68 (non-specific macrophages), anti-iNOS antibody (M1), and anti-CD163 antibody (M2). Inflammatory response was evaluated by determining the total number of CD68-stained macrophage relative to the overall number of cells based on DAPI staining. M1 and M2 polarization were quantified by counting the number of M2 macrophages divided by the combined number of M1 and M2 macrophages. Samples were imaged using an Olympus BX51 microscope. Samples were assessed from 3 different implants.
2.9 Statistical Analysis
Statistical analysis was performed using SigmaPlot. Student t-test and one way analysis of variance (ANOVA) were used to compare means of two or multiple groups, respectively. A p value less than 0.05 was considered statistically significant.
3. Results
3.1 Experimental design
PEG-D4 was chosen as in situ curable model polymer to study the generation of H2O2 during the oxidative crosslinking of terminal dopamine moieties. Due to the inert and highly predictable characteristics of PEG [26, 34], the observed changes in the physical and mechanical properties as well as biological responses can be fully attributed to the reactivity of the catechol side chain. When catechol is oxidized with the addition of NaIO4, PEG-D4 cured rapidly (under 30 seconds) as the branched architecture of PEG-D4 provides a junction point for rapid network formation when catechol moieties dimerize [26]. However, the oxidized catechol can continue to polymerize, forming oligomers consisting of up to 6 catechol residues over a period of 8 hours [12]. To observe the H2O2 generation during the process of catechol crosslinking, we compared PEG-D4 that was cured for 5 min with PEG-D4 that was cured for 16 hrs. The 5 min-cured hydrogel appeared light yellowish in color (Figure S3), which transitioned into dark reddish brown with time as catechol polymerizes through a mechanism that resembles quinone methide tanning [35]. Our experimental design aimed at minimizing the compositional differences between samples and the only variable that we examined here was the different time points after the initiation of the oxidative catechol crosslinking (e.g., 5 min- vs. 16 hrs-cured PEG-D4).
3.2 Rheological assessment of PEG-D4
Oscillatory rheometer was used to confirm the formation PEG-D4 hydrogel networks (Figure 1). For both 5 min- and 16 hrs-cured samples, the storage modulus (G′) values were significantly higher than those of the loss modulus (G″) and G′ was independent of frequency (< 25 Hz). Both observations indicated that PEG-D4 behaved as covalently crosslinked networks at both time points. The G′ values for the 16 hrs-cured hydrogel were significantly higher than those of 5 min-cured samples (~2 fold higher). Rheological data confirmed that PEG-D4 cured rapidly to form a chemically crosslinked network, but as the catechol continued to polymerize over time PEG-D4 hydrogels became stiffer and more densely crosslinked.
Figure 1.

Storage (G′, filled symbol) and loss (G″, open symbol) moduli for PEG-D4 tested at 5 min (■, □) and 16 hrs (●, ○) after mixing the precursor solutions.
3.3 H2O2 generated from PEG-D4 hydrogels
5 min-cured samples generated 24 μM of H2O2 within 1 hour and the concentration of H2O2 continued to increase for over 6 hours to reach a maximum of 40 μM (Figure 2). After which point, H2O2 concentration started to decrease, reaching a concentration of 9.5 μM at the 48 hr time point. For 16 hrs-cured samples, no H2O2 was captured in the hydrogel extract within 1 hour but the H2O2 concentration slowly increased to the peak concentration of 4.6 μM at the 6 hr time point and reduced to 2.9 μM after 48 hours of incubation. The peak concentration of H2O2 generated from the 16 hrs-cured sample was one order of magnitude lower compared to those detected from the 5 min-cured hydrogel. Despite having the same composition, the observed differences suggest that H2O2 is a byproduct generated during the oxidative crosslinking of catechol.
Figure 2.

H2O2 generated from 5 min- and 16 hrs-cured PEG-D4 hydrogels with a thickness of 0.7 mm.
PEG-D4 precursor solution generated a significantly higher H2O2 concentration when compared to PEG-D4 hydrogels. Over 110 μM of H2O2 was detected within 15 minutes, reaching a maximum of 1000 μM after 24 hours (Figure S4). The color of PEG-D4 precursor solutions turned dark red with time, indicating the oxidation of colorless catechol moiety into its quinone chromophore. Without the addition of NaIO4, catechol underwent autoxidation in a mildly basic and oxygenated cell culture medium [17], which likely generated H2O2 through a different mechanism as compared to that of IO4−-induced catechol oxidation. Additionally, H2O2 generated within the PEG-D4 hydrogel was trapped within a 3-dimentional polymer network, which slowed its release into the extract. On the other hand, the release of H2O2 from autoxidation of PEG-D4 polymer in solution was uninhibited and readily detectable. The large difference in the detected H2O2 between a PEG-D4 hydrogel and its precursor (a 25 fold difference) may also suggest that the H2O2 trapped within the hydrogel was consumed during the oxidative crosslinking process. Specifically, the maximum H2O2 concentration generated from the PEG-D4 hydrogel accounted for only a fraction of the catechol concentration (0.02 μM H2O2/μM catechol; Figure S5), whereas the maximum H2O2 concentration was 0.55 μM H2O2/ μM catechol (Figure S6) for PEG-D4 precursor. This is an indication that a large portion of the generated H2O2 were not released from the hydrogel network. There was no detectable H2O2 generated from the NaIO4 precursor solution (Figure S4), indicating that the source of H2O2 was the oxidation of catechol.
3.4 Effect of hydrogel thickness and PEG-D4 concentration on H2O2 generation and crosslinking density
The amount of H2O2 captured in the hydrogel extract was highly dependent on the thickness of the sample (Figure 3). Although thicker samples contained a higher amount of catechol (i.e., 1.3 and 2.2 mm-thick samples contained 1.9 and 3.1 fold higher catechol, respectively, when compared with a 0.7 mm-thick sample), the measured H2O2 concentration decreased with increasing hydrogel thickness. This indicates that H2O2 was potentially trapped within the thicker hydrogels due to a longer distance that H2O2 needed to travel and a reduced rate of solvent exchange due to reduced surface area. Additionally, a reduced amount of measured H2O2 in the thicker hydrogels suggests that the trapped H2O2 may be consumed, as it may contribute to oxidative crosslinking of catechol.
Figure 3.

H2O2 generated from 5 min-cured PEG-D4 hydrogels with different thicknesses. * p < 0.05 when compared to 2.2 mm-thick hydrogels. $ p < 0.05 when compared to both 1.3 and 2.2 mm-thick hydrogels.
Molecular weight between crosslinks (M̄c) of PEG-D4 was calculated to determine the effect of hydrogel thickness and cure time on the crosslinking density of the hydrogels. M̄c is the average molecular weight of polymer between crosslinking points in a hydrogel network, and is inversely proportional to its crosslinking density and mechanical properties [36, 37]. The calculated M̄c values ranged from 1,700 to 2,300 Da, which closely approximate the molecular weight of an individual PEG arm (2,500 Da). This implied that new elastic junction points were formed during the crosslinking and polymerization of the terminal catechol groups. The lower calculated M̄c values (i.e., less than 2000 Da) may be attributed to the inability of the modified Flory-Rehner equation to account for the changes in the functionality of the junction points as the terminal catechol group underwent polymerization over time [38].
Regardless of hydrogel thickness, M̄c values of 16 hrs-cured samples were significantly lower compared to those of 5 min-cured samples (Figure 4), confirming rheological results that crosslinking density increased with curing time. H2O2 generated during catechol oxidation is a mild oxidant [23]. For 16 hrs-cured samples, the generated H2O2 were trapped within the hydrogel network and contributed to catechol oxidation and crosslinking, resulting in a more densely crosslinked network. On the other hand, 5 min-cured samples were immersed into cell culture medium just 5 minutes after the initiation of crosslinking, and the generated H2O2 during the crosslinking process was continuously exchanged with the extracting fluid. For 5 min-cured samples, M̄c values also decreased with increasing hydrogel thickness. When combined with the H2O2 generation results where thicker sample generated reduced H2O2 (Figure 3), our data indicated that H2O2 was trapped within the thicker hydrogel to further promote crosslink formation resulting in a more densely crosslinked network. For 16 hrs-cured samples, changing hydrogel thickness did not significantly affect the calculated M̄c values, as the exchange of H2O2 did not begin until after the crosslinking process was complete.
Figure 4.

Molecular weight between crosslinks (M̄c) as a function of the thickness of PEG-D4 hydrogels. * p < 0.05 when compared to 16 hrs-cured hydrogels; $ p < 0.05 when compared to 5 min-cured hydrogels with a thickness of 2.2 mm.
Varying PEG-D4 concentration also greatly influenced the production of H2O2. Despite having a higher catechol concentration, hydrogels with a higher concentration of PEG-D4 produced less H2O2 (Figure S8). Based on the measured EWC values, the crosslinking density of the hydrogels decreased with increasing polymer concentration (Figure S9). These results indicated that the crosslinking density play a more prominent role in regulating the release of H2O2 and the release of H2O2 is not always proportional to the catechol concentration. Similar observation was reported for auto-oxidation of catechol, where H2O2 production was inversely proportional to the network crosslinking density despite having a higher catechol concentration [17]. On the other hand, H2O2 production increased with increased concentration of catecholic monomer in solution, where the released of the H2O2 was uninhibited by the polymer network [17].
3.5 Relative cell viability
Typically, cytotoxicity assays exposed cells to the extracts of biomaterials [6, 13, 17, 26, 39]. However, the extracts only represent byproducts generated during a certain time point (i.e., after the polymerization process is complete) and is only a one-time exposure of the byproduct to the cells. To capture the major byproduct generated during crosslinking process of catechol and to directly expose them to cells, PEG-D4 was fixated to a TransWell® insert and placed above L929 fibroblasts (Figure S2C). Relative cell viability for fibroblasts exposed to 5 min-cured sample (49%) was significantly lower when compared to those exposed to 16 hrs-cured hydrogels (77%) (Figure 5). Catalase is an enzyme that decomposes H2O2 into non-cytotoxic H2O and O2 [40]. Addition of catalase (100 U/mL) counteracted the cytotoxicity effect of H2O2 and significantly increased cell viability (67%) for cells exposed to 5 min-cured samples. These results indicated that relative cell viability is highly dependent on the concentration of H2O2 released into the cell culture media, and that the H2O2 generated during the oxidative crosslinking of catechol is a major source of cytotoxicity.
Figure 5.

Relative cell viability for L929 fibroblasts exposed directly to 5 min- and 16 hrs-cured hydrogels with or without 100 U/mL catalase based on MTT assay. * p < 0.05 when compared to 5 min-cured hydrogel without catalase.
For comparison purposes, the cytotoxicity of the PEG-D4 and NaIO4 precursor solutions was also determined. PEG-D4 and NaIO4 tested at concentrations (150 mg/mL and 27.3 mM, respectively) that were equivalent to those in a piece of hydrogel (diameter = 1 cm, thickness = 0.7 mm) were found to be cytotoxic (Figures S10 and S11, respectively). The source for the lower cell viability for PEG-D4 precursor (19%) when compared to the hydrogels extract is likely due to a higher concentration of H2O2 produced through autoxidation (Figure S4). PEG-D4 precursor became non-cytotoxic after a 1:2 dilution (Figure S10). On the other hand, NaIO4 required a 1:16 dilution to become non-cytotoxic (Figure S11). Although no H2O2 was generated from NaIO4, NaIO4 is a strong oxidant and induced cell death upon contact. In the current study, we did not characterize the concentration of unreacted NaIO4 released from the hydrogel, and the residual NaIO4 may also contribute to the observed cytotoxicity of the PEG-D4 hydrogel.
The cytotoxicity effect of H2O2 released from PEG-D4 hydrogels was highly localized, due to a short diffusion radius and half-life of H2O2 [41]. Based on live/dead staining, fibroblasts seeded directly under the 5 min-cured hydrogel were rounded in shape and mostly dead with a lower cell density (Figure 6A-1). There was a clear dead (red stained) and live (green stained) cell boundary at the edge of the TransWell® insert (Figure 6B-1), while cells located further away consisted mainly of live cells (Figure 6C-1). Both the cellular density and the percentage of live cells increased progressively when moving from directly underneath the hydrogel to edge of the cell culture well (Figure S12). On the contrary, cells exposed to 16 hrs-cured hydrogel did not show localized effects and cells were mostly stained green with similar cellular density and live cell percentages throughout the cell culture well (Figures 6 and S12).
Figure 6.

Live/dead staining of L929 fibroblasts exposed directly to 5 min- and 16 hrs-cured hydrogels, for cells located directly beneath the hydrogels (A), at the boundary of the hydrogel (B), and at the edge of culture well (C). The orange and black rings represent the edge of TransWell® insert and a well of the 12-wells plate, respectively. The diameter of the 12-wells plate well, the TransWell® insert, and the hydrogel are 2.2, 1.2, and 1 cm, respectively. Live and dead cells are stained green and red, respectively. Scale bar: 500 μm.
3.6 Prx 2 expression in rat dermal and tendon fibroblasts
Peroxiredoxin is a family of antioxidant enzymes that is upregulated in cells in response to oxidative stress, and Prx 2 catalyzes the reduction of H2O2 [42, 43]. Rat dermal fibroblasts exposed to 5 min-cured hydrogel expressed a higher amount of Prx 2 when compared to those exposed to 16 hrs-cured sample and the hydrogel-free media control (Figure 7). This indicated that cells exposed to a higher amount of H2O2 released by the 5 min-cured samples upregulated the expression of the antioxidant enzyme. On the other hand, Prx 2 was upregulated for rat tendon fibroblasts that were exposed to both 5 min- and 16 hrs-cured hydrogels. This indicated that tendon fibroblasts are extremely sensitive to H2O2 and a relatively small amount of H2O2 (~3 μM) released from 16 hrs-cured PEG-D4 upregulated Prx 2 expression. Regardless of the source of fibroblast, exposing cells to 5 min-cured samples resulted in reduced cellular density and the presence of Prx 2 in culture did not provide sufficient protection against the oxidative damage generated during the crosslinking of catechol.
Figure 7.


Peroxiredoxin 2 (Prx 2) antioxidant enzyme staining of rat dermal fibroblasts (A, C, and E) and rat tendon fibroblasts (B, D, and F) exposed directly to 5 min- or 16 hrs-cured hydrogels for 2 hours. Cell nuclei and Prx 2 were stained by DAPI (blue) and anti-Prx2 antibody (green), respectively. Fluorescent intensity of Prx 2 per cell in rat dermal and tendon fibroblasts (G). $ p < 0.05 when compared to both the media control and the 16 hrs-cured hydrogel groups. * p < 0.05 when compared to the media control. Scale bar: 100 μm.
3.7 Subcutaneous implantation of PEG-D4
PEG-D4 hydrogels were implanted subcutaneously in rat for up to 7 days to evaluate the effect of H2O2 generation on the in vivo biological responses. Freshly harvested tissues surrounding the implants were stained with DHE, which are oxidized into ethidium by superoxide generated in cell nuclei and has been widely used to monitor in vivo production of ROS [10]. There was no difference between DHE fluorescent intensity in tissues surrounding 5 min- and 16 hr-cured hydrogels after 3 days of implantation (Figure 8A and B). ROS is produced during the early phases of normal wound healing response [21], which may have masked the effect resulting from differences in the generated H2O2 between 5 min- and 16 hr-cured hydrogels. After 7 days post-implantation, the fluorescent intensity of DHE decreased in cells surrounding both implants when compared to the earlier time point (Figure 8C and D), signifying the elimination of acute inflammatory response. However, cells surrounding the 5 min-cured hydrogel exhibited a higher DHE fluorescent intensity (Figure 8E) when compared to its counterpart, indicating that elevated amount of H2O2 released from the 5 min-cured sample contributed to a more sustained production of ROS in the surrounding tissues.
Figure 8.


Dihydroethidium (DHE) staining of superoxide production surrounding 5 min- and 16 hrs-cured PEG-D4 hydrogels after 3 (A and B) and 7 (C and D) days subcutaneous implantation. The letter h indicates the location of the implanted hydrogel. Fluorescent intensity of DHE signals in the surrounding tissue (E). $ p < 0.05 when compared to 5 min-cured hydrogels after 7 days of subcutaneous implantation * p < 0.05 when compared to the 16 hrs-cured samples obtained after 7 days of subcutaneous implantation. Scale bar: 100 μm.
A higher percentage of cells surrounding 5 min-cured hydrogels (Figure 9A and C) were composed of macrophages with CD68 genotype when compared to those surrounding 16 hrs- cured hydrogel (Figure 9B and D) after both 3 and 7 days implantation. This indicated that an elevated H2O2 released from 5 min-cured implants attracted a higher macrophage infiltration into the surrounding tissues. H2O2 has a chemotactic role in recruiting inflammatory cells to the wound site [17, 44, 45]. For both implants, the percentage of macrophages decreased with time, signifying a reduced inflammatory response 7 days post-implantation (Figure 9E). However, the percentage of macrophages surrounding 5 min-cured samples was still significantly higher compared to those surrounding 16 hrs-cured hydrogels. Our data confirm published results where elevated macrophages number coincided with elevated ROS production [18, 46].
Figure 9.


Immunofluorescent staining of macrophages (CD 68) in tissue surrounding 5 min- and 16 hrs-cured PEG-D4 hydrogels after 3 (A, B) and 7 (C, D) days of subcutaneous implantation. Cell nuclei and macrophages were stained by DAPI (blue) and CD68 (red), respectively. The letter h indicates the location of the implanted hydrogel. The percentage of macrophages relative to the total number of cells stained with DAPI (E). * p < 0.05 when compared to 5 min-cured hydrogels after 3 days of subcutaneous implantation; $ p < 0.05 when compared to 5 min-cured hydrogels after 7 days of subcutaneous implantation; & p < 0.05 when compared to 16 hrs-cured hydrogels after 3 days of subcutaneous implantation. Scale bar: 100 μm.
Tissues surrounding the implants were further stained with iNOS (M1) and CD163 (M2) to determine the phenotype of the macrophages (Figures 10 and 11, respectively). After 3 days of implantation, M2 macrophages appeared immediately adjacent to the surface of the 5 min-cured hydrogel (Figure 10E), whereas these cells were more than 50 μm away from the 16 hrs-cured hydrogel (Figure 10F). This indicated that higher H2O2 near the 5 min-cured sample promoted M2 macrophage polarization near the interface of the implant. After 7 days of implantation, M2 macrophages disappeared from the interface of 5 min-cured hydrogel, likely because the hydrogel ceased to generate H2O2 (Figure 11E). For both time points, a significantly higher amount of M2 macrophages were found near the surface of 5 min-cured hydrogels when compared to those surrounding 16 hrs-cured samples (Table 1). For 5 min-cured samples, the percentage of M2 macrophages was 21% after 3 days of implantation and reaching 44% by day 7. These percentages are 1.6 and 2.4 folds higher when compared to cells surrounding 16 hrs-cured samples, respectively. H2O2 is necessary to activate M2 macrophage polarization [20] and H2O2 generated during oxidative crosslinking of catechol contributed to the activation of M2 macrophages at the PEG-D4-tissue interface. There was no difference in the amount of M1 macrophage density surrounding both hydrogels (Figure 10C and D; Figure 11C and D) at both time points. The total number of macrophages decreased over time, confirming results based on non-specific CD68 staining (Figure 8).
Figure 10.

Immunofluorescent staining of macrophages (iNOS and CD163) surrounding 5 min- (A, C, E and G) and 16 hrs- (B, D, F and H) cured PEG-D4 hydrogels after 3 days of subcutaneous implantation. Cell nuclei, macrophage type 1 (M1), and macrophage type 2 (M2) were stained by DAPI (blue), iNOS (green), and CD163 (red), respectively. The letter h indicates the location of the implanted hydrogel. The dash line indicates the interface between the hydrogel and the surrounding tissue. Scale bar: 100 μm.
Figure 11.

Immunofluorescent staining of macrophages (iNOS and CD163) surrounding 5 min- (A, C, E and G) and 16 hrs- (B, D, F and H) cured PEG-D4 hydrogels after 7 days of subcutaneous implantation. Cell nuclei, macrophage type 1 (M1), and macrophage type 2 (M2) were stained by DAPI (blue), iNOS (green), and CD163 (red), respectively. The letter h indicates the location of the implanted hydrogel. The dash line indicates the interface between the hydrogel and the surrounding tissue. Scale bar: 100 μm.
Table 1.
M1 and M2 type macrophages density and the percentage of M2 macrophage relative to the total number of macrophage in the surrounding tissue retrieved after 3 and 7 days of subcutaneous implantation.
| 3 days | 7 days | |||
|---|---|---|---|---|
| 5 min-cured | 16 hrs-cured | 5 min-cured | 16 hrs-cured | |
| M1 macrophage (×105 cells/mm2) | 4.7 ± 0.62 | 4.5 ± 0.67 | 0.86 ± 0.30 | 1.0 ± 0.45 |
| p = 0.97 | p = 0.89 | |||
|
| ||||
| M2 macrophage (×105 cells/mm2) | 1.2 ± 0.23 | 0.68 ± 0.08 | 0.59 ± 0.18 | 0.29 ± 0.11 51 |
| p < 0.0010 | p = 0.021 | |||
|
| ||||
| % M2 macrophage relative to the total number of macrophage | 21 ± 4.6 | 13 ± 2.8 | 44 ± 12 | 18 ± 3.9 |
| p = 0.0010 | p < 0.0010 | |||
4. Discussion
NaIO4-mediated crosslinking of catechol involves the formation of α,β-dehydro intermediates (Scheme 1) [26, 35, 47]. Addition of IO4− oxidizes dopamine to dopamine quinone (reaction A), which tautomerizes to form α,β-dehydrodopamine (reaction B).α,β-dehydrodopamine recovers a reduced catechol side chain, which is further oxidized to its corresponding quinone (reaction C). Crosslinking between α,β-dehydrodopamine and α,β-dehydrodopamine quinone resulted in the formation of dehydro dimer (reaction D), leading to subsequent polymerization (reaction E) [35, 47]. When catechol (i.e., dopamine and α,β-dehydrodopamine) was oxidized to its corresponding quinone (dopamine quinone and α,β-dehydrodopamine quinone, respectively) (reactions A and C, respectively), new oxidant species are generated during the process. During the fast oxidation process involving IO4−, IO4− was reduced to iodate (IO3− ) [48, 49] while molecular dioxygen (O2) was transformed into highly reactive O2•− (reaction F) [50]. IO3− can further oxidize catechol to form quinone [49]. At the same time, IO3− is reduced to a highly reactive intermediate product, iodite (IO2−), which is rapidly reduced to the final product iodine (I2) in the presence of phenolic compound [49, 51]. O2•− oxidizes catechol to generate H2O2 (reaction G) [52]. H2O2 can also be generated between O2•− and proton ions (H+) via water mediated dismutation reaction [48]. Finally, H2O2 is a mild oxidant which can oxidize catechol to form water (reaction H) [23].
Scheme 1.

Proposed mechanism of H2O2 generation during the IO4-induced crosslinking of catechol. R = 4 armed PEG.
Although PEG-D4 cured rapidly through the dimer formation of terminal dopamine moieties, polymerization of catechol may take over 8 hours to complete [12]. The newly generated oxidant species (i.e., IO3−, O2•−, and H2O2) during the oxidation of catechol were trapped within the PEG network, which further contributed to oxidative crosslinking and polymerization reactions. As such, PEG-D4 cured for over 16 hours were significantly stiffer compared to those cured for only 5 minutes (Figure 1). Similarly, thicker samples with reduced surface to volume ratio resulted in stiffer and more densely crosslinked networks (Figure 4). The amount of released H2O2 decreased with increasing hydrogel thickness (Figure 3), indicating that the trapped H2O2 contributed to reducing hydrogel swelling while increasing the crosslinking density of the network. Hydrogels with higher PEG-D4 concentrations also generated less H2O2 despite having a higher catechol concentration (Figure S8), likely due to a higher crosslinking density that limit the diffusion of the generated H2O2 into the extraction media (Figure S9). Additionally, a PEG-D4 hydrogel (Figure S4) generated more than an order of magnitude less H2O2 when compared to the PEG-D4 precursor (Figure S6), further support that H2O2 trapped within a network was consumed. We previously reported that network crosslinking density plays a role in limiting the release of H2O2 from a hydrogel network [17]. This “cage effect” also explains why autoxidation of PEG-D4 resulted in significantly higher H2O2 measurement, as the generated oxidant is not trapped and consumed within a polymer network.
During each successive oxidation and crosslinking reaction, the more potent oxidants (i.e., IO4− ) were consumed and progressively converted to the less reactive oxidants (i.e., H2O2). H2O2 is relatively more stable compared to other oxidants and oxidizes catechol at a significantly reduced rate [53]. As such, H2O2 accumulated within PEG-D4 hydrogels and were captured in the hydrogel extracts. In our experiment, the catechol moieties within the 5 min-cured samples continued to undergo oxidative crosslinking and generated H2O2 during the process (Figure 2). On the other hand, the crosslinking process within 16 hrs-cured samples was complete and generated an order of magnitude lower amount of H2O2. By comparing PEG-D4 hydrogels at these two different time points, we were able to characterize the effect of H2O2 generated during oxidative crosslinking while using samples with the same composition and preparation method.
A typical culture condition is highly oxidative due to elevated oxygen levels and is deficient of antioxidant enzymes when compared to in vivo conditions [54, 55]. H2O2 released during the oxidative crosslinking of catechol was cytotoxic (Figure 5). We previously demonstrated that a H2O2 concentration of 100 μM and lower to be non-cytotoxic [17]. Although the 5 min-cured samples generated a maximum of 40 μM of H2O2 (Figure 2), a continuous generation of H2O2 during the crosslinking process likely contributed to reduced cell viability. Doping the culture medium with antioxidant enzyme, catalase, counteracted the cytotoxic effect, indicating that H2O2 is the main source of cytotoxicity for PEG-D4. Due to the instability of H2O2 in a typical culture condition (~1 hour) [56], H2O2 exhibited a short diffusion radii [41] and highly localized effect (Figure 6). This is potentially beneficial in minimizing undesirable systemic effect for in vivo applications.
Cells exposed to an oxidative environment secret a series of antioxidant enzymes (i.e., catalase, glutathione peroxidases and Prx) to conserve the normal redox homeostasis [57]. Prx 2 is ubiquitously expressed in rat skin cells including fibroblasts and keratinocytes [58]. H2O2 generated by PEG-D4 upregulated the expression of Prx 2 in the cytoplasm of primary dermal and tendon fibroblasts (Figure 7). However, the efficiency of Prx in catalyzing H2O2 (~105 M−1 S−1) is significantly lower when compared to catalase (~106 M−1 S−1) and glutathione peroxidase (~108 M−1 S−1) [43, 59, 60]. Cells exposed to relatively higher amount of H2O2 (i.e., 5 min-cured hydrogel) also resulted in a lower cell density, despite an increase in Prx 2 expression. Due to ischemia induced by localized tissue compression in tendons, tendon fibroblasts are typically exposed to a lower concentrations of oxygen when compared to dermal fibroblasts [61, 62]. Therefore, tendon fibroblasts were more sensitive to ROS. Exposing tendon fibroblasts to a significantly lower amount of H2O2 (i.e., 16 hrs-cured hydrogel) resulted in an increased expression of Prx 2. This result indicates the importance when considering the application of catechol-containing biomaterials to different tissues and biological systems, as the biological responses to the released H2O2 will likely be significantly different.
Subcutaneous implantation revealed that higher amount of H2O2 released from the 5 min-cured samples increased the production of O2•− in the surrounding tissues (Figure 8) and recruited macrophages to the tissue-implant interface (Figure 9). Concentrations of O2•−, H2O2 and macrophages are closely regulated in a wound site [63]. The mitochondria in respiring cells utilize oxygen to produce O2•−, which is catalyzed by superoxide dismutase to form H2O2. H2O2 could be decomposed by catalase to form H2O and O2 which increases the local oxygen concentration and further be used by cells to produce more O2•−. H2O2 and O2•− also function as chemotactic agent to attract macrophage infiltration, which secretes more ROS and activates phagocytes as a part of natural foreign body response to prevent infection [64, 65]. Although 5 min-cured hydrogel recruited more macrophages to the interface when compared to 16 hrs-cured samples, a higher percentage of these cells were M2 macrophages (Table 1). ROS has been reported to play an important role in M2 polarization [20], which is supported by our findings. Unlike the pro-inflammatory response demonstrated by M1 macrophages, M2 macrophages are responsible for regulatory and anti-inflammatory responses, which are critical in promoting tissue regeneration [66, 67].
Taken together, our report highlights the importance of H2O2 production during the oxidative crosslinking of catechol. H2O2 plays an important in promoting chemical crosslinking, which affects the physical (i.e., swelling behavior) and mechanical properties for injectable biomaterials and bioadhesive designed for load bearing applications. Additionally, H2O2 is responsible for a number of biological responses and these effects are highly dependent on the concentration of H2O2, duration of release, as well as the biological system (i.e., source of cell, in vitro vs. in vivo) that H2O2 comes in contact with. As such, it is paramount to monitor and tailor the production of H2O2 generated from catechol-containing biomaterials for a given application.
5. Conclusion
PEG-D4 was used as a model system to study the generation of H2O2 during the oxidative crosslinking of catechol. H2O2 trapped within the hydrogel network contributed to catechol polymerization, creating a more densely crosslinked and stiffer hydrogel. For in vitro cell assays, H2O2 generation resulted in localized cytotoxicity response and upregulated the expression of Prx 2 in primary cell lines. In the subcutaneous implantation model, the released H2O2 also promoted superoxide production, macrophage recruitment and M2 macrophage differentiation in the surrounding tissue of the PEG-D4 implant. These results indicated that H2O2 generation is one of the main contributors to the cytotoxicity and inflammatory responses during the oxidative crosslinking of the catechol moiety.
Supplementary Material
Statement of Significance.
Remarkable underwater adhesion strategy employed by mussels has been utilized to design a wide variety of biomaterials ranging from tissue adhesives to drug carrier and tissue engineering scaffolds. Catechol is the main adhesive moiety that is widely incorporated to create an injectable biomaterials and bioadhesives. However, the biocompatibility and biological responses associated with the byproducts generated during the curing process of catechol has never been characterized. In this manuscript, we design a model system to systemically characterize the release of hydrogen peroxide (H2O2) during the crosslinking of catechol. Given the multitude of biological responses associated with H2O2 (i.e., wound healing, antimicrobial, chronic inflammation), its release from catechol-containing biomaterials need to be carefully monitored and controlled for a desired application.
Acknowledgments
This project was supported by National Institutes of Health under Award number R15GM104846. HM and YL were support in part by the Doctoral Finishing Fellowship provided by the Graduate School at Michigan Technological University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waite JH. Nature’s underwater adhesive specialist. International Journal of Adhesion and Adhesives. 1987;7(1):9–14. [Google Scholar]
- 2.Waite JH. Adhesion a la moule. Integrat Comparat Bio. 2002;42(6):1172–1180. doi: 10.1093/icb/42.6.1172. [DOI] [PubMed] [Google Scholar]
- 3.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Mussel-Inspired Adhesives and Coatings. Annu Rev Mater Res. 2011;41:99–132. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaushik NK, Kaushik N, Pardeshi S, Sharma JG, Lee SH, Choi EH. Biomedical and Clinical Importance of Mussel-Inspired Polymers and Materials. Marine Drugs. 2015;13(11):6792–6817. doi: 10.3390/md13116792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krogsgaard M, Nue V, Birkedal H. Mussel-Inspired Materials: Self-Healing through Coordination Chemistry. Chemistry – A European Journal. 2016;22(3):844–857. doi: 10.1002/chem.201503380. [DOI] [PubMed] [Google Scholar]
- 6.Mehdizadeh M, Weng H, Gyawali D, Tang L, Yang J. Injectable citrate-based mussel-inspired tissue bioadhesives with high wet strength for sutureless wound closure. Biomaterials. 2012;33(32):7972–7983. doi: 10.1016/j.biomaterials.2012.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haller CM, Buerzle W, Kivelio A, Perrini M, Brubaker CE, Gubeli RJ, Mallik AS, Weber W, Messersmith PB, Mazza E, Ochsenbein-Koelble N, Zimmermann R, Ehrbar M. Mussel-mimetic tissue adhesive for fetal membrane repair: An ex vivo evaluation. Acta Biomaterialia. 2012;8(12):4365–4370. doi: 10.1016/j.actbio.2012.07.047. [DOI] [PubMed] [Google Scholar]
- 8.Brodie M, Vollenweider L, Murphy JL, Xu F, Lyman A, Lew WD, Lee BP. Biomechanical properties of Achilles tendon repair augmented with bioadhesive-coated scaffold. Biomed Mater. 2011;6(1):015014. doi: 10.1088/1748-6041/6/1/015014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brubaker CE, Kissler H, Wang L-j, Kaufman DB, Messersmith PB. Biological performance of mussel-inspired adhesive in extrahepatic islet transplantation. Biomaterials. 2010;31(3):420–427. doi: 10.1016/j.biomaterials.2009.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu JH, Lee Y, Do MJ, Jo SD, Kim JS, Kim BS, Im GI, Park TG, Lee H. Chitosan-g-hematin: Enzyme-mimicking polymeric catalyst for adhesive hydrogels. Acta Biomaterialia. 2014;10(1):224–233. doi: 10.1016/j.actbio.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Kastrup CJ, Nahrendorf M, Figueiredo JL, Lee H, Kambhampati S, Lee T, Cho SW, Gorbatov R, Iwamoto Y, Dang TT, Dutta P, Yeon JH, Cheng H, Pritchard CD, Vegas AJ, Siegel CD, MacDougall S, Okonkwo M, Thai A, Stone JR, Coury AJ, Weissleder R, Langer R, Anderson DG. Painting blood vessels and atherosclerotic plaques with an adhesive drug depot. Proc Natl Acad Sci USA. 2012;109(52):21444–21449. doi: 10.1073/pnas.1217972110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee BP, Dalsin JL, Messersmith PB. Synthesis and Gelation of DOPA-Modified Poly(ethylene glycol) Hydrogels. Biomacromol. 2002;3(5):1038–47. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R, Lee BP. Injectable Dopamine-Modified Poly(Ethylene Glycol) Nanocomposite Hydrogel with Enhanced Adhesive Property and Bioactivity. ACS Appl Mater Interfaces. 2014;6(19):16982–92. doi: 10.1021/am504566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brubaker CE, Messersmith PB. Enzymatically Degradable Mussel-Inspired Adhesive Hydrogel. Biomacromolecules. 2011;12(12):4326–4334. doi: 10.1021/bm201261d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Meng H, Liu Y, Narkar A, Lee BP. Gelatin Microgel Incorporated Poly(ethylene glycol)-Based Bioadhesive with Enhanced Adhesive Property and Bioactivity. ACS Applied Materials & Interfaces. 2016;8(19):11980–11989. doi: 10.1021/acsami.6b01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ISO 10993–12. Biological evaluation of medical devices, Part 12: Sample preparation and reference materials. International Organization for Standardization; 2012. [Google Scholar]
- 17.Meng H, Li Y, Faust M, Konst S, Lee BP. Hydrogen peroxide generation and biocompatibility of hydrogel-bound mussel adhesive moiety. Acta Biomaterialia. 2015;17:160–169. doi: 10.1016/j.actbio.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brian N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive Oxygen Species (ROS)-A Family of Fate Deciding Molecules Pivotal in Constructive Inflammation and Wound Healing. European Cells and Materials. 2012;24:249–265. doi: 10.22203/ecm.v024a18. [DOI] [PubMed] [Google Scholar]
- 19.Sen CK, Khanna S, Babior BM, Hunt TK, Ellison EC, Roy S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J Biol Chem. 2002;277(36):33284–33290. doi: 10.1074/jbc.M203391200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23(7):898–914. doi: 10.1038/cr.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy S, Khanna S, Nallu K, Hunt TK, Sen CK. Dermal Wound Healing Is Subject to Redox Control. Mol Ther. 2006;13(1):211–220. doi: 10.1016/j.ymthe.2005.07.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldry MGC. The bactericidal, fungicidal and sporicidal properties of hydrogen peroxide and peracetic acid. Journal of Applied Bacteriology. 1983;54(3):417–423. doi: 10.1111/j.1365-2672.1983.tb02637.x. [DOI] [PubMed] [Google Scholar]
- 23.Mastore M, Kohler L, Nappi AJ. Production and utilization of hydrogen peroxide associated with melanogenesis and tyrosinase-mediated oxidations of DOPA and dopamine. FEBS Journal. 2005;272(10):2407–2415. doi: 10.1111/j.1742-4658.2005.04661.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee F, Chung JE, Kurisawa M. An injectable enzymatically crosslinked hyaluronic acid-tyramine hydrogel system with independent tuning of mechanical strength and gelation rate. Soft Matter. 2008;4(4):880–887. doi: 10.1039/b719557e. [DOI] [PubMed] [Google Scholar]
- 25.Sakai S, Hirose K, Moriyama K, Kawakami K. Control of cellular adhesiveness in an alginate-based hydrogel by varying peroxidase and H2O2 concentrations during gelation. Acta Biomaterialia. 2010;6(4):1446–1452. doi: 10.1016/j.actbio.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Cencer MM, Liu Y, Winter A, Murley M, Meng H, Lee BP. Effect of pH on the rate of curing and bioadhesive properties of dopamine functionalized poly(ethylene glycol) hydrogels. Biomacromol. 2014;15(8):2861–2869. doi: 10.1021/bm500701u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clement MV, Long LH, Ramalingam J, Halliwell B. The cytotoxicity of dopamine may be an artefact of cell culture. Journal of Neurochemistry. 2002;81(3):414–421. doi: 10.1046/j.1471-4159.2002.00802.x. [DOI] [PubMed] [Google Scholar]
- 28.Andreopoulos FM, Beckman EJ, Russell AJ. Light-induced tailoring of PEG-hydrogel properties. Biomaterials. 1998;19(15):1343–1352. doi: 10.1016/s0142-9612(97)00219-6. [DOI] [PubMed] [Google Scholar]
- 29.Arlie PJP, Spegt P, Skoulios A. Etude de la cristallisation des polymères. II. Structure lamellaire et repliement des chaînes du polyoxyéthylène. Die Makromolekulare Chemie. 1967;104(1):212–229. [Google Scholar]
- 30.Peppas NA, Merrill EW. Poly(vinyl alcohol) hydrogels: Reinforcement of radiation-crosslinked networks by crystallization. Journal of Polymer Science: Polymer Chemistry Edition. 1976;14(2):441–457. [Google Scholar]
- 31.Merrill EW, Dennison KA, Sung C. Partitioning and diffusion of solutes in hydrogels of poly(ethylene oxide) Biomaterials. 1993;14(15):1117–1126. doi: 10.1016/0142-9612(93)90154-t. [DOI] [PubMed] [Google Scholar]
- 32.ISO 10993–5. Biological evaluation of medical devices, Part 5: Tests for cytotoxicity: in vitro methods. International Organization for Standardization; 2012. [Google Scholar]
- 33.McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13(9):1400–1412. doi: 10.4161/cc.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cencer M, Murley M, Liu Y, Lee BP. Effect of nitro-functionalization on the cross-linking and bioadhesion of biomimetic adhesive moiety. Biomacromol. 2015;16(1):404–410. doi: 10.1021/bm5016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abebe A, Zheng D, Evans J, Sugumaran M. Reexamination of the mechanisms of oxidative transformation of the insect cuticular sclerotizing precursor, 1,2-dehydro-N-acetyldopamine. Insect Biochemistry and Molecular Biology. 2010;40(9):650–659. doi: 10.1016/j.ibmb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Anseth KS, Bowman CN, Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996;17(17):1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 37.Peppas NA, Bures P, Leobandung W, Ichikawa H. Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics and Biopharmaceutics. 2000;50(1):27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 38.Ding X, Vegesna GK, Meng H, Winter A, Lee BP. Nitro-Group Functionalization of Dopamine and its Contribution to the Viscoelastic Properties of Catechol-Containing Nanocomposite Hydrogels. Macromolecular Chemistry and Physics. 2015;216(10):1109–1119. doi: 10.1002/macp.201500010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Meng H, Liu Y, Lee BP. Fibrin Gel as an Injectable Biodegradable Scaffold and Cell Carrier for Tissue Engineering. The Scientific World Journal. 2015;(2015):10. doi: 10.1155/2015/685690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cellular and Molecular Life Sciences CMLS. 2004;61(2):192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takano M, Meneshian A, Sheikh E, Yamakawa Y, Wilkins KB, Hopkins EA, Bulkley GB. Rapid upregulation of endothelial P-selectin expression via reactive oxygen species generation. American Journal of Physiology - Heart and Circulatory Physiology. 2002;283(5):H2054–H2061. doi: 10.1152/ajpheart.01001.2001. [DOI] [PubMed] [Google Scholar]
- 42.Immenschuh S, Baumgart-Vogt E. Peroxiredoxins, Oxidative Stress, and Cell Proliferation. Antioxidants & Redox Signaling. 2005;7(5–6):768–777. doi: 10.1089/ars.2005.7.768. [DOI] [PubMed] [Google Scholar]
- 43.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The High Reactivity of Peroxiredoxin 2 with H2O2 Is Not Reflected in Its Reaction with Other Oxidants and Thiol Reagents. Journal of Biological Chemistry. 2007;282(16):11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 44.Lee CF, Ullevig S, Kim HS, Asmis R. Regulation of Monocyte Adhesion and Migration by Nox4. PLoS ONE. 2013;8(6):e66964. doi: 10.1371/journal.pone.0066964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loo AEK, Wong YT, Ho R, Wasser M, Du T, Ng WT, Halliwell B. Effects of Hydrogen Peroxide on Wound Healing in Mice in Relation to Oxidative Damage. PLoS ONE. 2012;7(11):e49215. doi: 10.1371/journal.pone.0049215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jay Forman H, Torres M. Redox signaling in macrophages. Molecular Aspects of Medicine. 2001;22(4–5):189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 47.Sugumaran M, Semensi V, Kalyanaraman B, Bruce JM, Land EJ. Evidence for the formation of a quinone methide during the oxidation of the insect cuticular sclerotizing precursor 1,2-dehydro-N-acetyldopamine. J Biol Chem. 1992;267(15):10355–61. [PubMed] [Google Scholar]
- 48.Bokare AD, Choi W. Singlet-Oxygen Generation in Alkaline Periodate Solution. Environmental Science & Technology. 2015;49(24):14392–14400. doi: 10.1021/acs.est.5b04119. [DOI] [PubMed] [Google Scholar]
- 49.Harrison CR, Hodge P. Polymer-supported periodate and iodate as oxidizing agents. Journal of the Chemical Society, Perkin Transactions. 1982;1(0):509–511. [Google Scholar]
- 50.Munoz-Munoz JL, García-Molina F, Varón R, Tudela J, García-Cánovas F, Rodríguez-López JN. Generation of hydrogen peroxide in the melanin biosynthesis pathway. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2009;1794(7):1017–1029. doi: 10.1016/j.bbapap.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Gupta YK, Sharma DN. Kinetics and mechanism of the reduction of iodate to iodite by bromide in the presence of phenol. The Journal of Physical Chemistry. 1971;75(16):2516–2522. [Google Scholar]
- 52.Sawyer DT, Calderwood TS, Johlman CL, Wilkins CL. Oxidation by superoxide ion of catechols, ascorbic acid, dihydrophenazine, and reduced flavins to their respective anion radicals. A common mechanism via a combined proton-hydrogen atom transfer. The Journal of Organic Chemistry. 1985;50(9):1409–1412. [Google Scholar]
- 53.Mochizuki M, Yamazaki S-i, Kano K, Ikeda T. Kinetic analysis and mechanistic aspects of autoxidation of catechins. Biochimica et Biophysica Acta (BBA) - General Subjects. 2002;1569(1–3):35–44. doi: 10.1016/s0304-4165(01)00230-6. [DOI] [PubMed] [Google Scholar]
- 54.Halliwell B. The wanderings of a free radical. Free Radical Biology and Medicine. 2009;46(5):531–542. doi: 10.1016/j.freeradbiomed.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 55.de Groot H, Littauer A. Hypoxia, reactive oxygen, and cell injury. Free Radical Biology and Medicine. 1989;6(5):541–551. doi: 10.1016/0891-5849(89)90047-6. [DOI] [PubMed] [Google Scholar]
- 56.Reznikov K, Kolesnikova L, Pramanik A, Tan-No K, Gileva I, Yakovleva T, Rigler R, Terenius L, Bakalkin G. Clustering of apoptotic cells via bystander killing by peroxides. The FASEB Journal. 2000;14(12):1754–1764. doi: 10.1096/fj.99-0890com. [DOI] [PubMed] [Google Scholar]
- 57.Tavender TJ, Bulleid NJ. Peroxiredoxin IV protects cells from oxidative stress by removing H(2)O(2) produced during disulphide formation. Journal of Cell Science. 2010;123(15):2672–2679. doi: 10.1242/jcs.067843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JE, Kwon BD, Lee JB, Won YH, Kim YP, Lee SC, Chae HZ, Ahn KY. Peroxiredoxin is Ubiquitously Expressed in Rat Skin: Isotype-Specific Expression in the Epidermis and Hair Follicle. Journal of Investigative Dermatology. 2000;115(6):1108–1114. doi: 10.1046/j.1523-1747.2000.00177.x. [DOI] [PubMed] [Google Scholar]
- 59.Yuan J, Murrell GAC, Trickett A, Landtmeters M, Knoops B, Wang MX. Overexpression of antioxidant enzyme peroxiredoxin 5 protects human tendon cells against apoptosis and loss of cellular function during oxidative stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2004;1693(1):37–45. doi: 10.1016/j.bbamcr.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 60.Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends in Biochemical Sciences. 2003;28(1):32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 61.Rempel D, Abrahamsson SO. The effects of reduced oxygen tension on cell proliferation and matrix synthesis in synovium and tendon explants from the rabbit carpal tunnel: an experimental study in vitro. Journal of Orthopaedic Research. 2001;19(1):143–148. doi: 10.1016/S0736-0266(00)00005-X. [DOI] [PubMed] [Google Scholar]
- 62.Chen JH, Jones RH, Tarry-Adkins J, Smith NH, Ozanne SE. Adverse effects of reduced oxygen tension on the proliferative capacity of rat kidney and insulin-secreting cell lines involve DNA damage and stress responses. Experimental Cell Research. 2008;314(16):3075–3080. doi: 10.1016/j.yexcr.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Seminars in Immunology. 2008;20(2):86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rieger S, Sagasti A. Hydrogen Peroxide Promotes Injury-Induced Peripheral Sensory Axon Regeneration in the Zebrafish Skin. PLoS Biol. 2011;9(5):e1000621. doi: 10.1371/journal.pbio.1000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seifried HE, Anderson DE, Fisher EI, Milner JA. A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem. 2007;18(9):567–579. doi: 10.1016/j.jnutbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 66.Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, Vunjak-Novakovic G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35(15):4477–4488. doi: 10.1016/j.biomaterials.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marchetti V, Yanes O, Aguilar E, Wang M, Friedlander D, Moreno S, Storm K, Zhan M, Naccache S, Nemerow G, Siuzdak G, Friedlander M. Differential Macrophage Polarization Promotes Tissue Remodeling and Repair in a Model of Ischemic Retinopathy. Sci Rep. 2011;1 doi: 10.1038/srep00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.