

SUMMARY
Bacterial CRISPR-Cas systems utilize sequence-specific RNA-guided nucleases to defend against bacteriophage infection. As a counter-measure, numerous phages are known that produce proteins to block the function of Class 1 CRISPR-Cas systems. However, currently no proteins are known to inhibit the widely used Class 2 CRISPR-Cas9 system. To find these inhibitors, we searched cas9-containing bacterial genomes for the co-existence of a CRISPR spacer and its target, a potential indicator for CRISPR inhibition. This analysis led to the discovery of four unique type II-A CRISPR-Cas9 inhibitor proteins encoded by Listeria monocytogenes prophages. More than half of L. monocytogenes strains with cas9 contain at least one prophage-encoded inhibitor, suggesting widespread CRISPR-Cas9 inactivation. Two of these inhibitors also blocked the widely used Streptococcus pyogenes Cas9 when assayed in Escherichia coli and human cells. These natural Cas9-specific “anti-CRISPRs” present tools that can be used to regulate the genome engineering activities of CRISPR-Cas9.
Keywords: CRISPR-Cas, Cas9, dCas9, anti-CRISPR, Listeria monocytogenes, bacteriophage, prophage, gene editing, Cas9 inhibitor
Graphical Abstract
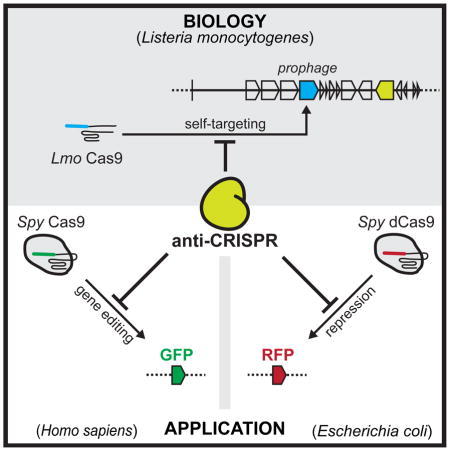
Four CRISPR-Cas9 inhibitor proteins encoded by Listeria monocytogenes prophages prevent Cas9 binding and gene editing in bacteria and human cells, including currently the most widely used Cas9 from Streptococcus pyogenes.
INTRODUCTION
The ability to prevent attack from viruses is a hallmark of cellular life. Bacteria employ multiple mechanisms to resist infection by bacterial viruses (phages), including restriction enzymes and CRISPR-Cas systems (Labrie et al., 2010). CRISPR arrays consist of the DNA remnants of previous phage encounters (spacers), located between Clustered Regularly Interspaced Short Palindromic Repeats (Mojica et al., 2005). These spacers are transcribed to generate CRISPR RNAs (crRNAs) that direct the binding and cleavage of specific nucleic acid targets (Brouns et al., 2008; Garneau et al., 2010). The CRISPR-associated (cas) genes required for immune function are often found adjacent to the CRISPR array (Marraffini, 2015; Wright et al., 2016). Cas proteins perform many functions, including destroying foreign genomes (Garneau et al., 2010), mediating the acquisition of foreign sequences into the CRISPR array (Nuñez et al., 2014; Yosef et al., 2012) and facilitating the production of mature CRISPR RNAs (crRNAs) (Deltcheva et al., 2011; Haurwitz et al., 2010).
CRISPR-Cas adaptive immune systems are both common and diverse in the bacterial world. Two distinct classes, encompassing six CRISPR types (I–VI) have been identified across bacterial genomes (Abudayyeh et al., 2016; Makarova et al., 2015), each with the ability to cleave target DNA or RNA molecules with sequence specificity directed by the RNA guide. The facile programmability of CRISPR-Cas systems has been widely exploited, opening the door to an array of novel genetic technologies, most prominently gene editing in animal cells (Barrangou and Doudna, 2016). Most technologies are based on Cas9 (Class 2, type II-A) from Streptococcus pyogenes (Spy), together with an engineered single guide RNA (sgRNA) because of the simplicity of the system (Jinek et al., 2012). Gene editing in animal cells has been successful with Spy Cas9 (Cong et al., 2013; Mali et al., 2013), Cas9 orthologs within the II-A subtype (Ran et al., 2015), and new Class 2 single protein effectors such as Cpf1 (Type V (Zetsche et al., 2015)). Applications are also being developed through the characterization of Type VI CRISPR-Cas systems, represented by C2c2, which naturally cleave RNA (Abudayyeh et al., 2016; East-Seletsky et al., 2016). In contrast, the complex Class 1 CRISPR-Cas systems (Type I, III, and IV), consisting of RNA-guided multi-protein complexes and thus have been overlooked for most genomic applications. These systems are, however, the most common in nature, comprising ~75% of all bacterial CRISPR-Cas systems and nearly all systems in archaea (Makarova et al., 2015).
In response to the bacterial war on phage infection, phages, in turn, often encode inhibitors of bacterial immune systems that enhance their ability to either lyse their host bacterium or integrate into its genome (Samson et al., 2013). The first examples of phage-encoded “anti-CRISPR” proteins came for the Class 1 type I-F and I-E systems in Pseudomonas aeruginosa (Bondy-Denomy et al., 2013; Pawluk et al., 2014). Remarkably, ten type I-F anti-CRISPR and four type I-E anti-CRISPR genes have been discovered to date (Pawluk et al., 2016), all of which encode distinct, small proteins (50–150 amino acids), previously of unknown function. Our biochemical investigation of four I-F anti-CRISPR proteins revealed that they directly interact with different Cas proteins in the multi-protein CRISPR-Cas complex to prevent either the recognition or cleavage of target DNA (Bondy-Denomy et al., 2015). Anti-CRISPR proteins have distinct sequences (Bondy-Denomy et al., 2013), structures (Maxwell et al., 2016; Wang et al., 2016), and modes of action (Bondy-Denomy et al., 2015). These findings support the independent evolution of CRISPR-Cas inhibitors and suggests that many more are yet to be discovered. Indeed, a recent investigation exploited the conservation of signature anti-CRISPR associated (aca) gene with a predicted helix-turn-helix (HTH) motif to identify anti-CRISPRs across proteobacteria, broadly spanning the type I-F CRISPR-Cas phylogeny (Pawluk et al., 2016).
Although anti-CRISPRs are both prevalent and diverse within proteobacteria, it is presently unknown whether anti-CRISPR proteins occur in other bacterial phyla. Likewise, it is also unclear if anti-CRISPRs exist for systems other than types I-E and I-F. In P. aeruginosa, type I anti-CRISPRs are expressed from integrated phage genomes (prophages) and caused the constitutive inactivation of the host CRISPR-Cas system (Bondy-Denomy et al., 2013). In such cases the prophage can possess a DNA target with perfect identity to a CRISPR spacer in the same cell, as the CRISPR-Cas system is inactivated. The genomic co-occurrence of a perfect spacer and its target DNA is called “self-targeting” (Figure 1A). Bacteria with self-targeting require CRISPR-Cas inactivation for survival: in the absence of anti-CRISPR genes, the host genome will be cleaved in the act of targeting the prophage (Bondy-Denomy et al., 2013; Edgar and Qimron, 2010). Expression of an anti-CRISPR, therefore, neutralizes this risk. We surmised that genomes possessing a CRISPR system with apparent self-targeting would be candidates for the identification of new CRISPR-Cas inhibitors. Here, we describe the identification of four previously unknown phage-encoded CRISPR-Cas9 inhibitors in Listeria monocytogenes using a bioinformatics approach to identify incidents of self-targeting. We also demonstrate that two of these inhibitors can block the activity of S. pyogenes Cas9 in bacterial and human cells.
Figure 1. A survey for CRISPR-Cas9 genomic self-targeting (ST) in Listeria monocytogenes.

(A) A schematic depicting the principle of genomic self-targeting, where a mobile genetic element (MGE) possesses a target sequence for a spacer in a CRISPR array in the same genome. CRISPR-Cas9 function in this “self-targeting genome” is presumably inactive for continued cell viability.
(B) The abundance of genomes with (red) and without (gray) cas9-linked self-targeting (ST), in L. monocytogenes genomes. See Table S1 for a list of self-targeting strains.
(C) An example of an ST event, where spacer 16 in the type II-A CRISPR array of strain J0161 has a perfect PAM and protospacer match with a resident prophage (ϕJ0161a). See Figure S1B for the entire CRISPR array.
RESULTS
CRISPR-Cas9 in Listeria monocytogenes targets foreign DNA
Listeria monocytogenes is a facultative intracellular food-borne pathogen with a well characterized phage population. Many L. monocytogenes isolates have type II-A CRISPR-Cas systems (Sesto et al., 2014) and their CRISPR spacers possess identity to many virulent, temperate, and integrated phages (Di et al., 2014; Sesto et al., 2014). However, there is no experimental evidence of canonical CRISPR-Cas function. We analyzed 275 genomes of L. monocytogenes and identified type II-A CRISPR-Cas9 systems (Lmo Cas9) in 15% (n = 41) of them (Figure 1B). Interestingly, we found eight genomes (3% of the total), with examples of self-targeting (ST; Figure 1B and 1C and Table S1), although the CRISPR-Cas9 system is anticipated to be functional as all requisite genes are present with no obvious mutations (Figure S1A). Many self-targeted protospacers were found in prophages, and thus we predicted that these prophages encode inhibitors of CRISPR-Cas9 that allow the stable co-existence of a spacer-protospacer pair.
To test whether inhibitors were encoded by the prophages of L. monocytogenes, we first established the functionality of CRISPR-Cas9 in an L. monocytogenes strain (10403s) that does not exhibit self-targeting. To test the activity of this system we designed a plasmid (pT) possessing a targeted protospacer (i.e. a sequence that is complementary to a natural spacer in the CRISPR array) along with a cognate protospacer adjacent motif (PAM), a three base motif that is necessary for Cas9 binding (Figure 2A). We measured the transformation efficiency of 10403s with either pT or a control plasmid possessing a non-targeted sequence with an identical plasmid backbone (pNT). Transformation with pT yielded miniscule colonies relative to pNT (Figure 2B, leftmost panel), although the number of colonies that emerged upon prolonged incubation were the same (see Discussion for further analysis). To determine whether the 10403s prophage (ϕ10403s) was inhibiting CRISPR-Cas9 function in any way, a prophage-cured version of this strain (ϕcure) was tested, yielding the same tiny colonies (Figure 2B). The ϕcure strain was used for all subsequent experiments since it was indistinguishable from wt10403s in this assay. To confirm that the observed transformation inhibition was the result of CRISPR-Cas9 interference, we constructed a cas9-deletion strain. Transformation of this strain with pT and pNT produced colonies of indistinguishable size (Figure 2B). However, adding back cas9 to the L. monocytogenes chromosome under a constitutively active promoter completely prevented transformation with pT (Figure 2B, rightmost panel). Together, these experiments demonstrate that Cas9 is functional in L. monocytogenes 10403s at both endogenous and overexpressed levels, and limits transformation with a plasmid bearing a protospacer.
Figure 2. A prophage from L. monocytogenes J0161 inhibits CRISPR-Cas9 function.

(A) The type II-A CRISPR-Cas locus in L. monocytogenes 10403s. Four cas genes and the upstream tracrRNA are indicated, along with a CRISPR array containing 30 spacers. The predicted direction of transcription is indicated with black arrows. Subsequent experiments utilize a non-targeted plasmid (pNT) and a targeted plasmid (pT) that has a protospacer matching spacer 1 in this strain.
(B) Representative pictures of colonies of Lmo 10403s wild type (wt), prophage-cured (ϕcure), cas9-deletion strain (Δcas9), and a cas9 overexpression strain (Δcas9 + cas9) after being transformed with pT or pNT plasmids. Bar graphs below the plates show the calculated transformation efficiency (colony forming units per μg of plasmid). Data are represented as the mean of three biological replicates +/− SD. L.D. limit of detection, transformants with small colonies denoted with #.
(C) Plasmid-targeting assay with wild type J0161 (contains the ϕJ0161a prophage; experiment conducted as in (B), except with pTJ0161 as the targeted plasmid) is shown in red to denote self-targeting (as in Figure 1).
(D) A schematic demonstrating the construction of a 10403s strain containing the prophage ϕJ0161a (10403s::ϕJ0161a). See STAR Methods for details. (E) Plasmid-targeting assay with 10403s lysogenized with the ϕJ0161a prophage (10403s::ϕJ0161a) with endogenous (no mod) or overexpressed cas9 (Δcas9 + cas9; experiment conducted as in (B)).
Resident prophages inactivate CRISPR-Cas9 in L. monocytogenes
To determine whether CRISPR-Cas9 may be disabled in a strain with self-targeting spacers, we examined immunity function in L. monocytogenes strain J0161, whose spacer 16 perfectly matches a prophage (ϕJ0161a) in the same genome (Figure 1C). We could not detect any clearly deleterious CRISPR-Cas mutations in the CRISPR repeat, PAM, tracrRNA, Cas9, and the associated promoters of strain J0161 (Figures S1B-F and S2), suggesting that this self-targeting scenario was the result of inhibition and not loss of function. Since the type II-A CRISPR array of J0161 is distinct from that of 10403s, a J0161-specific targeted plasmid (pTJ0161) was used to test the function CRISPR-Cas9 in J0161. Consistent with the inactivation implied by self-targeting, there were no significant differences in transformation efficiency or colony size to distinguish pTJ0161 from pNT (Figure 2C). Thus, we reasoned that the J0161 genome may encode Cas9 inhibitors.
In search of the genetic basis for CRISPR-Cas9 inactivation in J0161, we focused on the prophage ϕJ0161a as a likely source of an inhibitor gene because it contained the self-targeted sequence in this strain. To determine whether ϕJ0161a contained an inhibitor, the prophage-cured strain of 10403s was lysogenized with ϕJ0161a and assayed for CRISPR-Cas9 functionality by plasmid transformation (Figure 2D). The acquisition of ϕJ0161a was sufficient to inactivate CRISPR-Cas9 function (Figure 2E, left panels), suggesting that this prophage encodes an inhibitor of CRISPR-Cas9. The ϕJ0161a prophage also inactivated plasmid targeting in a strain constitutively expressing cas9, suggesting that the inhibitory mechanism does not operate by disrupting natural regulation of the cas9 promoter (Figure 2E, right panels).
Given that the ϕJ0161a prophage inhibited CRISPR-Cas9 function in 10403s, and the endogenous ϕ10403s prophage did not, we compared the genomes of these two closely related phages to identify the regions of difference (Figure 3A). In addition to sharing 39 core phage genes with >40% protein sequence identity, ten non-overlapping unique clusters of genes were identified (cluster boundaries were chosen based on predicted operon structure, with 1–12 genes per cluster). Each cluster was cloned and integrated into the genome of prophage-cured 10403s and assayed for CRISPR-Cas9 function. Of the ten fragments, seven were successfully introduced into L. monocytogenes, while three fragments could not be inserted in the L. monocytogenes genome and were presumably toxic in isolation. Plasmid transformation assays revealed that ϕJ0161a fragment 1 was the only fragment capable of inhibiting CRISPR-Cas9, indicating that this fragment encoded at least one CRISPR-Cas9 inhibitor (Figure 3B). Expressing the individual genes from this four-gene fragment led to the conclusive identification of two anti-CRISPR genes, LMOG_03146 and LMOG_03147 (herein referred to as acrIIA1 and acrIIA2, respectively) while LMOG_03145 and LMOG_03148 (orfB and orfA, respectively) had no anti-CRISPR activity (Figure 3B). Deletion of both acrIIA1 and acrIIA2 from a 10403s::ϕJ0161a lysogen restored CRISPR-Cas9 function, confirming that these are the only anti-CRISPR genes in ϕJ0161a (Figure 3B, rightmost panels).
Figure 3. Identification of four distinct anti-CRISPR proteins.

(A) Comparison of the open reading frames from two similar prophages from L. monocytogenes 10403s and J0161. Unique genes (red) comprising ten fragments of ϕJ0161 were tested for CRISPR-Cas9 inhibition in 10403s. n.e., No effect on CRISPR-Cas9 activity, tox., fragment toxic when expressed, t., location of self-targeted protospacer. The encircled fragment exhibited anti-CRISPR activity with two genes (acrAII1, acrAII2) independently capable of inhibiting CRISPR-Cas activity. Conserved (grey) genes were not tested. For reference, phage genes involved in cell lysis, capsid assembly and host integration (int.) are labeled.
(B) Representative colony pictures of Lmo 10403s ϕcure strains constitutively expressing “fragment 1” (as shown in (A)) or the indicated individual genes from ϕJ0161a transformed with pNT or pT. The rightmost panels show a 10403s lysogen of ϕJ0161a with CRISPR-Cas9 inhibitor genes deleted (::ϕJ0161aΔacrIIA1-2). See Figure S2 for data from the other ϕJ0161a fragments and Figure S2/S3 for full plates.
(C) Representative colony pictures of Lmo 10403s ϕcure strains constitutively expressing acrIIA3, acrIIA4, or orfD transformed with pNT or pT.
Anti-CRISPR genes are widespread in L. monocytogenes prophages
To identify additional type II-A anti-CRISPRs, we examined the genomic position analogous to that of acrIIA1 and acrIIA2 in related L. monocytogenes prophages. A recurring anti-CRISPR (acr) locus containing acrIIA1 within a small operon (2–5 genes) of highly conserved gene order was identified between the “left” integration site and the genes involved in cell lysis (Figure 4A). We identified five additional protein families conserved within acr loci. To test these families for anti-CRISPR function, we cloned and integrated representatives into the 10403s genome and assayed for transformation efficiency of pT and pNT. Two new genes were identified that were capable of CRISPR inactivation (acrIIA3 and acrIIA4), while the remaining three (orfC, D, E) were not (Figure 3C, Figure S3).
Figure 4. Genomic organization and prevalence of acrIIA genes.

(A) The genomic context of acrIIA1 (1) and its homolog from L. monocytogenes (orfD) are depicted to scale as cartoons with acrIIA1 homologs in vertical alignment. Typically, acrIIA genes are encoded within prophages adjacent to or near the phage lysin (ply) gene. Genomic neighbors of acrIIA1 and orfD (acrIIA1-4, orfA-E) are shown. Individual genes (***) were assayed for CRISPR-Cas9 inhibition in L. monocytogenes 10403s (see Figure 3 and Figure S3). Helix-turn-helix (HTH) and AP2 DNA binding motifs were detected in some proteins using hidden markov model (HMM) prediction software (Söding et al., 2005).
(B) Pie-graph representation of the frequency of each acrIIA gene co-occurrences
(C) Pie-graph representation of the prevalence of acrIIA and cas9 genes in the L. monocytogenes pangenome. See Table S1 for relevant accession numbers.
To determine whether CRISPR-Cas9 inactivation in L. monocytogenes is pervasive, we next analyzed the conservation pattern for each anti-CRISPR. Although each acrIIA gene was sufficient to inactivate CRISPR-Cas9 in isolation, we observed a common presence of acrIIA1 in most acr loci. Nearly all instances (88%) of acrIIA2-4 were found upstream of the helix-turn-helix (HTH) motif-containing acrIIA1, suggesting that this gene may be a marker for acr loci (Figure 4A and 4B). The most common scenario in 119 acr loci was either acrIIA1-2 or acrIIA1-2-3, together representing 66% of acr loci (Figure 4B). In total, acrIIA genes were identified in 25% of L. monocytogenes genomes, with 53% of L. monocytogenes cas9-containing strains possessing at least one anti-CRISPR in the same genome (Figure 4C). Many instances of L. monocytogenes genomes possessing multiple acrIIA-encoding prophages were also identified (Supplementary Table 1). Furthermore, at least one acrIIA gene was found in the genomes of all eight instances of self-targeting that were initially identified (Figure 1B, Supplementary Table 1), explaining how these scenarios are stable. Together, these data suggest widespread prophage-mediated inactivation of CRISPR-Cas9 in L. monocytogenes.
Previous HTH-containing anti-CRISPR associated (aca) genes were used as markers to identify novel type I anti-CRISPR genes (Pawluk et al., 2016), although the aca genes did not have anti-CRISPR activity themselves. We hypothesized that acrIIA1 could fulfill the role of such a marker. A comprehensive phylogenetic analysis of acrIIA1 revealed that homologs were conserved widely across Firmicutes, in both mobile elements and core genomes (Figure 5A). A family of distantly related acrIIA1 homologs was identified in Listeria genomes, as exemplified by the orfD gene, which had been independently identified as an acr locus member that also occurs upstream of acrIIA4 homologs in contexts outside of prophages (Figure 4A and Table S1). While orfD lacked anti-CRISPR activity in a functional assay (Figure 3B), its co-occurrence with a bona fide acr gene suggests that the broad acrIIA1/orfD superfamily could be used as a marker to identify new acr genes. Future work will be necessary to determine whether the HTH-containing genes in these systems serve as effective markers for novel anti-CRISPR discovery.
Figure 5. Phylogenetic analysis of AcrIIA1-4 homologs.

An unrooted phylogenetic reconstruction of full-length protein sequences identified following an iterative psi-BLASTp search to query all non-redundant protein sequences within GenBank for
(A) AcrIIA1.
BLASTp was used to identify sequences for similar phylogenetic reconstructions of
(B) AcrIIA2,
(C) AcrIIA3,
(D) AcrIIA4 (see STAR Methods). Selected bootstrapping support values are denoted with filled ovals (≥90%), open rectangles (≥70%) or dashed lines (<70%). The sequence family that is boxed-in represents the family that was tested for anti-CRISPR function. Other homologs reflect distinct sub-families present in the genomes described under the tree.
To determine the homology landscape of acrIIA2-4, additional phylogenetic analyses were performed. Unlike acrIIA1, which was widespread across Firmicutes core genomes, the other three acr genes were mostly restricted to prophages in Listeria. Three distinct sequence families of acrIIA2 were identified, all restricted to Listeria siphophages (a family of longtailed, non-contractile phages) (Figure 5B), while two acrIIA3 families were observed in the genomes of siphophages infecting Listeria and Streptococcus (Figure 5C). Lastly, acrIIA4 was observed in two distinct sequence families, one in Listeria siphophages and plasmids, and the other in a group of obligate virulent myophages (long contractile-tailed phages) (Figure 5D). While acrIIA2 and acrIIA3 were nearly always found with acrIIA1, acrIIA4 often occurred in the absence of acrIIA1 homologs in phages and mobile elements of Listeria. For example, the family of acrIIA4 in virulent phages are distinct from the other family of acrIIA4 homologs in that they have an ~70 amino acid C-terminal extension in the predicted protein and do not occur with the HTH-containing genes acrIIA1 or orfD, suggesting potential mechanistic and evolutionary distinctions between these acrIIA4 families. Together, these analyses reveal ample sequence space for surveying homologous acr genes for specificity determinants and suggest an active arms race between cas9 and mobile elements in L. monocytogenes.
AcrIIA2 and AcrIIA4 inhibit S. pyogenes Cas9
To determine the versatility of the Lmo Cas9 AcrIIA proteins, we asked whether these inhibitors were functional on the related Cas9 protein from S. pyogenes (Spy, 53% identical to Lmo Cas9). This ortholog has been used widely for biotechnological applications as an RNA-guided nuclease (Barrangou and Doudna, 2016), as well as for programmable gene repression by a catalytically deactivated mutant (dCas9) (Gilbert et al., 2013; Qi et al., 2013). Using an E. coli strain that carries Spy dCas9, we tested whether AcrIIA proteins block dCas9 from interfering with transcription of a chromosomal RFP reporter gene (Figure 6A). In a genetic background lacking inhibitors, the presence of an sgRNA and dCas9 reduced RFP fluorescence ~40-fold (2.6% relative to that of a strain with no sgRNA). acrIIA1 had no impact on dCas9-mediated transcriptional repression, nor did orfA, orfC, or orfD, negative controls that had no anti-CRISPR activity in L. monocytogenes. acrIIA2 partially blocked dCas9 function, with fluorescence reduced only 4-fold (25% relative to the no guide control), while acrIIA4 nearly completely blocked dCas9, with fluorescence at 85% of the no guide control (Figure 6B). We could not obtain meaningful data from acrIIA3 because the protein was toxic to E. coli. This lowered the recorded cell count during flow cytometry (see Figure S4a) and lead to large variability in the fluorescence measurements. A homolog of acrIIA3 from S. pyogenes (accession number: AND04610.1) with 45% sequence identity to Lmo_acrIIA3 was tested, but also resulted in impaired growth of E. coli (Figure S4b). The mechanism of acrIIA3 toxicity in E. coli remains to be determined. We conclude that the acrIIA2 and acrIIA4 inhibit Spy dCas9 in E. coli to different degrees.
Figure 6. Inhibition of Streptococcus pyogenes dCas9 and Cas9.

(A) A schematic outlining the experimental setup, where single-cell fluorescence of E. coli BW25113 expressing Streptococcus pyogenes (Spy) dCas9 and a sgRNA targeted towards a chromosomal red fluorescent protein (RFP) gene was measured by flow cytometry.
(B) Candidate (orf) and validated (acr) acrIIA genes were tested for their ability to inhibit dCas9-based gene repression. Measurements taken reflect the median RFP fluorescence value of a single cell in a unimodal population normalized for each candidate gene to a sgRNA-free control. Error bars represent the mean +/− SD of at least three biological replicates. See Figure 3 and Figure S3 for gene-identification information. See Figure S4 for raw flow cytometry data.
(C) A schematic outlining the experimental setup, where HEK293T cells with a chromosomally-integrated, doxycycline-inducible eGFP cassette were transfected with a plasmid encoding a single transcript tracrRNA/eGFP-targeting sgRNA and NLS-SpyCas9 alongside expression constructs encoding one of five codon-optimized phage genes at different ratios. The percent of eGFP positive cells was measured 12 hours after induction by flow cytometry.
(D) Candidate (orf) and validated (acr) acrIIA genes were tested for their ability to inhibit dCas9-based gene editing. An increasing amount of inhibitor plasmid (in ng) was added from left to right, at a ratio to the Cas9/sgRNA plasmid of 1:1 and 3:1. Data were normalized to transfection with no phage ORF as the baseline. Average percent of eGFP positive cells is depicted +/− SD across biological triplicates. See Figure S5 for raw flow cytometry data.
Given the common application of Spy Cas9 in eukaryotic cells, we next tested the AcrIIA proteins for their ability to block gene editing in human cells. HEK293T cells with an inducible, chromosomally-integrated eGFP reporter gene were transiently transfected with a plasmid expressing both Spy Cas9 and an sgRNA targeting eGFP in the presence or absence of vectors expressing human codon optimized acrIIA genes. After allowing gene editing to proceed for 36 h, eGFP was induced for 12 h, and cellular fluorescence was then measured by flow cytometry (Figure 6C). In the presence of Cas9 and the eGFP sgRNA, gene editing resulted in a 25% decrease in the number of GFP positive cells, while co-expression with acrIIA2 or acrIIA4 prevented Cas9-based gene editing (Figure 6D). We additionally tested the S. pyogenes homolog of acrIIA3 (Spy_acrIIA3), which was not toxic in human cells, but it had no impact on Cas9 function in this assay. acrIIA1 was non-functional in human cells, as was the negative control, orfA. Taken together with dCas9 experiments in E. coli, these data demonstrate the utility of the AcrIIA2 and AcrIIA4 proteins to inhibit the function of an orthologous Cas9 in heterologous hosts. These reagents, therefore, represent new tools in the CRISPR-Cas9 genome engineering toolkit.
DISCUSSION
Phage-encoded inhibitors of bacterial immune systems emerge due to the strong selective pressures in the evolutionary arms race between these two entities (Samson et al., 2013). The first identification of phage encoded anti-CRISPRs in type I CRISPR-Cas systems hinted that more CRISPR-Cas inhibitors existed, but methods were lacking for their discovery. Here, we present a bioinformatics strategy that uses “self-targeting” as a genomic marker for CRISPR-Cas inhibitor genes (Figure 1A). This approach led to the identification of four different type II-A CRISPR-Cas9 inhibitors (Figure 3 and 4A), which are collectively present in half of all Cas9-encoding L. monocytogenes genomes, including all genomes with self-targeting (Figure 4C). We anticipate that this approach will be helpful for identifying acr genes in other CRISPR-Cas systems, although a distinct mechanism for tolerance of self-targeting has been described for type III systems (Goldberg et al., 2014; Samai et al., 2015).
To facilitate the identification of AcrIIA proteins, we first demonstrate a functional CRISPR-Cas9 system in L. monocytogenes (Figure 2B). Previous studies of CRISPR-Cas in this organism have focused on the type I-B system and an associated orphan CRISPR array lacking cas genes (Mandin et al., 2007; Sesto et al., 2014). Although no canonical CRISPR-Cas function had been established for either system previously, the orphan array was shown to be processed by a host ribonuclease to generate non-coding RNAs (Mandin et al., 2007; Sesto et al., 2014). To observe function for the type II-A CRISPR-Cas system, we used a standard transformation efficiency assay, showing that CRISPR-Cas9 function in strain 10403s is able to limit transformation of a plasmid in a sequence specific manner (Figure 2A and 2B). Given the small colony phenotype observed during transformation of 10403s with the targeted plasmid (pT), we suspect that endogenous levels of cas9 expression are not sufficient to totally clear the plasmid. Either a small fraction of cells retain the plasmid, or alternatively, cells temporarily possess the plasmid at a reduced copy number, resulting in the small colony phenotype. Consistent with low endogenous expression of cas9 leading to either form of incomplete plasmid clearance, increased expression of cas9 resulted in an elimination of detectable transformants in this assay (Figure 2B).
Among the strains with self-targeting, we selected J0161 for further analysis. Using the transformation efficiency assay, we observed no plasmid targeting in this strain (Figure 2C), an observation consistent with the presence of an inhibitor. Indeed, the immune system was inactivated when the ϕJ0161a prophage was transferred to the CRISPR-Cas9-active strain 10403s (Figure 2D). Furthermore, we observed that ϕJ0161a can inactivate CRISPR-Cas9 function in a strain that overexpresses Cas9 (Figure 2E). Mechanistically, this demonstrates that inhibitors are unlikely to function by disrupting the transcriptional regulation of Cas9 and are sufficiently expressed from the integrated prophage to cope with enhanced Cas9 levels.
To identify candidate anti-CRISPR genes, related prophages from CRISPR-active strain 10403s and a prophage that inhibits CRISPR-Cas9 from strain J0161 were compared, and a process-of-elimination cloning approach was taken (Figure 3A). Two isolated acr genes (acrIIA1 and acrIIA2) were first identified in ϕJ0161a (Figure 3B). In searching for more anti-CRISPRs, we find that conserved genomic positioning in related phages is a good proxy for identifying distinct type II-A Cas9 inhibitor proteins, despite a lack of sequence conservation between the proteins themselves (Figure 4A). This has been observed previously in studies of Type I-F and I-E anti-CRISPRs (Bondy-Denomy et al., 2013; Pawluk et al., 2014). In L. monocytogenes, the high prevalence of Cas9 inhibitors in prophages suggests the widespread inactivation of CRISPR-Cas9 function (Figure 4C). At present, we do not understand whether there is a mechanistic link to explain the common co-occurrence of acrIIA1 with other anti-CRISPRs (Figure 4A and 4B). Although this gene is sufficient to inactivate CRISPR-Cas9 function in a plasmid challenge assay, we speculate that it could act as a co-factor or regulator of other acrIIA genes during infection or lysogeny, thus explaining the genomic associations observed. Future work will be necessary to understand whether AcrIIA1 is, in fact, a bi-functional protein in this regard and more broadly, whether its superfamily is a marker for acr genes.
Phylogenetic analyses demonstrate common occurrences of acrIIA2-4 in mobile elements in Listeria mobile elements (Figure 5). Inhibiting the adaptive immune system likely aids horizontal gene transfer in this organism by blocking Cas9-based targeting and adaptation (Heler et al., 2015). In addition to the family of prophages where these acrIIA genes were first identified, homologs were also found in distant siphophages, myophages and plasmids (Supplementary Table 1). Most notably, the acrIIA4 homologs encoded by virulent myophages did not have acrIIA1 superfamily homologs in their vicinity. Furthermore, the presence of acrIIA1 and acrIIA3 homologs in genera outside of Listeria demonstrates that CRISPR-Cas9 inactivation may be common-place in the Firmicutes.
Many potential mechanisms could explain CRISPR-Cas9 inactivation. In their native hosts, L. monocytogenes, we have defined anti-CRISPRs by their ability to inhibit Lmo Cas9-based targeting of a plasmid. Furthermore, by demonstrating the efficacy of acrIIA2 and acrIIA4 in heterologous hosts with engineered elements (i.e. cas9 promoter, sgRNA design and promoter) we conclude acr-mediated transcriptional repression of the CRISPR-Cas9 system is unlikely. Using the orthologous Spy Cas9, it is clear that AcrIIA2 and AcrIIA4 have broad specificity, given that Lmo Cas9 and Spy Cas9 only share 53% sequence identity. AcrIIA2 and AcrIIA4 likely target regions conserved between the two Cas9 proteins. Type I anti-CRISPRs function by binding directly to the Cas proteins required for target interference and preventing DNA binding or DNA cleavage (Bondy-Denomy et al., 2015). By extension, we expect a similar mechanism for AcrIIA2 and AcrIIA4, given their ability to function in heterologous hosts. The enhanced efficacy of acrIIA2 in the cleavage-based Cas9 assay relative to the dCas9 based assay suggests that it may inhibit both binding and cleavage to some degree, with cleavage inhibition manifesting as a full inactivation of Cas9 function. However, comparing the results of these two experiments, it is important to note the differences between the stability of Cas9 and dCas9 interactions with the mammalian and bacterial genomes, respectively. Given the efficacy of AcrIIA4 in blocking dCas9-based function (Figure 6B), stable DNA-binding is likely inhibited, although whether this is through a direct interaction with Cas9 remains to be seen.
The identification and future mechanistic dissection of type II-A inhibitors will provide valuable new reagents for studying canonical CRISPR-Cas9 function in natural and engineered settings. The ability of AcrIIA proteins to block Spy Cas9 in E. coli and human cells suggests that these proteins can provide a post-translational “off-switch” for Cas9. This could add a layer of regulation on this powerful system that can be applied in eukaryotic systems to control genome engineering. This new addition to the CRISPR-Cas9 toolbox could enable new applications, such as specifically reversing the effects of dCas9 binding to a genomic locus, or limiting the amount of time that Cas9 is active in the nucleus to reduce off-target gene editing. It will be important to continue to exploit the abundant tools provided to us from the phage-bacteria arms race as we expand the search for inhibitor proteins.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the lead contact, Joseph Bondy-Denomy (joseph.bondy-denomy@ucsf.edu), Department of Microbiology and Immunology, University of California, San Francisco.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
Listeria monocytogenes strains were cultured on Brain-Heart Infusion (BHI) medium. Escherichia coli strains were cultured on LB medium.
Cell lines
Human Embryonic Kidney 293 plus T cell antigen (HEK293T, CRL-3216, ATCC) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and 50μg/mL penicillin/streptomycin (P/S, UCSF CCF).
METHOD DETAILS
Assay of CRISPR-Cas9 in L. monocytogenes
Plasmid-transformation assay of CRISPR-Cas9
Targeted (pT; pNT for J0161; pRAU31) and non-targeted (pNT; pTJ0161; pRAU29) plasmids for L. monocytogenes 10403s were constructed by ligating annealed primer pairs into the HindIII and BamHI sites of pKSV7. See Table S3 for plasmid-insert sequences. L. monocytogenes strains were transformed with 0.5–1.0 μg pT or pNT by electroporation. Electrocompotent cells were prepared and transformed as described (Park and Stewart, 1990; Zemansky et al., 2009). Transformations were diluted 10-fold into BHI and recovered for two hours, shaking at 30°C. Recovered cultures were plated on BHI with 1.5% agar and 7.5 μg/ml chloramphenicol to select for pT or pNT. For pPL2oexL integrants, tetracycline selection (2 μg/ml) was maintained throughout the procedure, with exception to recovery cultures, which were performed without selection. Whereas plates that contained only chloramphenicol were incubated at 30°C for 36–40 hours prior to imaging, plates that also contained tetracycline were incubated at 30°C for 64–72 hours. Plate images were collected using the Gel Doc™ EZ Gel Documentation System (BioRad) and Image Lab (BioRad) software.
Construction of pPL2oexL-integrants in L. monocytogenes 10403s
The pPL2oexL plasmid for constitutive chromosomal expression of genes in L. monocytogenes was derived from pPL2 (Lauer et al., 2002) (See Figure S6). Individual genes or phage fragments were PCR-amplified from genomic DNA and cloned into pPL2oexL by Gibson Assembly. pPL2oexL-derivative plasmids were electroporated into nonlysogenic 10403s, using a procedure like that which was employed for the plasmid-transformation assay of CRISPR-Cas9 (see text under previous heading). Transformations were recovered for two hours, shaking at 37°C and were plated on BHI-agar with 2 μg/ml tetracycline. Colonies emerged after 36–48 hours incubating at 37°C, and were re-streaked once on the same selective medium to ensure genotypic homogeneity.
Construction of a 10403s::ϕJ0161a lysogen
Phage was induced from L. monocytogenes strain J0161 by exposure to ultraviolet radiation as described previously (Loessner and Busse, 1990). 10403s::ϕJ0161a lysogens were isolated from plaques that resulted from spotting amplified J0161 phage stock on a lawn of nonlysogenic 10403s (suspended in BHI with 0.7% agar and 2.5 mM CaCl2). Plaques emerged after 16 hours incubation at 30°C. Lysogeny was confirmed by PCR, as described (Lauer et al., 2002).
Construction of markerless chromosomal deletion strains
Markerless deletions of cas9 and acrIIA1-2 were constructed by allelic exchange in nonlysogenic 10403s and 10403s::ϕJ0161a, respectively. Up- and down-stream (700–1000 base pairs) regions flanking the genes to be deleted were fused by overlap-extension PCR and ligated into pKSV7. The Δcas9 genotype was inserted between the HindIII and BamHI restriction sites, whereas the ΔacrA1-2 genotype was inserted between the SacI and BamHI restriction sites. Knockout vectors were transformed by electroporation. Subsequent manipulations were performed as previously reported (Camilli et al., 1993).
Bioinformatic analyses
Identification of self-targeting CRISPR-Cas systems
L. monocytogenes genome sequences were downloaded from NCBI. Type-IIA CRISPR arrays were identified within individual genomes using CRISPRfinder (Grissa et al., 2007) or CRISPRDetect (Biswas et al., 2016) web utilities. See Figure S1B for a representative L. monocytogenes type II-A CRISPR array. Self-targeting CRISPR-Cas systems were identified using the CRISPRtarget web utility (Biswas et al., 2013) by searching individual L. monocytogenes genomes with their own CRISPR arrays. Bona fide self-targeting events were defined as perfect matches lacking spacer-protospacer mutations in the PAM-proximal region (20 bp), concurrent with a cognate PAM sequence (5′-NGG-3′). See Table S1 for a list of self-targeting strains.
Phylogenetic reconstruction of AcrIIA protein families
AcrIIA2- (AEO04363.1), AcrIIA3- (CBY03209.1) and AcrIIA4- (AEO04689.1) homologous protein sequences were acquired by BLASTp searches of all the non-redundant protein sequence database of NCBI on November 5, 2016. Full length (>78% query coverage) sequences of high homology (E value < 1e-04) were downloaded and aligned using Muscle (Edgar, 2004) in MEGA6 (Tamura et al., 2013). Phylogenetic reconstructions of each protein family were performed in MEGA6 using the neighbor-joining method with the Poisson model for amino acid substitution, uniform rates among sites and pairwise deletion of gaps. Reconstructions were tested using the bootstrap method (1000 replications). Reconstruction images were then edited for clarity in Illustrator (Adobe). AcrIIA1- (AEO04364.1) homologous protein sequences were acquired by four iterations of psiBLASTp searches of the non-redundant protein sequence database of NCBI on October 26, 2016. The position-specific scoring matrix (PSSM) was enchriched with all full-length (>80%) protein sequences. Sequences were downloaded, aligned and reconstructed using the same methodology that was employed for the analysis of AcrIIA2, 3 and 4 (see above). However, in the case of AcrIIA1, sequences with large insertions (>30 amino acids) were removed from the sequence alignments, prior to phylogenetic reconstruction.
Analysis of gene-conservation patterns
The conservation of acrIIA1, acrIIA2, acrIIA3, acrIIA4 and cas9 were catalogued in reference to a control gene (cysteinyl-tRNA synthetase) that occurs once in all L. monocytogenes genomes. BLASTp searches were performed to acquire lists of genome-specific accession numbers for encoded proteins. These were used as surrogates for genes to assess conservation. Lists were compiled into a single table and sorted so that individual rows of data included accession numbers for all proteins of interest encoded within a single genome.
Inhibition of Spy-CRISPRi in E. coli
Reporter strain construction
Our E. coli Spy-CRISPRi reporter system uses integrated components of the previously reported CRISPRi system (Qi et al., 2013) with minor modifications. The promoter for mrfp was modified in the entry vector by changing the promoter from PLlacO-1 to a minimal synthetic promoter (BBa_J23119) (http://parts.igem.org/), PCR amplified, and integrated into BW25113 at nfsA by recombineering as described. The mrfp-targeting sgRNA was cloned into the site-specific integrating plasmid pCAH63 under control of PLlacO-1 to generate pCs550-r, and integrated at lambda att using the helper plasmid pINT-ts (Haldimann and Wanner, 2001), selecting for chloramphenicol resistance. Conjugation was used to move a chromosomal dcas9 cassette into recipient strains harboring mrfp, sgRNA or both. A “pseudo-Hfr” strain isogenic with BW25113, carries the transfer region from F and a spectinomycin marker integrated downstream of rhaM (4086kb) (Typas et al., 2008). A “pseudo-Hfr” dcas9 donor strain was constructed by integrating dcas9 and a gentamycin resistance marker at the Tn7 att site (Choi et al., 2005), adjacent to the origin of transfer. dcas9 was cloned from pdCas9-bacteria (Addgene #44249) under control of BBa_J23105 (http://parts.igem.org/). Putative Cas9 inhibitor proteins were cloned into pBAD24 (Guzman et al., 1995) by Gibson Assembly (NEB) and transformed into the Spy-CRISPRi strains by electroporation.
Flow cytometry
Strains were grown overnight in LB with arabinose in deep 96-well plates, and then back-diluted 1:400 into fresh LB with arabinose (to maintain expression of the inhibitor) and IPTG (to induce expression of the sgRNA). After 2.5hr growth (OD~0.4) cultures were fixed using 1.5% final formaldehyde and quenched with glycine, and then diluted 1:30 into phosphate buffered saline. Red fluorescence levels were measured using an LSRII flow cytometer (BD Biosciences) using the yellow/green laser (561 nm) and the PE-Texas Red® detector (610/20 nm). Data for at least 20,000 cells were collected, and median fluorescence values were extracted using FlowJo (FlowJo, LLC). Error bars represent the standard deviation from 3 or more biological replicates. Data from representative samples were plotted as histograms using FlowJo.
Inhibition of Cas9 cleavage in human cells
An eGFP-targeting crRNA was ordered as complementary single-stranded DNA oligos (IDT) and cloned into BbsI linearized pX330 (Addgene, Zhang lab) to generate a single vector expressing S. pyrogenes Cas9-NLS and an eGFP-targeting CRISPR cassette. One candidate (orf) and three validated (acr) acrIIA genes were codon-optimized for human cell expression, synthesized in vitro (IDT, GeneBlock), and cloned into BamHI/EcoRI linearized pcDNA3.1(+) by Gibson assembly. Similarly, the gene encoding enhanced Green Fluorescent Protein (eGFP) was synthesized and cloned into BamHI/EcoRI linearized pLVX-TetOne-Puro (Clontech). Doxycycline-inducible eGFP lentivirus was produced in Human Embryonic Kidney (HEK)293T cells (ATCC) by cotransfection (polyJet, SignaGen) with Gag-Pol packaging construct and VSV-G envelope (pMD2.G, Addgene). Lentiviruses were precipitated from the cellular supernatant at 4°C by incubation in a final concentration of 8.5% Poly(ethylene glycol) average Mn 6000 (PEG-6000) and 0.3M NaCl for 4 hours. Viruses were concentrated at 3500 RPM for 20 minutes in a spinning bucket rotor, suspended in 1 mL 1xPBS, and preserved at −80. One-thousandth viral preparation by volume was used to transduce 250,000 HEK293T cells and successful integrants purified by selection in 1 μg/mL puromycin for 48 hours.
Polyclonal HEK293T cells with a chromosomally-integrated, inducible eGFP cassette were expanded, plated, and transfected with the eGFP-targeting CRISPR construct and each of the bacteriophage genes at different ratios in triplicate (Trans-IT, Mirus). An empty vector was used to equalize the total mass of transfected plasmid across each sample. 36 hours after transfection, cells were treated with 2 μg/mL doxycycline to induce eGFP expression. 12 hours later, cells were suspended by incubation in PBS-EDTA, fixed in 1% formaldehyde PBS, and percent eGFP-positive cells monitored by flow cytometry (FACSCalibur, Gladstone Flow Cytometry Core). Data was normalized to no sgRNA controls and presented as the average percent eGFP-positive cells +/− standard deviation.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were conducted with at least three biological replicates (N >= 3). Statistical parameters are reported in the Figures and the Figure Legends. Additional statistical tests were not performed.
DATA AND SOFTWARE AVAILABILITY
The accession numbers, locus tags and coding sequences for individual genes tested for CRISPR-Cas9 inhibition activity are disclosed in Figure S3. Additional accession numbers for AcrIIA homologs are reported in Table S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| Listeria monocytogenes 10403s | Laboratory of Daniel Portnoy | ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Info&id=393133&lvl=3&lin=f&keep=1&srchmode=1&unlock |
| Listeria monocytogenes 10403s derivatives | this paper | see Table S2 |
| Listeria monocytogenes J0161 | Laboratory of Martin Wiedmann | ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=393130 |
| Listeria monocytogenes SLCC2482 | Ariane Pietzka | ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=863767 |
| Listeria monocytogenes SLCC2540 | Ariane Pietzka | ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=879089 |
| Escherichia coli BW25113 derivatives | this paper | see Table S2 |
| Recombinant DNA | ||
| pBAD24 | Laboratory of Carol Gross | ncbi.nlm.nih.gov/nuccore/X81837.1 |
| pBAD24-derivative plasmids | this paper | see Table S2 |
| pdCas9-bacteria | Addgene | addgene.org/vector-database/44249/ |
| pLVX-TetOne-Puro | Clontech | clontech.com/US/Products/Inducible_Systems/TetSystems_Product_Overview/Tet-One_Overview |
| pMD2.G | Addgene | addgene.org/12259/ |
| pX330 | Addgene | addgene.org/vector-database/42230/ |
| pcDNA3.1(+) | Addgene | addgene.org/vector-database/2093/ |
| pKSV7 | Laboratory of Daniel Portnoy | addgene.org/26686/ |
| pKSV7-derivative plasmids | this paper | see Table S2 |
| pPL2oexL | Laboratory of Daniel Portnoy | see Figure S6 |
| pPL2oexL-derivative plasmids | this paper | see Table S2 |
| Sequence-Based Reagents | ||
| GeneBlocks for HEK293T expression of phage proteins | IDT | see Table S3 |
| Software and Algorithms | ||
| Prism 5 | GraphPad | graphpad.com/scientific-software/prism/ |
| CRISPRfinder | I2BC | crispr.i2bc.paris-saclay.fr/Server/ |
| CRISPRDetect | Univsersity of Otago | brownlabtools.otago.ac.nz/CRISPRDetect/predict_crispr_array.html |
| CRISPRtarget | Univsersity of Otago | bioanalysis.otago.ac.nz/CRISPR Target/crispr_analysis.html |
| illustrator | adobe | adobe.com/Illustrator |
| MEGA6 | MEGA | megasoftware.net/ |
| Image Lab 5.2.1 | BioRad | bio-rad.com/en-cn/product/image-lab-software |
| FlowJo | FlowJo LLC | flowjo.com/ |
Supplementary Material
(A) Comparison of type II-A CRISPR-cas loci from Streptococcus pyogenes SF370 (Spy_SF370), Listeria monocytogenes 10403s (Lmo_10403s), Listeria monocytogenes J0161 (Lmo_J0161) and Listeria innocua (Lin_Clip11262). Percent identity between Cas9 protein sequences is shown.
(B) The CRISPR array of self-targeting strain Lmo J0161. A type II-A CRISPR array, predicted by the CRISPRDetect web utility is shown. The self-targeting spacer (number 16) is boxed. In bold, are the RNA-coding nucleotides responsible for target recognition.
(C) Alignment of tracrRNA loci.
(D) Alignment of CRISPR loci.
Alignment of Cas9 protein sequences. Residues with essential chemical functionalities are boxed.
Representative plates depicting colonies after transformation and selection for targeted (pT; pRAU31) or non-targeted (pNT; pRAU29) plasmids. Given names, and strain numbers are provided. See Table S2 for additional information pertinent to plasmid and strain design and nomenclature. See Table S3 for gene sequences.
(A) Distribution of single-cell RFP fluorescence values for E. coli CRISPRi reporter strains with and without expression of AcrIIA proteins. Expression of AcrIIA proteins leads to unimodal shift in population fluorescence towards the sgRNA (no CRISPRi knockdown) state, indicating a uniform disruption of CRISPRi activity. Strains were grown for 2.5hr in the presence of IPTG to induce CRISPRi, with or without expression of the AcrIIA inhibitor.
(B) Expression of Spy AcrIIA3 is toxic in E. coli. In the presence of IPTG (CRISPRi induction) and arabinose (AcrIIA3 induction), Spy AcrIIA3 is toxic in the presence or absence of sgRNA, indicating that its toxicity is independent of CRISPRi activity.
(C) Expression of either acrIIA3 ortholog is toxic in E. coli. Indicated strains express all components for CRISPRi and were grown in microtiter plates with or without 0.0005% arabinose (AcrIIA3 expression induction) and 1mM IPTG (CRISPRi induction), and optical density was monitored every 4 minutes. Plots depict the mean of three biological replicates +/− SD.
HEK293T cells were analyzed for eGFP expression 12 hours post-induction by flow cytometry. Representative flow plots from one biological replicate are depicted for cells that received Cas9 with no guide RNA (No sgRNA), Cas9 with an eGFP-targeting guide RNA (eGFP sgRNA), and Cas9 with an eGFP-targeting guide RNA and an independent AcrII4 expression vector (eGFP sgRNA + AcrII4). Forward scatter is depicted on the horizontal axis and eGFP fluorescence on the vertical axis. Vertical gating was set with cells that did not receive induction with doxycycline
Genes and phage fragments to be tested for CRISPR-Cas9 inhibition in L. monocytogenes 10403s were cloned into pPL2oexL between pHyper and the FLAG tag. Native stop codons were included in pPL2oexL derivatives.
HIGHLIGHTS.
Bacteriophage anti-CRISPR proteins AcrIIA1-4 inactivate CRISPR-Cas9
Half of L. monocytogenes isolates possess inhibited CRISPR-Cas9 systems
AcrIIA2 and AcrIIA4 prevent target binding by dCas9 in bacteria
AcrIIA2 and AcrIIA4 inhibit Cas9-mediated gene editing in human cells
Acknowledgments
We would like to thank Aaron T. Whiteley and Daniel A. Portnoy (UC Berkeley) for providing the prophage-cured and wild type strains of L. monocytogenes 10403s as well as plasmids pKSV7 and pPL2oexL. We also acknowledge Martin Wiedmann (Cornell University) for providing strain J0161 and Ariane Pietzka (Austrian Agency for Health and Food Safety) for providing SLCC2482 and SLCC2540. We acknowledge Carol Gross’ lab (UCSF) for providing the pBAD24 plasmid and for productive conversations about the project. We specifically thank Carol Gross and Adair Borges for critical reading of the manuscript and thoughtful advice. J.B.D. was supported by the University of California, San Francisco Program for Breakthrough in Biomedical Research, funded in part by the Sandler Foundation, and an NIH Office of the Director Early Independence Award (DP5-OD021344). M.R.S. was supported by National Science Foundation No. 1144247. J.F.H. was supported by an amFAR Mathilde Krim Fellowship, an NIH Human Immunology Project Consortium Infrastructure Pilot (N.J.K, U19 AI118610), and NIH funding for the UCSF-Gladstone Institute of Virology & Immunology Center for AIDS Research (N.J.K., P30 AI027763).
Footnotes
AUTHOR CONTRIBUTIONS
B.J.R., M.R.S., J.F.H, and J.B.D. designed the experiments; B.J.R., M.R.S., J.F.H, C.W., and M.J.M. prepared strains and performed experiments; N.J.K and J.B.D. supervised experiments; B.J.R. and J.B.D. wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nature Biotechnology. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Staals RHJ, Morales SE, Fineran PC, Brown CM. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, Davidson AR. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015;526:136–139. doi: 10.1038/nature15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Molecular Microbiology. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di H, Ye L, Yan H, Meng H, Yamasak S, Shi L. Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem Biophys Res Commun. 2014;454:399–403. doi: 10.1016/j.bbrc.2014.10.018. [DOI] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JHD, Tjian R, Doudna JA. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Qimron U. The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J Bacteriol. 2010;192:6291–6294. doi: 10.1128/JB.00644-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, Marraffini LA. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. 2014;514:633–637. doi: 10.1038/nature13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Research. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann A, Wanner BL. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001;183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and Structure-Specific RNA Processing by a CRISPR Endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Micro. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner MJ, Busse M. Bacteriophage typing of Listeria species. Applied and Environmental Microbiology. 1990;56:1912–1918. doi: 10.1128/aem.56.6.1912-1918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Micro. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Research. 2007;35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- Maxwell KL, Garcia B, Bondy-Denomy J, Bona D, Hidalgo-Reyes Y, Davidson AR. The solution structure of an anti-CRISPR protein. Nature Communications. 2016;7:13134. doi: 10.1038/ncomms13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, Doudna JA. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat Struct Mol Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SF, Stewart GS. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene. 1990;94:129–132. doi: 10.1016/0378-1119(90)90479-b. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, Davidson AR. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. mBio. 2014;5:e00896–14. doi: 10.1128/mBio.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Staals RHJ, Taylor C, Watson BNJ, Saha S, Fineran PC, Maxwell KL, Davidson AR. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nature Microbiology. 2016;1:1–6. doi: 10.1038/nmicrobiol.2016.85. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson JE, Magadán AH, Sabri M, Moineau S. Revenge of the phages: defeating bacterial defences. Nat Rev Micro. 2013;11:675–687. doi: 10.1038/nrmicro3096. [DOI] [PubMed] [Google Scholar]
- Sesto N, Touchon M, Andrade JM, Kondo J, Rocha EPC, Arraiano CM, Archambaud C, Westhof E, Romby P, Cossart P. A PNPase dependent CRISPR System in Listeria. PLoS Genet. 2014;10:e1004065. doi: 10.1371/journal.pgen.1004065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yao D, Xu J-G, Li A-R, Xu J, Fu P, Zhou Y, Zhu Y. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat Struct Mol Biol. 2016;23:868–870. doi: 10.1038/nsmb.3269. [DOI] [PubMed] [Google Scholar]
- Wright AV, Nuñez JK, Doudna JA. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Research. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J Bacteriol. 2009;191:3950–3964. doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Comparison of type II-A CRISPR-cas loci from Streptococcus pyogenes SF370 (Spy_SF370), Listeria monocytogenes 10403s (Lmo_10403s), Listeria monocytogenes J0161 (Lmo_J0161) and Listeria innocua (Lin_Clip11262). Percent identity between Cas9 protein sequences is shown.
(B) The CRISPR array of self-targeting strain Lmo J0161. A type II-A CRISPR array, predicted by the CRISPRDetect web utility is shown. The self-targeting spacer (number 16) is boxed. In bold, are the RNA-coding nucleotides responsible for target recognition.
(C) Alignment of tracrRNA loci.
(D) Alignment of CRISPR loci.
Alignment of Cas9 protein sequences. Residues with essential chemical functionalities are boxed.
Representative plates depicting colonies after transformation and selection for targeted (pT; pRAU31) or non-targeted (pNT; pRAU29) plasmids. Given names, and strain numbers are provided. See Table S2 for additional information pertinent to plasmid and strain design and nomenclature. See Table S3 for gene sequences.
(A) Distribution of single-cell RFP fluorescence values for E. coli CRISPRi reporter strains with and without expression of AcrIIA proteins. Expression of AcrIIA proteins leads to unimodal shift in population fluorescence towards the sgRNA (no CRISPRi knockdown) state, indicating a uniform disruption of CRISPRi activity. Strains were grown for 2.5hr in the presence of IPTG to induce CRISPRi, with or without expression of the AcrIIA inhibitor.
(B) Expression of Spy AcrIIA3 is toxic in E. coli. In the presence of IPTG (CRISPRi induction) and arabinose (AcrIIA3 induction), Spy AcrIIA3 is toxic in the presence or absence of sgRNA, indicating that its toxicity is independent of CRISPRi activity.
(C) Expression of either acrIIA3 ortholog is toxic in E. coli. Indicated strains express all components for CRISPRi and were grown in microtiter plates with or without 0.0005% arabinose (AcrIIA3 expression induction) and 1mM IPTG (CRISPRi induction), and optical density was monitored every 4 minutes. Plots depict the mean of three biological replicates +/− SD.
HEK293T cells were analyzed for eGFP expression 12 hours post-induction by flow cytometry. Representative flow plots from one biological replicate are depicted for cells that received Cas9 with no guide RNA (No sgRNA), Cas9 with an eGFP-targeting guide RNA (eGFP sgRNA), and Cas9 with an eGFP-targeting guide RNA and an independent AcrII4 expression vector (eGFP sgRNA + AcrII4). Forward scatter is depicted on the horizontal axis and eGFP fluorescence on the vertical axis. Vertical gating was set with cells that did not receive induction with doxycycline
Genes and phage fragments to be tested for CRISPR-Cas9 inhibition in L. monocytogenes 10403s were cloned into pPL2oexL between pHyper and the FLAG tag. Native stop codons were included in pPL2oexL derivatives.