

Abstract
UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-PT) is an α2β2γ2 hexamer that mediates the initial step in the formation of the mannose 6-phosphate targeting signal on newly synthesized lysosomal acid hydrolases. The GNPTAB gene encodes the 1256 amino acid long α/β precursor which is normally cleaved at K928 in the early Golgi by Site-1 protease (S1P). Here, we show that removal of the so-called `spacer-1´ domain (residues 86–322) results in cleavage almost exclusively at a second S1P consensus sequence located upstream of K928. In addition, GlcNAc-1-PT lacking spacer-1 exhibits enhanced phosphorylation of several non-lysosomal glycoproteins, while the phosphorylation of lysosomal acid hydrolases is not altered. In view of these effects on the maturation and function of GlcNAc-1-PT, we suggest renaming `spacer-1´ the `regulatory-1´ domain.
Keywords: GlcNAc-1-phosphotransferase, lysosomal enzyme, site-1 protease, mannose 6-phosphate, spacer domain
INTRODUCTION
The cis-Golgi enzyme UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-PT) performs the initial step in the generation of the mannose 6-phosphate (Man-6-P) targeting signal on lysosomal acid hydrolases. It does so by selectively binding to conformation-dependent protein determinants on these enzymes and catalyzing the transfer of GlcNAc-1-P from UDP-GlcNAc to mannose residues on high mannose-type N-linked glycans of the hydrolases (1). The GlcNAc is then excised by a second enzyme to generate Man-6-P monoesters, which mediate high affinity binding to Man-6-P receptors (MPRs) in the trans-Golgi network followed by transport of the hydrolases to the endo-lysosomal system (2).
GlcNAc-1-PT is an α2β2γ2 hexameric protein encoded by two genes. The smaller γ subunit is encoded by the GNPTG gene, whereas the α and β subunits are encoded as a single α/β precursor by the GNPTAB gene (3–6). Mutations in the GNPTAB gene gives rise to the severe lysosomal storage disorder mucolipidosis II (I-cell disease) or the attenuated mucolipidosis III (pseudo-Hurler polydystrophy), while mutations in the GNPTG gene cause the least severe phenotype known as mucolipidosis III γ (7). Proteolytic cleavage of the human 1256 aa α/β precursor at K928 is mediated by the Site-1 protease (S1P) in the Golgi and this cleavage is essential for catalytic competency of the protein (8). The α and β subunits harbor four Stealth domains that together form the catalytic core of the protein (Fig. 1A). The Stealth domains of all eukaryotic GlcNAc-1-PTs are highly conserved and resemble sequences within bacterial genes that encode sugar-phosphate transferases involved in cell wall polysaccharide biosynthesis (9). The α subunit also contains two Notch modules and a DNA methyltransferase-associated protein (DMAP) interaction domain that we have demonstrated have roles in the specific recognition of protein determinants on lysosomal acid hydrolases, resulting in phosphorylation of their high mannose oligosaccharides (10). In addition, the α/β precursor has four so-called “spacer” domains of which only one, spacer-2, has been characterized as the γ-subunit binding site (10, 11). Hitherto, no function has been ascribed to the other spacer regions.
Fig. 1. Spacer-1 domain regulates site of cleavage of α/β precursor.
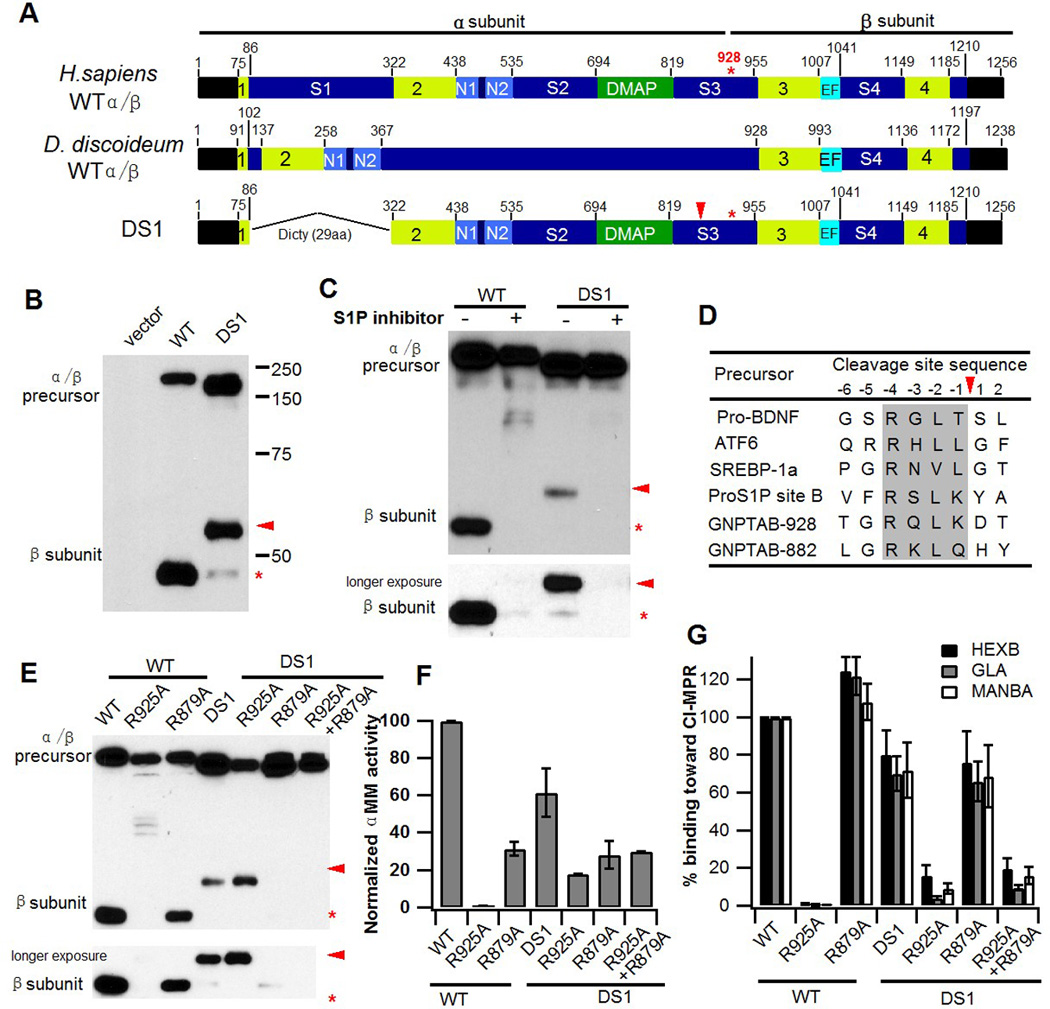
A. Schematic of human and D. discoideum GlcNAc-1-PT α/β subunit modular arrangement. It is not known whether the D. discoideum protein undergoes proteolytic processing like the human protein. The 4 regions shown in lime together comprise the Stealth, an evolutionarily conserved domain first identified in bacterial proteins involved in capsular polysaccharide biosynthesis (9). In the construct DS1, the human spacer-1 sequence was replaced with 29 aa of the corresponding region from D. discoideum. B. Immunoblot analysis of WT α/β versus the DS1 deletion mutant expressed in GNPTAB−/− HeLa cells and probed with anti-V5 antibody. C. Inhibition of S1P activity of GNPTAB−/− HeLa cells transfected with either WT α/β precursor or the DS1 mutant cDNA. 24 h post-transfection, PF-429242 was added to the cells at a final concentration of 10 µM and cells were incubated for a further 24 h before being harvested. Cell extracts were prepared and 20 µg of each lysate was separated by SDS-PAGE and subject to western blotting. D. Amino acid alignment of the two GlcNAc-1-PT α subunit S1P substrate sites with other known S1P sites. The shaded box shows the conserved consensus cleavage motif (19). E. Immunoblot analysis of the point mutants, R925A, R879A, and R925A/R879A in the context of either WT α/β or the DS1 deletion mutant. Proteins expressed in GNPTAB−/− HeLa cells were separated by SDS-PAGE gel, transferred to nitrocellulose and probed with anti-V5 antibody. F. Phosphotransferase activity toward the simple sugar αMM, using extracts of GNPTAB−/− cells transfected with WT or DS1 α/β precursors, or the various point-mutation cDNAs of these constructs. Activity was normalized to total protein concentration. G. Transfection of GNPTAB−/− HeLa cells with either WT or DS1 α/β precursors, or the various mutants shown in F to evaluate the degree of lysosomal enzyme phosphorylation. Cell extracts were incubated with CI-MPR affinity beads to bind the phosphorylated lysosomal enzymes. The beads were then assayed for the activity of these lysosomal enzymes. The values obtained with cells transfected with WT α/β are set to 100%. In all 3 instances, the values obtained with cells transfected with cDNAs for the lysosomal enzymes alone were less than 1 % of the values obtained by co-transfection with the WT α/β precursor.
In this study, we investigated the role of spacer-1 in the maturation and function of GlcNAc-1-PT. We found that spacer-1 dictates cleavage of the α/β precursor precisely at K928 by the site-1 protease (S1P) so as to allow for full catalytic activity as removal of spacer-1 results in cleavage at an alternate site (Q882). In addition, deletion of spacer-1 gives rise to an enzyme with enhanced ability to phosphorylate a number of non-lysosomal glycoproteins. Together these findings reveal a novel and unexpected role for spacer-1 in the maturation of the GlcNAc-1-PT α/β precursor and the minimization of phosphorylation of non-lysosomal proteins.
Materials & Methods
Cell lines
The GNPTAB−/− HeLa cell line has been described in detail elsewhere (10). Cells were maintained in DMEM (Life Technologies, Carlsbad, CA) containing 0.11 g/L sodium pyruvate and 4.5 g/L glucose, supplemented with 10% (vol/vol) FBS (Atlanta Biologicals, Flowery Branch, GA), 100,000 U/L penicillin, 100 mg/L streptomycin (Life Technologies) and 2 mM L-glutamine (Life Technologies).
DNA constructs
Human GNPTAB-V5/His in pcDNA6 has been described (12). In order to generate the DS1 and ΔS1 constructs, 0.5 kb gBlocks gene fragments were synthesized (IDT Inc., Coralville, IA) that encoded either the D. discoideum spacer-1 sequence or a Gly/Ser linker sequence together with the human Stealth1 and Stealth 2 sequences. The gBlocks gene fragments were utilized in the first step of a 2-step overlap-extension-PCR to generate the desired constructs. Point mutations were generated by the QuikChange site-directed mutagenesis method and all sequences were confirmed to be correct by DNA sequencing.
The LIF cDNA construct was kindly provided by Richard Steet (University of Georgia. Athens, GA), while the Renin-HA cDNA was purchased from Addgene (Cambridge, MA). DNase I, glycopepsinogen, CathD-myc, GLA, and NPC2-myc have been described (10, 13, 14).
Immunofluorescence microscopy
To visualize the subcellular localization of WT α/β and the various mutants, the different constructs were transfected into GNPTAB−/− HeLa cells using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s protocol. 24 h post-transfection, the cells were fixed and the α/β subunits were detected with mouse anti-V5 monoclonal antibody (Life Technologies). The Golgi marker, GOLPH4, was detected with rabbit anti-GOLPH4 polyclonal antibody (Abcam, Cambridge, MA). The processed cells were mounted in ProLong® Gold antifade mounting medium (Life Technologies), and the images were acquired with an LSM880 confocal microscope (Carl Zeiss Inc., Peabody, MA). Images were analyzed by Image J software (Fiji).
CI-MPR affinity chromatography and enzyme assays
Soluble bovine CI-MPR was purified from fetal bovine serum and covalently conjugated to Cyanogen bromide-activated-Sepharose 4B (Sigma-Aldrich, St. Louis, MO) as described (15). Cell extracts from 2-day transfected GNPTAB−/− HeLa were incubated with CI-MPR affinity beads, which were washed twice with buffer and then assayed for bound lysosomal enzymes as previously described (12). The extracts were also assayed for GlcNAc-1-PT activity using the α-methylmannoside (αMM) assay (12). Enzyme activity was normalized to the total protein concentration of the different cell extracts.
Western blotting
Proteins resolved by SDS-PAGE under reducing conditions were transferred to nitrocellulose membrane and detected with antibodies as described in the figure legends. Equal amount of whole cell extracts were loaded.
[2-3H]Mannose labeling experiments
Labeling experiments were performed with transfected GNPTAB−/− HeLa cells as follows: 48 h post-transfection of WT or DS1 cDNA, along with cDNAs for the various acid hydrolases or non-lysosomal glycoproteins, cells in 60-mm tissue culture plates were incubated with 50–150 µCi of [2-3H]mannose (Perkin Elmer, Waltham, MA) for 2 h, followed by the addition of complete medium containing 5 mM glucose, 5 mM mannose and 10 mM NH4Cl to stop mannose uptake and induce secretion. The cells were incubated for an additional 3 h before the media was collected. In several experiments, cell extracts were prepared and equal amounts of lysates were subjected to Western blotting for β subunit content to confirm that the constructs were being expressed at comparable levels (not shown).
Immunoprecipitation and oligosaccharide analysis
The proteins of interest secreted into the media were immunoprecipitated, and oligosaccharides isolated and analyzed essentially as described in detail previously (16). For the CathD-myc, NPC2-myc and Renin-HA experiments, 20 µl anti-myc monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or 5 µl anti-HA monoclonal antibody (Sigma-Aldrich) was pre-bound to 100 µl Protein G-agarose-PLUS beads (Santa Cruz Biotechnology) prior to immunoprecipitation of labeled proteins from the media. In the case of GLA, DNase I, and LIF, the secreted enzymes were immunoprecipitated with Protein G-agarose-PLUS beads pre-bound to anti-GLA antibody (Amicus Therapeutics, Cranbury, NJ), and rProteinA-agarose beads (RepliGen, Waltham, MA) pre-bound to anti-DNase I antibody (Sigma-Aldrich), or anti-LIF antibody (generously provided by Frederic Blanchard, University of Nantes, Nantes, France). Immunoprecipitated material was treated with Endo H (NEB, Ipswich, MA) and filtered with Ultracel-10K (EMD Millipore, Billerica, MA). The filtrate containing released neutral and phosphorylated high mannose glycans was treated with mild acid to remove any N-acetylglucosamine residues still attached to the phosphate moieties and applied to a QAE-column matrix to separate the oligosaccharides bearing zero, one or two Man-6-P residues. The retentate containing Endo H-resistant complex-type oligosaccharides was treated with Pronase (Roche Diagnostics, Indianapolis, IN) and fractionated on ConA-sepharose 4B (GE Healthcare, Pittsburgh, PA) to separate the complex-type glycans from the small amount of high mannose glycans not released by Endo H. The [2-3H]-mannose content of each fraction was determined and the percent phosphorylation was calculated as described (16). In all cases, values obtained with the mock transfection were subtracted.
Results
Deletion of spacer-1 results in GlcNAc-1-PT α/β cleavage at an alternate site
As a first-step toward analyzing the function of the spacer-1 domain of the α/β subunit of GlcNAc-1-PT, we performed a sequence alignment between the human protein and that of the lower eukaryote D. discoideum. While it has been demonstrated that the D. discoideum GlcNAc-1-PT is responsible for the transfer of GlcNAc-1-P residues from the donor UDP-GlcNAc to specific mannoses on the N-linked high mannose glycans of the acceptor glycoproteins (17), we noted that the human spacer-1 sequence is 200 aa longer than the short sequence present in D. discoideum (Fig. 1A), as is the case with all mammalian GlcNAc-1-PT spacer-1 regions for which sequence data is available. This suggested that the mammalian spacer-1 region might play a role not associated with the D. discoideum spacer-1 sequence. To examine this possibility, the 236 aa human spacer-1 sequence was replaced with 29 aa of the D. discoideum sequence at the DNA level and the resulting construct (Fig. 1A, DS1) was transfected into GNPTAB−/− HeLa cells generated by the CRISPR/Cas9 method (10). Western blot analysis of whole cell extracts expressing the WT and DS1 mutant was performed to determine if replacement of human spacer-1 with the D. discoideum sequence allowed for efficient folding of the mutant protein and its exit from the endoplasmic reticulum (ER) to the cis-Golgi where the α/β precursor is cleaved to the α and β subunits. As shown in Fig. 1B, the mutant protein is indeed expressed well and cleaved, but the bulk of the β subunit product migrated slower on an SDS-PAGE gel than the WT β subunit (Fig. 1B, arrowhead), indicating that most of the DS1 mutant is being cleaved at an alternate upstream site relative to the K928 cleavage site of the WT protein (Fig. 1B, *). A small amount of the normal β subunit was also seen with DS1 (Fig. 1B *). This raised the question as to whether the alternate cleavage resulting from removal of spacer-1 is due to the same protease, S1P, that cleaves WT α/β precursor at K928, or if a different protease might be involved. To address this issue, we treated cells with an inhibitor of S1P, the aminopyrrolidineamide PF-429242 (18). The presence of the inhibitor resulted in loss of the β subunit formation in both the WT and the DS1 mutant precursors (Fig. 1C), demonstrating that cleavage at the alternate site is mediated by S1P. If this is the case, an additional consensus S1P cleavage site should exist N-terminal to the original cleavage site.
An examination of GlcNAc-1-PT α/β amino acid sequence revealed this to be true with the consensus key arginine residue, R879, occurring at the invariant −4 position, and cleavage postulated to occur at Q882 (Fig. 1D) (19). Cleavage at Q882 is consistent with the increase in molecular mass of the β subunit seen with DS1 by SDS-PAGE. As expected, mutation of R925 abolishes cleavage of WT α/β precursor at K928 (Fig. 1E, lane 2). Mutation of R879, on the other hand, did not affect the normal processing of the full-length α/β precursor at K928 (Fig. 1E, lane 3), but abolished cleavage at Q882 for the DS1 mutant, as shown by loss of the slower migrating β subunit (Fig. 1E, lane 6). The trace amount of K928 cleaved β in this case was not affected (Fig. 1E, lane 6, longer exposure). Mutation of both R925 and R879 resulted in complete loss of β formation (Fig. 1E, lane 7). These data identify Q882 as a novel S1P cleavage site in the α/β precursor that is rarely utilized except in the absence of spacer-1.
Proteolytic processing of the WT α/β precursor at residue K928 is essential for generation of a catalytically active enzyme (4, 12). This raises the question as to whether cleavage at the Q882 site instead of at K928 also results in an active enzyme. In order to address this question, we tested the activity of the point mutants shown in Fig. 1E toward the simple sugar αMM (Fig. 1F) and a number of lysosomal enzymes, (Fig. 1G), both in the context of WT α/β precursor as well as the DS1 mutant. The various constructs were expressed in GNPTAB−/− HeLa cells and 48 h post-transfection, cells extracts were prepared and an aliquot of each was saved to perform the αMM activity assay (Fig. 1F). The remaining extracts were incubated with beads containing immobilized cation-independent (CI)-MPR to bind the lysosomal enzymes that had been phosphorylated. The beads were washed and assayed for the extent of binding of three lysosomal enzymes as described in Materials & Methods (Fig. 1G). As shown in Figs. 1F & 1G, the R925A mutant in the context of WT α/β precursor had only background activity toward both αMM and lysosomal enzymes, in concordance with previous findings (12). R879A/WT, on the other hand, exhibited 30% of the activity toward αMM and between 110–125% of the activity toward the three lysosomal enzymes compared to the WT α/β precursor. The basis for the discrepancy observed with the R879A mutant between αMM and lysosomal enzyme activity is not clear at this time.
The DS1 mutant exhibited approximately 60% of WT activity toward both αMM (Fig. 1F) and the panel of lysosomal enzymes (Fig. 1G). In contrast to the findings with the WT, the R925A mutant in the DS1 background retained almost 20% of WT activity toward αMM, as did the R925A/R879A double mutant (Fig. 1F). These mutants also exhibited low levels of activity toward the lysosomal enzyme panel (Fig. 1G). Since the double mutant lacks any proteolytic processing, it demonstrates that in the absence of spacer-1, the uncleaved α/β precursor is partially active. Interestingly, the R879A single mutant retained good activity toward the lysosomal enzyme panel, most likely the result of a combination of the activity of the α/β precursor plus the trace amount of β originating from cleavage at K928 (Fig. 1G).
Since the 236 aa human spacer-1 sequence was replaced with 29 aa of the D. discoideum sequence, we considered the possibility that utilization of the alternate cleavage site is a consequence of introducing the D. discoideum sequence as opposed to removal of the human spacer-1 sequence. To exclude this, another spacer-1 deletion mutant was constructed in which human spacer-1 was replaced with a 26 aa linker comprising of the small residues Gly and Ser (Fig. 2A, ΔS1). Δspacer-1 (ΔS1) behaved in every respect similar to DS1 in that the proteolytic processing mediated by S1P resulted in cleavage for the most part at the new site (Q882) (Fig. 2B), and ΔS1 had 40% of WT activity toward αMM (Fig. 2C). Moreover, the S1P inhibitor, PF-429242, blocked formation of the β subunit with ΔS1, as it did with WT and the DS1 mutant α/β precursors (Fig. 2D). We ascertained that both DS and ΔS1, along with the point mutants shown in Fig. 1E had identical Golgi localization as WT GlcNAc-1-PT (Fig. 3), ruling out mislocalization of these mutants as a possible cause for the altered cleavage. These results clearly show that the presence of the 236aa spacer-1 sequence in human GlcNAc-1-PT ensures cleavage at K928 instead of Q882.
Fig. 2. Replacement of human spacer-1 with a GGS linker results in cleavage at Q882.
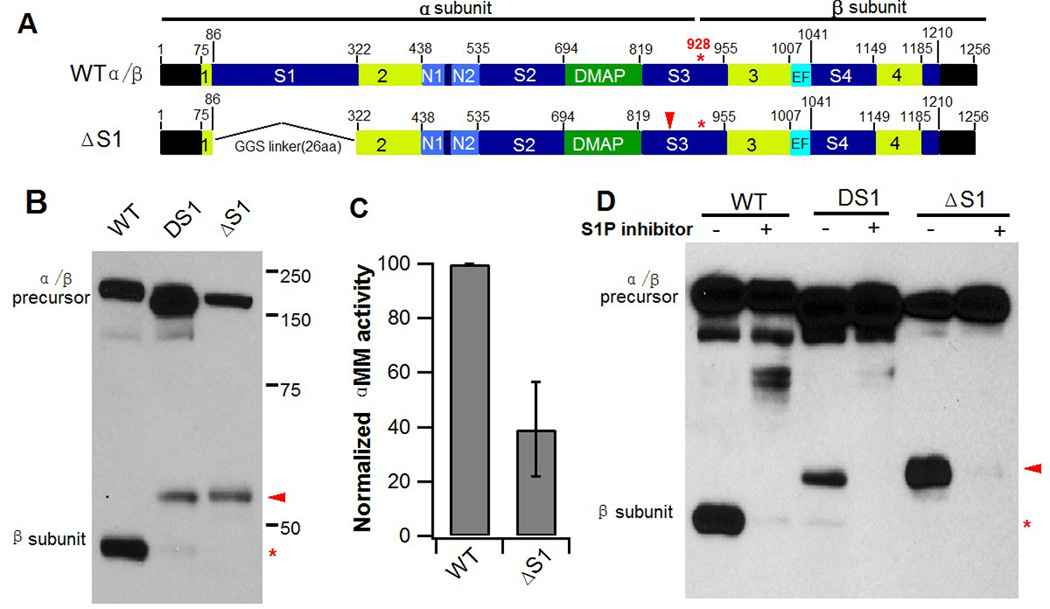
A. Schematic of GlcNAc-1-PT α/β subunit modular arrangement and replacement of the human spacer-1 sequence with a 26 aa linker sequence comprising of Gly and Ser residues. B. Immunoblot analysis of WT α/β versus the DS1 and ΔS1 deletion mutants expressed in GNPTAB−/− HeLa cells and probed with anti-V5 antibody. C. Phosphotransferase activity toward the simple sugar αMM, using extracts of GNPTAB−/− cells transfected with WT α/β precursor or the ΔS1 mutant cDNA. Activity was normalized to total protein concentration. D. Inhibition of S1P activity of GNPTAB−/− HeLa cells transfected with either WT α/β precursor, the DS1 or the ΔS1 mutant cDNA. 24 h post-transfection, PF-429242 was added to the cells at a final concentration of 10 µM and cells were incubated for a further 24 h before being harvested. Cells extracts were prepared and 20 µg of each lysate was separated by SDS-PAGE and subject to western blotting.
Fig. 3. Golgi localization of WT α/β precursor and mutants.
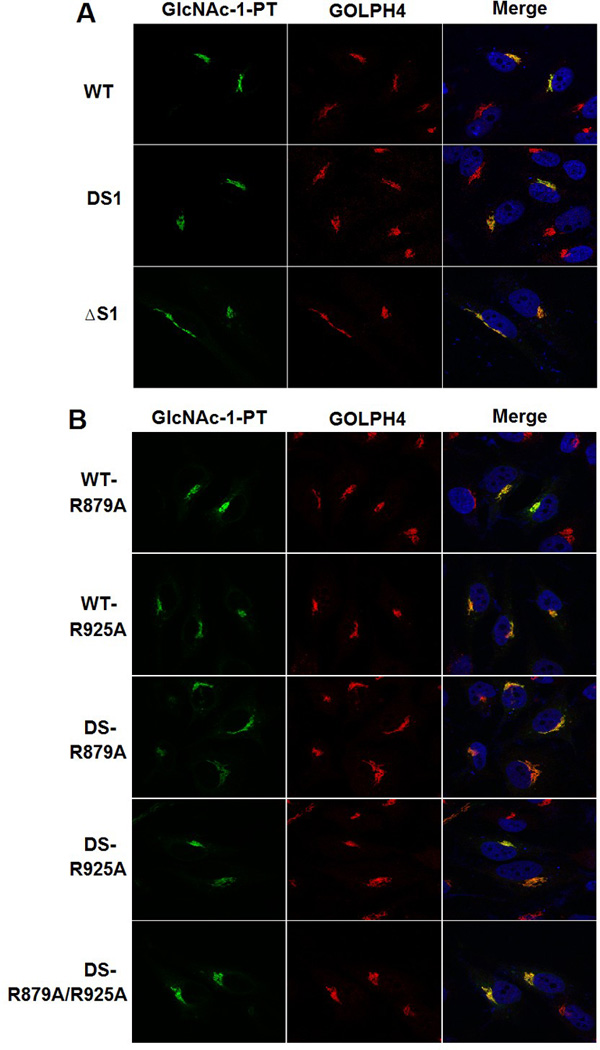
Confocal immunofluorescence images of GNPTAB−/− HeLa cells transfected with either WT α/β precursor, or the indicated mutant cDNAs, and colocalized with the Golgi markers GOLPH4, respectively (see Materials & Methods).
Deletion of spacer-1 enhances phosphorylation of several non-lysosomal glycoproteins
We next sought to determine the consequence of spacer-1 deletion on the phosphorylation of a number of non-lysosomal glycoproteins that have been shown to acquire low levels of the Man-6-P tag (13, 20–22). For this purpose, cDNAs for the non-lysosomal glycoproteins DNase1, Renin and leukemia inhibitory factor (LIF) were co-transfected along with either WTα/β precursor or the DS1 mutant cDNA, and the degree of phosphorylation quantitated using [2-3H]mannose-labeling, immunoprecipitation and direct glycan analysis as described in Material & Methods. In all three cases, the extent of mannose phosphorylation mediated by DS1 was 1.4–2.3 fold higher than that achieved with the WT construct (Fig. 4A). There was also a substantial increase in the percent oligosaccharides with two Man-6-P residues in the case of DNase1 and Renin. The lysosomal proteins, α-Galactosidase (GLA), Niemann-Pick disease, type C2 (NPC2) and Cathepsin D (CathD), on the other hand, showed a similar degree of phosphorylation irrespective of whether WT or the DS1 construct was co-transfected into the GNPTAB−/− HeLa cells along with expression vectors for the individual enzymes (Fig. 4A), in agreement with our previous findings with the three endogenous lysosomal enzymes (Fig. 1G). There was, however, a small increase in the content of glycans with two man-6-P residues in the case of GLA. Consistent with these data, Renin but not NPC2 displayed increased binding to immobilized CI-MPR when the cDNAs for these two proteins were co-transfected with the DS1 construct compared to the WT α/β precursor (Fig. 4B). The enhanced binding of Renin likely reflects its increased content of glycans with two Man-6-P residues. Neither glycopepsinogen (GP) nor the membrane glycoproteins, Lamp1 and Lamp2, showed any detectable binding under these conditions (Fig. 4B). These results demonstrate that spacer-1 limits the extent of phosphorylation of a number of non-lysosomal glycoproteins.
Fig. 4. Deletion of spacer-1 enhances phosphorylation of several non-lysosomal glycoproteins.
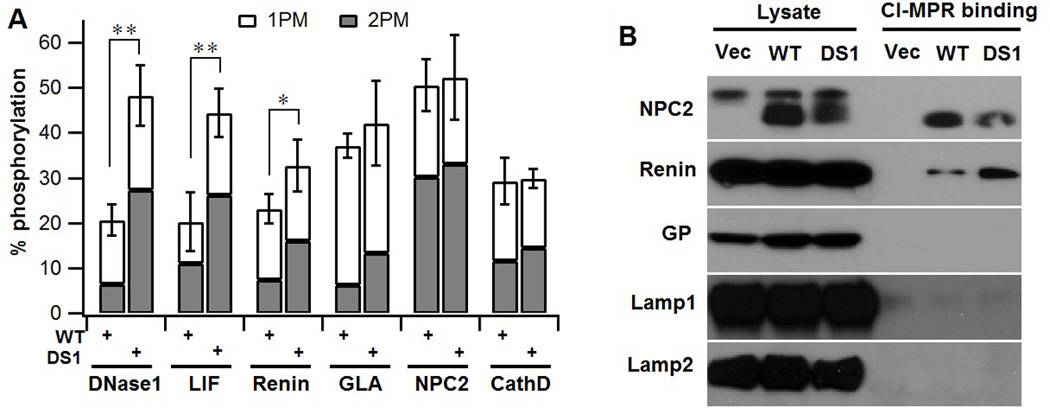
A. GNPTAB−/− HeLa cells were co-transfected with plasmids encoding either the 3 non-lysosomal proteins or 3 lysosomal proteins along with WT α/β precursor or the DS1 mutant cDNA. Cells were labeled with [2-3H]-mannose, followed by immunoprecipitation of the proteins secreted into the media and determination of the percent N-glycans containing either one (1PM) or two (2 PM) Man-6-P residues. In all instances, the lysosomal proteins secreted by the cells transfected with plasmids encoding only the lysosomal proteins contain less than 1 % phosphorylated glycans (10). N= 2–5. B. Western blot of GNPTAB−/− HeLa cells co-transfected with the expression plasmids for the indicated proteins along with empty vector, WT α/β precursor or the DS1 mutant cDNA. Cell lysates were incubated with CI-MPR-affinity beads and the binding of the various proteins was determined by probing the blot with the following antibodies: Renin - anti-HA; NPC2, GP, Lamp1 and Lamp2 with antibodies generated against the native protein.
Discussion
The findings presented in this study provide the first evidence for a role of spacer-1 in the biosynthesis and function of the α/β subunits of GlcNAc-1-PT. With regards to biosynthesis, we found that deletion of spacer-1 results in cleavage of the α/β precursor by the S1P occurring predominantly at Q882 rather than at K928, the normal cleavage site. The reason why S1P cleaves the enzyme at Q882 rather than at K928 in the absence of spacer-1 has not been resolved at this point. One possible explanation is that deletion of spacer-1 leads to a conformational change in the α/β precursor such that the Q882 cleavage site becomes exposed to the S1P while the K928 site becomes inaccessible to this protease. Interestingly, Velho et al. have reported that an in-frame deletion of residues Y937 to M972 of the α/β precursor identified in a mucolipidosis II patient results in cleavage by S1P at an alternate upstream site within the α subunit (23). While the new cleavage site was not identified, it is likely to be Q882, with the in-frame deletion leading to a conformational change that exposes the Q882 site. An alternate but less likely explanation for the altered cleavage site is that spacer-1 directly shields the Q882 site from the action of S1P. The spacer-1 region has a defined structure (PDB ID:2N6D) and could potentially cover the Q882 site (24). However, this would not explain why cleavage at the normal K928 site does not occur even when the S1P motif at Q882 is mutated.
Another surprising finding to emerge from these studies is that under some circumstances, cleavage of the α/β precursor is not required for catalytic activity. We first noted this with the DS1 construct that was mutated at both sites to prevent cleavage by S1P. This mutant had about 30% of WT catalytic activity toward αMM. Presumably, there is more than one path to the formation of a catalytically active enzyme. The other major finding in this study is that spacer-1 serves to minimize phosphorylation of the high mannose glycans of a group of non-lysosomal glycoproteins known to be substrates of the transferase. The deletion of spacer-1 led to an increase in the phosphorylation of DNase1, Renin, and LIF by 1.4–2.3 fold over that achieved by the WT enzyme. The basis for this effect awaits further investigation. Nevertheless, the findings presented in this study provide evidence for spacer-1 modulating the maturation and function of GlcNAc-1-PT. In view of this, we propose that the “spacer-1” domain be renamed the “regulatory-1” domain.
Acknowledgments
This work was supported by National Institutes of Health grant CA-008759 and the Yash Gandhi Foundation.
Footnotes
Author contributions: LL, WSL, BD and SK designed research; LL, WSL, and BD performed research; LL, WSL, BD and SK analyzed data; and LL, WSL, BD and SK wrote the paper.
References
- 1.Lang L, Reitman M, Tang J, Roberts RM, Kornfeld S. Lysosomal enzyme phosphorylation. Recognition of a protein-dependent determinant allows specific phosphorylation of oligosaccharides present on lysosomal enzymes. The Journal of biological chemistry. 1984;259(23):14663–14671. [PubMed] [Google Scholar]
- 2.Kornfeld S. Trafficking of lysosomal enzymes in normal and disease states. The Journal of clinical investigation. 1986;77(1):1–6. doi: 10.1172/JCI112262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao M, Booth JL, Elmendorf BJ, Canfield WM. Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. The Journal of biological chemistry. 1996;271(49):31437–31445. doi: 10.1074/jbc.271.49.31437. [DOI] [PubMed] [Google Scholar]
- 4.Kudo M, et al. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. The Journal of biological chemistry. 2005;280(43):36141–36149. doi: 10.1074/jbc.M509008200. [DOI] [PubMed] [Google Scholar]
- 5.Raas-Rothschild A, et al. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC) The Journal of clinical investigation. 2000;105(5):673–681. doi: 10.1172/JCI5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiede S, et al. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nature medicine. 2005;11(10):1109–1112. doi: 10.1038/nm1305. [DOI] [PubMed] [Google Scholar]
- 7.Braulke T, Raas-Rothschild A, Kornfeld S. I-Cell Disease and Pseudo-Hurler Polydystrophy: Disorders of Lysosomal Enzyme Phosphorylation and Localization. In: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G, editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw-Hill Companies, Inc.; 2014. [Google Scholar]
- 8.Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333(6038):87–90. doi: 10.1126/science.1205677. [DOI] [PubMed] [Google Scholar]
- 9.Sperisen P, Schmid CD, Bucher P, Zilian O. Stealth proteins: in silico identification of a novel protein family rendering bacterial pathogens invisible to host immune defense. PLoS computational biology. 2005;1(6):e63. doi: 10.1371/journal.pcbi.0010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Meel E, et al. Multiple Domains of GlcNAc-1-phosphotransferase Mediate Recognition of Lysosomal Enzymes. The Journal of biological chemistry. 2016;291(15):8295–8307. doi: 10.1074/jbc.M116.714568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Pace R, et al. Subunit interactions of the disease-related hexameric GlcNAc-1-phosphotransferase complex. Human molecular genetics. 2015;24(23):6826–6835. doi: 10.1093/hmg/ddv387. [DOI] [PubMed] [Google Scholar]
- 12.Qian Y, et al. Analysis of Mucolipidosis II/III GNPTAB Missense Mutations Identifies Domains of UDP-GlcNAc:lysosomal Enzyme GlcNAc-1-phosphotransferase Involved in Catalytic Function and Lysosomal Enzyme Recognition. The Journal of biological chemistry. 2015;290(5):3045–3056. doi: 10.1074/jbc.M114.612507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishikawa A, Nanda A, Gregory W, Frenz J, Kornfeld S. Identification of amino acids that modulate mannose phosphorylation of mouse DNase I, a secretory glycoprotein. The Journal of biological chemistry. 1999;274(27):19309–19315. doi: 10.1074/jbc.274.27.19309. [DOI] [PubMed] [Google Scholar]
- 14.Steet R, Lee WS, Kornfeld S. Identification of the minimal lysosomal enzyme recognition domain in cathepsin D. The Journal of biological chemistry. 2005;280(39):33318–33323. doi: 10.1074/jbc.M505994200. [DOI] [PubMed] [Google Scholar]
- 15.Valenzano KJ, Remmler J, Lobel P. Soluble insulin-like growth factor II/mannose 6-phosphate receptor carries multiple high molecular weight forms of insulin-like growth factor II in fetal bovine serum. The Journal of biological chemistry. 1995;270(27):16441–16448. doi: 10.1074/jbc.270.27.16441. [DOI] [PubMed] [Google Scholar]
- 16.Dustin ML, Baranski TJ, Sampath D, Kornfeld S. A novel mutagenesis strategy identifies distantly spaced amino acid sequences that are required for the phosphorylation of both the oligosaccharides of procathepsin D by N-acetylglucosamine 1-phosphotransferase. The Journal of biological chemistry. 1995;270(1):170–179. doi: 10.1074/jbc.270.1.170. [DOI] [PubMed] [Google Scholar]
- 17.Qian Y, West CM, Kornfeld S. UDP-GlcNAc:Glycoprotein N-acetylglucosamine-1-phosphotransferase mediates the initial step in the formation of the methylphosphomannosyl residues on the high mannose oligosaccharides of Dictyostelium discoideum glycoproteins. Biochemical and biophysical research communications. 2010;393(4):678–681. doi: 10.1016/j.bbrc.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay BA, et al. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorganic & medicinal chemistry letters. 2007;17(16):4411–4414. doi: 10.1016/j.bmcl.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 19.Elagoz A, Benjannet S, Mammarbassi A, Wickham L, Seidah NG. Biosynthesis and cellular trafficking of the convertase SKI-1/S1P: ectodomain shedding requires SKI-1 activity. The Journal of biological chemistry. 2002;277(13):11265–11275. doi: 10.1074/jbc.M109011200. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard F, et al. The mannose 6-phosphate/insulin-like growth factor II receptor is a nanomolar affinity receptor for glycosylated human leukemia inhibitory factor. The Journal of biological chemistry. 1998;273(33):20886–20893. doi: 10.1074/jbc.273.33.20886. [DOI] [PubMed] [Google Scholar]
- 21.Faust PL, Chirgwin JM, Kornfeld S. Renin, a secretory glycoprotein, acquires phosphomannosyl residues. The Journal of cell biology. 1987;105(5):1947–1955. doi: 10.1083/jcb.105.5.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sleat DE, Zheng H, Qian M, Lobel P. Identification of sites of mannose 6-phosphorylation on lysosomal proteins. Molecular & cellular proteomics : MCP. 2006;5(4):686–701. doi: 10.1074/mcp.M500343-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Velho RV, et al. Analyses of disease-related GNPTAB mutations define a novel GlcNAc-1-phosphotransferase interaction domain and an alternative site-1 protease cleavage site. Human molecular genetics. 2015;24(12):3497–3505. doi: 10.1093/hmg/ddv100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano P, Geralt M, Wuthrich K. NMR structure of the 140–315 fragment of the N-acetylglucosamine-1-phosphate transferase, alpha and beta subunits. 2015 http://www.rcsb.org/pdb/explore/explore.do?structureId=2N6D. [Google Scholar]