

SUMMARY
The removal of unwanted or damaged mitochondria by autophagy, a process called mitophagy, is essential for key events in development, cellular homeostasis, tumor suppression, and prevention of neurodegeneration and aging. However, the precise mechanisms of mitophagy remain uncertain. Here, we identify the inner mitochondrial membrane protein, prohibitin 2 (PHB2), as a crucial mitophagy receptor involved in targeting mitochondria for autophagic degradation. PHB2 binds the autophagosomal membrane-associated protein LC3 through an LC3-interaction region (LIR) domain upon mitochondrial depolarization and proteasome-dependent outer membrane rupture. PHB2 is required for Parkin-induced mitophagy in mammalian cells and for the clearance of paternal mitochondria after embryonic fertilization in C. elegans. Our findings pinpoint a conserved mechanism of eukaryotic mitophagy and demonstrate a function of prohibitin 2 that may underlie its roles in physiology, aging, and disease.
Graphical abstract
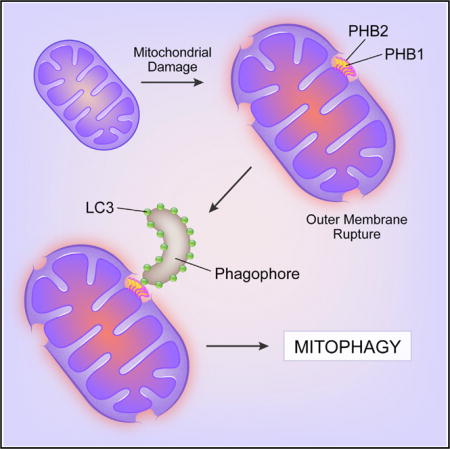
INTRODUCTION
The selective removal of mitochondria by autophagy (mitophagy) is the major pathway by which eukaryotic cells degrade damaged or unwanted mitochondria. Mitophagy is crucial for protecting cells against deleterious effects of damaged mitochondria and for fundamental processes in eukaryotic development, including the uniparental inheritance of mtDNA (Green and Levine, 2014; Redmann et al., 2014). Damaged mitochondria release reactive oxygen intermediates, leading to inflammasome activation, genotoxic stress, promotion of tumorigenesis, and aging. Accordingly, defects in mitophagy likely contribute to neurodegenerative diseases, aberrant inflammation and inflammatory diseases, cancer, and decreased lifespan. Maternal inheritance of mtDNA occurs in most animals, and in C. elegans and Drosophila, paternal mitochondria are eliminated by mitophagy (Levine and Elazar, 2011; Politi et al., 2014; Wang et al., 2016; Zhou et al., 2016). The precise reason for maternal mitochondrial inheritance has remained elusive; however, mtDNA heteroplasmy (i.e., the presence of both maternal and paternal mtDNA) is associated with reduced fitness in nematodes (Liau et al., 2007; Zhou et al., 2016) and mice (Sharpley et al., 2012). Thus, the proper removal of mitochondria is essential for organismal health in diverse eukaryotic species (Green and Levine, 2014; Redmann et al., 2014).
Prior research on mitophagy has largely focused on the role of events at the outer mitochondrial membrane (OMM) (Bingol and Sheng, 2016; Pickrell and Youle, 2015; Yamaguchi et al., 2016). When mitochondria are damaged, the mitochondrial kinase, PINK1, is stabilized, resulting in the recruitment of the Parkin E3 ligase to damaged mitochondria and ubiquitination of several OMM proteins to trigger mitophagy. Adaptor proteins, such as p62, NBR1, NDP52, Tax1BP1, and optineurin that bind ubiquitin and the autophagosome-associated protein, LC3, may direct the isolation membrane/phagophore of growing autophagosomes to surround damaged mitochondria, although the significance of some of these adaptors in mitophagy remains unclear (Pickrell and Youle, 2015; Lazarou et al., 2015). Other OMM events, including endoplasmic reticulum-associated degradation (ERAD)-like extraction of proteins (mitofusins) by p97 and promotion of mitochondrial fission, may be required for certain forms of mitophagy (Pickrell and Youle, 2015). In addition, some yeast and mammalian OMM mitophagy receptors directly interact with LC3 through LC3-interacting region (LIR) domains (Yamaguchi et al., 2016). The PINK1-Parkin pathway is a crucial amplifying mechanism that renders mitophagy more efficient, and mutations in this pathway contribute to the pathogenesis of neurodegenerative diseases (Pickrell and Youle, 2015). However, mitophagy occurs in cells that lack detectable Parkin and mitophagy-dependent developmental events, such as NIX-mediated mitochondrial clearance in mammalian erythrocyte maturation and paternal mitochondrial clearance in C. elegans embryogenesis, are Parkin-independent (Green and Levine, 2014), suggesting the existence of alternative mitophagy receptors.
The current paradigm is that the ubiquitinated mitochondrial outer membrane is the primary target of engulfment by autophagosomes during mitophagy. However, the types of organelle dysfunction that trigger mitophagy (e.g., depolarization, presence of paternal mtDNA) localize to the inner mitochondrial membrane (IMM) and matrix. Thus, there may exist mechanisms to target and ensure destruction of the inner mitochondrial compartment. Indeed, proteasomal-dependent rupture of the OMM may be required for Parkin-dependent mitophagy (Chan et al., 2011; Yoshii et al., 2011), which could allow the autophagic machinery direct access to the intermembrane space. This led us to consider the possibility of an IMM receptor in mitophagy.
We therefore used an unbiased biochemical approach to reveal IMM proteins that function as mitophagy receptors during Parkin-mediated mitophagy. Our results identify an IMM protein, prohibitin 2 (PHB2), as a key mitophagy receptor for both Parkin-mediated mitophagy in mammalian cells and for paternal mitochondrial clearance in C. elegans. As PHB2 is a conserved IMM protein that functions in development, lifespan regulation, and diverse cellular processes (including mitochondrial dynamics) (Merkwirth and Langer, 2009), these findings have implications for understanding both the molecular mechanisms of mitophagy and the mechanisms by which PHB2 regulates normal physiology, aging, and disease.
RESULTS
The Mitochondrial Inner Membrane Protein PHB2 Binds to LC3
We hypothesized that an IMM protein serves as an essential mitophagy receptor. To investigate this, we used immunoprecipitation:mass spectrometry to identify IMM protein(s) that bind to LC3 upon mitochondrial membrane protein depolarization. We used HeLa cells that stably express both Flag.StrepII-tagged LC3 (an autophagosomal membrane protein whose cleaved and lipidated form, LC3-II, binds to cargo) (Mizushima et al., 2010) and Parkin, an E3 ligase that translocates to depolarized mitochondria and promotes their degradation by mitophagy (Pickrell and Youle, 2015).
Upon treatment with the mitochondrial uncoupling agent carbonyl cyanide 3-chlorophenylhydrazone (CCCP), we identified two unique bands that immunoprecipitated with LC3 (Figure 1A). One was the IMM protein, prohibitin 2 (PHB2), an evolutionarily conserved ~34 kDa protein that forms a large heterodimeric IMM complex with prohibitin 1 (PHB1) (Merkwirth and Langer, 2009). Western blot analysis confirmed the presence of both PHB2 and PHB1 in the Flag.StrepII-LC3 immunoprecipitate samples used for mass spectrometry analysis from CCCP-treated (but not DMSO control-treated) HeLa/Parkin cells (Figure 1A). LC3-II (but not LC3-I, the uncleaved non-lipidated form of LC3) immunoprecipitated with both endogenous PHB1 and PHB2 in HeLa cells in the presence, but not absence, of Parkin and treatment with two inhibitors of mitochondrial respiration (oligomycin + antimycin [OA]) (Figure 1B). Thus, Parkin-mediated mitochondrial damage results in the binding of the PHB1/PHB2 complex to the autophagosomal membrane-associated protein LC3-II.
Figure 1. PHB2 Interacts with LC3-II during Mitophagy.
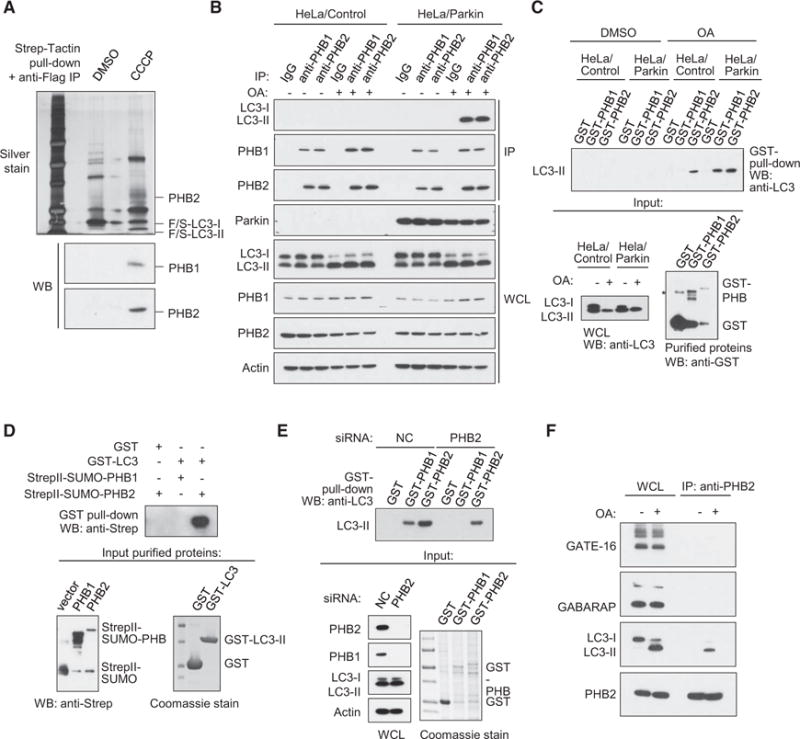
(A) Isolation of Flag-StrepII-LC3 complexes by Strep-Tactin pull-down followed by anti-Flag immunoprecipitation from HeLa/Parkincells stably expressingFlag-StrepII-LC3 and treated with either DMSO (control) or 10μM CCCP (mitochondrial uncoupling agent) for 4 hr. Top gel:silver stainofFlag-StrepII-LC3 complexes. Labeled bands were identified by mass spectrometry analysis. F/S, Flag-StrepII. Bottom gels: western blot analyses of Flag-StrepII-complexes with indicated antibodies.
(B) Co-immunoprecipitation of LC3-II with anti-PHB1 or anti-PHB2 in HeLa/Control or HeLa/Parkin cells treated with either DMSO or OA (oligomycin, 2.5 μM; antimycin A, 250 nM) (mitochondrial electron transport chain inhibitors) for 4 hr.
(C) Recombinant GST-PHB1 or GST-PHB2 pull-down of LC3-II from HeLa/Control or HeLa/Parkin cell lysates treated with DMSO or OA for 4 hr. Top: western blot analysis using anti-LC3 of GST-PHB pull-downs. Bottom left: western blot analysis of LC3 in whole cell lysates used for experiment on top. Bottom right: western blot analysis using anti-GST of purified recombinant GST proteins used for experiment on top. Asterisk denotes non-specific band.
(D) In vitro interaction of purified GST-LC3 with StrepII-SUMO-PHB1 or StrepII-SUMO-PHB2. Top gel: western blot analysis using anti-Strep after GST pull-down. Bottom left gel: western blot using anti-Strep of purified StrepII-SUMO-PHB proteins used for experiment on top. Bottom right gel: Coomassie stain of purified GST proteins used for experiment on top.
(E) Recombinant GST-PHB1 or GST-PHB2 pull-down of LC3-II from OA-treated HeLa/Parkin cell lysates treated with non-targeting control (NC) or PHB2 dicer-substrate siRNA. Top: western blot analysis with anti-LC3 of GST-PHB pull-downs. Bottom left: western blot analysis of LC3 in whole cell lysates used for experiment on top. Bottom right: western blot analysis using anti-GST of purified recombinant GST proteins used for experiment on top. WCL, whole cell lysates.
(F) Co-immunoprecipitation of GATE-16, GABARAP, or LC3-II with anti-PHB2 in HeLa/Parkin cells treated with either DMSO or OA for 4 hr.
See also Figure S1.
We investigated whether PHB1 and/or PHB2 interact directly with LC3. Purified GST-PHB1 and GST-PHB2 pulled down LC3-II in OA-treated HeLa/Parkin but not vector control-transfected HeLa cell lysates; GST-PHB2 pulled down a small amount of LC3-II even in the absence of Parkin expression, whereas both GST-PHB1 and GST-PHB2 pulled down LC3-II in the presence of Parkin (Figure 1C). The increase in GST-PHB1 and GST-PHB2 pull down of LC3-II in the presence of Parkin and OA treatment may reflect unknown post-translational modification(s) in LC3-II required for binding and/or the higher levels of LC3-II observed in these mitophagy-inducing conditions. Further analyses revealed that PHB2 interacts directly with LC3-II, whereas PHB1 interacts indirectly with LC3-II as a result of being part of a heterodimeric complex with PHB2. In support of this conclusion, purified StrepII-SUMO-PHB2, but not StrepII-SUMO-PHB1, bound in vitro to purified GST-LC3 (truncated at amino acid 120, the location of the Atg4 cleavage site) (Figure 1D). Moreover, GST-PHB1 was unable to pull-down LC3-II in lysates of HeLa cells with small interfering RNA (siRNA)-mediated depletion of PHB2 (Figure 1E). Consistent with previous reports (Merkwirth and Langer, 2009), PHB2 siRNA also knocked down expression of PHB1 (Figure 1E) and exogenous PHB2 expression was difficult to detect unless exogenous PHB1 was co-expressed (Figures S1A and S1B), as PHB2 are PHB1 are only stable when bound to each other in a heterodimeric state.
Taken together, these results indicate that during mitophagy, the autophagosome-associated protein LC3-II directly binds to the inner mitochondrial protein PHB2 and indirectly binds to the heterodimeric PHB2 binding partner PHB1. Other mammalian ATG8/LC3 orthologs, such as GABARAP and GATE-16, did not co-immunoprecipitate with PHB2 during mitophagy-inducing conditions (Figure 1F). Thus, given the direct binding of LC3-II to PHB2, we focused on PHB2.
PHB2 Is Required for Parkin-Mediated Mitophagy
We evaluated whether PHB2 is required for Parkin-mediated mitophagy in HeLa cells and murine embryonic fibroblasts (MEFs). PHB2 knockdown with three different dicer-substrate siRNAs blocked Parkin-mediated OA-induced mitophagy, as measured by quantitative image analysis of ATP5B (a mitochondrial matrix protein) puncta (Figures 2A and 2B) or western blot analysis of degradation of the IMM protein TIM50 or the mitochondrial matrix protein HSP60 (Figures 2C–2E). Mitophagy inhibition with PHB2 knockdown was similar to that observed with knockdown of an essential autophagy gene, ATG7. In addition, OA-induced mitophagy resulted in marked degradation of PHB1 and PHB2 that was blocked by ATG7 siRNA, indicating that levels of PHB1 and PHB2 can be used as markers of mitophagy.
Figure 2. PHB2 Is Essential for Parkin-Mediated Mitophagy.
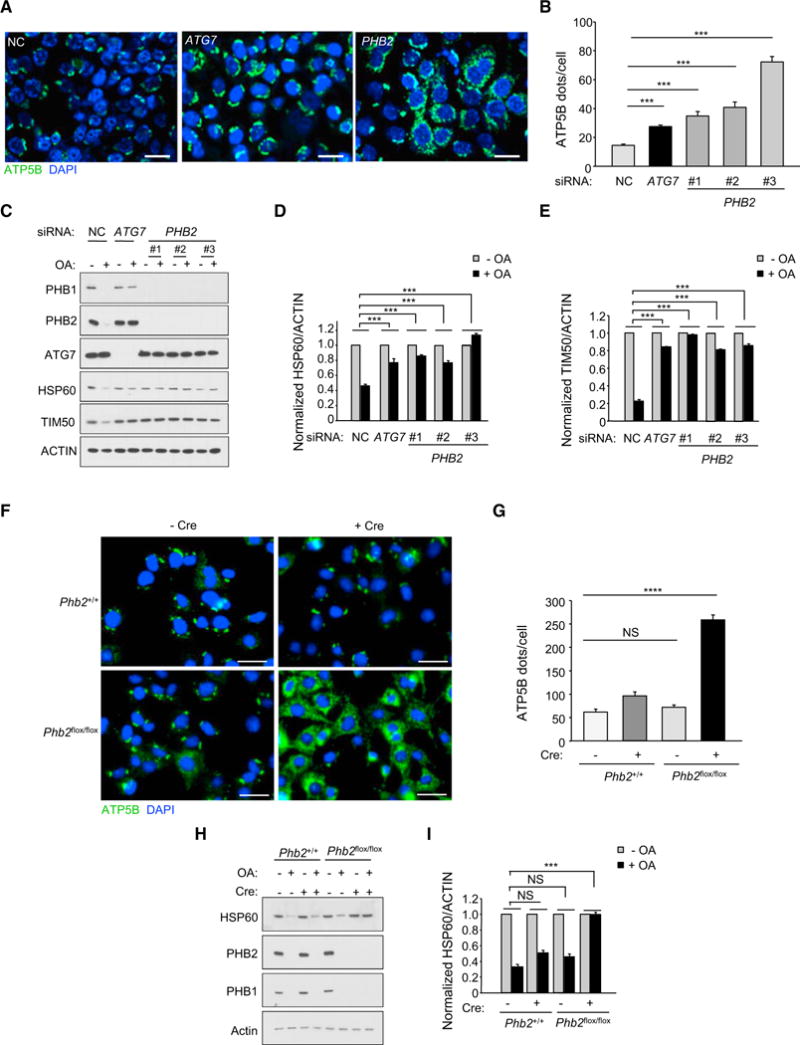
(A) Representative images of ATP5B immunofluorescence staining of HeLa/Parkin cells transfected with indicated siRNA and treated with OA for 16 hr prior to imaging and automated image analysis. Scale bars, 20 μm.
(B) Quantitation of cytoplasmic ATP5B puncta (as shown in representative images in A) in cells transfected with indicated siRNA and treated with OA. Shown are mean ± SEM of 150 cells analyzed per condition. Similar results were observed in three independent experiments. ***p < 0.001, one-way ANOVA with the Sidak test.
(C–E) Western blot analysis of indicated mitophagy markers (C) in cells treated as shown in (A) and densitometric quantification of HSP60/ACTIN (D) and TIM50/ACTIN (E) ratios for western blot analyses of cells treated as in (A). Shown are mean ± SEM values from western blot analyses of three independent experiments. ***p < 0.001, two-way ANOVA for comparison of magnitude of change ± OA in indicated siRNA group versus NC RNA control.
(F) Representative images of ATP5B immunofluorescence staining of Phb2+/+/Parkin or Phb2flox/flox/Parkin MEFs transduced with empty vector or a Cre-expressing lentivirus and treated with OA for 16 hr prior to imaging and automated image analysis. Scale bars, 20 μm.
(G) Quantitation of cytoplasmic ATP5B puncta in cells of indicated genotype treated as shown in representative images displayed in (D). Shown are mean ± SEM of 150 cells analyzed per condition. Similar results were observed in three independent experiments. **p < 0.01; ****p < 0.0001; NS, not significant; one-way ANOVA with the Sidak test.
(H and I) Western blot analysis of indicated mitophagy markers (H) in MEFs treated as shown in (D) and densitometric quantification of HSP60/actin ratios (I). Shown are mean ± SEM values from western blot analyses of three independent experiments. NS, not significant; ***p < 0.001; Two-way ANOVA for comparison of magnitude of change ± OA in indicated group versus Cre (−) Phb2+/+ group.
We verified the essential role of PHB2 in Parkin-mediated mitophagy using MEFs lacking Phb2. As Phb2 null mice are early embryonic lethal (Merkwirth et al., 2008), we used Phb2+/+ or Phb2flox/flox MEFs (Merkwirth et al., 2008) stably transfected with a Parkin-expressing plasmid and transduced with a lentivirus expressing Cre recombinase (Pfeifer et al., 2001). Cre expression resulted in efficient depletion of endogenous PHB2 (and PHB1) protein (Figure 2H) and a block in OA-induced mitophagy, as measured by ATP5B puncta clearance (Figures 2F and 2G) or western blot analysis of HSP60 degradation (Figures 2H and 2I). Thus, PHB2 is essential for Parkin-mediated mitophagy.
Proteasomal-Dependent Outer Mitochondrial Membrane Rupture Is Required for the PHB2 and LC3 Interaction
The binding of PHB2, an IMM protein, to LC3-II during mitophagy suggested that OMM rupture was necessary for binding to occur. Previous studies have shown that proteasome-dependent OMM rupture occurs during Parkin-mediated mitophagy (Chan et al., 2011; Yoshii et al., 2011). We therefore sought to determine whether proteasomal-dependent OMM rupture is required for the interaction between PHB2 and LC3-II.
To evaluate whether the PHB1/PHB2 complex is exposed to the cytoplasm during mitophagy, we compared protease sensitivity of PHB1 and PHB2 in purified mitochondrial fractions from HeLa cells in the presence or absence of Parkin expression and OA (Figure 3A). In all conditions, the OMM protein TOMM20 was degraded upon trypsin protease treatment. However, PHB1 and PHB2 were resistant to trypsin digestion unless cells both expressed Parkin and were treated with OA, indicating the cytoplasmic exposure of this IMM protein complex during mitophagy.
Figure 3. Proteasomal-Dependent Outer Mitochondrial Membrane Rupture Is Required for PHB2/LC3 Interaction and Mitophagy.
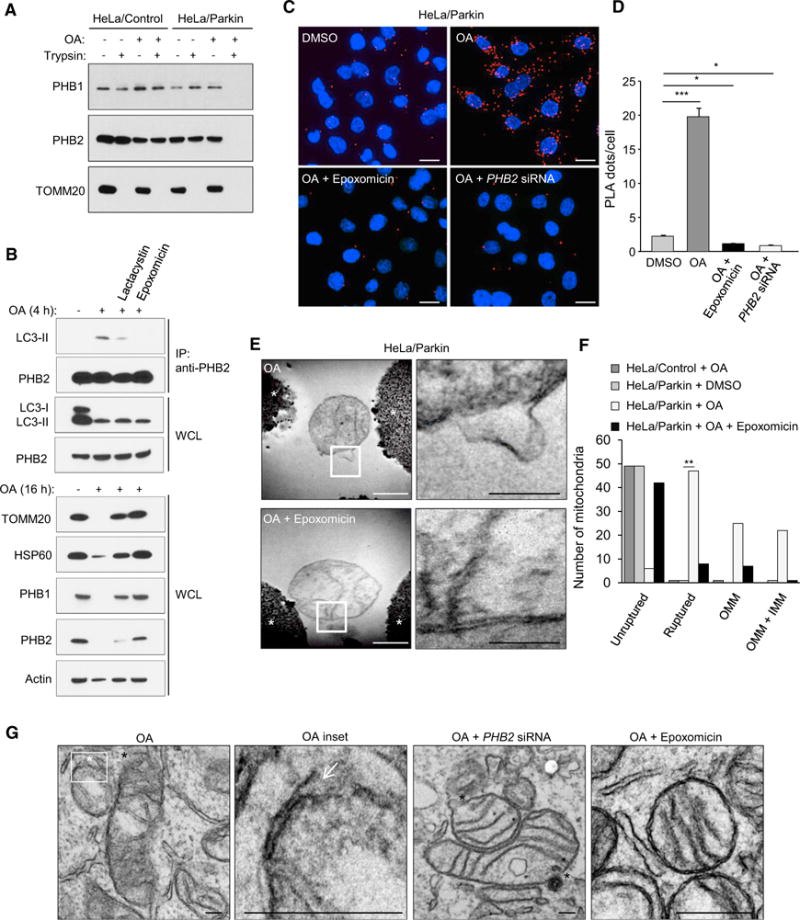
(A) Protease protection assay of mitochondrial fractions purified from HeLa/Control or HeLa/Parkin cells treated with DMSO or OA for 4 hr.
(B) Effects of proteasome inhibitors (5 mM lactacystin or 100 nM epoxomicin) on PHB2/LC3-II interaction, OMM rupture and mitophagy in HeLa/Parkin cells treated with OA for 4 hr (top gels assess co-immunoprecipitation of PHB2 and LC3) or 16 hr (bottom gels show western blots of indicated mitochondrial proteins). WCL, whole cell lysates. Similar results were observed in two independent experiments.
(C and D) Representative images (C) and quantification (D) of Duolink in situ PLA assay demonstrating the interaction between LC3 and PHB2 in HeLa/Parkin/Flag.StrepII-LC3 cells in cells transfected with NC or PHB2 siRNA for 48 hr and then treated for 2 hr with either DMSO, OA, or OA + epoxomicin. In (D), bars are mean ± SEM for triplicate samples of >50 cells per sample. Similar results were obtained in three independent experiments. ***p < 0.001, *p < 0.05;one-way ANOVA.
(E) Electron micrographs of mitochondria immunoprecipitated from HeLa/Parkin cells treated for 4 hr with OA in the presence or absence of epoxomicin. Right panels: higher magnification images of regions outlined in left panels. See also Figure S2. Asterisk, Dynabead. White scale bar, 500 nm. Black scale bar, 200 nm.
(F) Quantitation of mitochondria with unruptured or ruptured membranes (further subdivided into mitochondria with OMM rupture only or rupture of both OMM and IMM [OMM + IMM]) in the experiment shown in (E). Fifty mitochondria per condition were examined by an observer blinded to experimental condition. **p < 0.01, Chi-square test.
(G) Representative electron micrographs showing mitochondrial rupture in HeLa/Parkin cells transfected with NC or PHB2 siRNA and treated with OA with or without epoxomicin for 2 hr. White asterisk and white arrow, site of focal OMM rupture; black asterisks, sites of focal rupture of OMM and IMM. Scales bars, 100 μm.
To evaluate whether binding of the IMM protein PHB2 to LC3-II during mitophagy requires proteasome-dependent degradation of the OMM, we co-immunoprecipitated PHB2 and LC3 in HeLa/Parkin cells treated with OA and the proteasome inhibitors, lactacystin and epoxomicin (that is a more potent inhibitor) (Meng et al., 1999) (Figure 3B). The binding of LC3-II to PHB2 in mitophagy conditions was partially decreased by lactacystin, which partially blocked TOMM20 degradation, but was abrogated by epoxomicin that completely blocked Parkin-mediated OA-induced TOMM20 degradation. Epoxomicin also blocked OA-induced mitophagy, as evidenced by the lack of degradation of the IMM proteins, PHB1 and PHB2, and the mitochondrial matrix protein HSP60 (Figure 3B).
We used a proximity ligation assay (PLA) to further assess whether PHB2 and LC3 interact during Parkin-mediated mitophagy in a proteasome-dependent manner. In HeLa/Parkin cells, there was a dramatic increase in numbers of PHB2 and LC3 PLA dots per cell in response to OA (Figures 3C and 3D). Consistent with the PHB2/LC3-II co-immunoprecipitation results, this increase was blocked by the proteasome inhibitor, epoxomicin. PHB2 knockdown also blocked this increase, decreasing PLA dots to numbers less than those observed in DMSO-treated controls, confirming the specificity of the PLA for detecting PHB2/LC3 interaction in intact cells.
To confirm whether epoxomicin blocks OMM rupture during Parkin-mediated mitophagy, we performed a quantitative analysis of electron micrographs of intact mitochondria immunoprecipitated from HeLa cells (Sumpter et al., 2016) in the presence and absence of Parkin expression, OA, and epoxomicin (Figures 3E and 3F). In HeLa/Control cells treated with OA or HeLa/Parkin cells treated with DMSO, nearly all mitochondria were unruptured (Figure S2). In contrast, in HeLa/Parkin cells treated with OA, the majority of mitochondria had focal sites of rupture; about half of the mitochondria had rupture of the OMM alone and half had rupture of both the OMM and IMM. This focal mitochondrial membrane rupture was nearly completely blocked by epoxomicin (Figure 3F).
We also performed transmission electron microscopy (TEM) of intact OA-treated HeLa/Parkin cells in the presence or absence of epoxomicin or PHB2 knockdown (Figure 3G). In OA-treated HeLa/Parkin cells, the majority of mitochondria showed focal membrane rupture. As with TEM analysis of immunoprecipitated mitochondria, some mitochondria had rupture of the OMM alone whereas some had rupture of both the OMM and IMM. siRNA targeting PHB2 did not block mitochondrial membrane rupture. In contrast, the majority of mitochondria contained intact membrane structure in cells treated with epoxomicin.
Taken together, our results indicate that PHB2 binds to LC3-II upon proteasome-dependent mitochondrial OMM rupture during mitophagy.
Phagophores Localize to Damaged Mitochondria at Focal Sites of Outer Mitochondrial Membrane Rupture
Our results suggest a model in which phagophores capture mitochondria at the sites of OMM rupture through an interaction between LC3 and PHB2. We used super-resolution structured illumination microscopy (SIM) and immune EM to better characterize the localization of LC3 during mitophagy.
We examined whether LC3 colocalizes with TOMM20, an OMM protein, and/or PHB2, an IMM protein, during baseline and mitophagy-inducing conditions in HeLa/Parkin cells. In control conditions, TOMM20 and PHB2 were closely aligned in spherical shapes (as expected with intact mitochondria), and we did not observe GFP-LC3 near mitochondria (Figure 4A, left panel). In the presence of OA, intact mitochondrial structure was lost and GFP-LC3 was in close proximity to PHB2 but not TOMM20 (Figure 4A, middle panels). When HeLa/Parkin cells were treated with both OA and epoxomicin, mitochondrial structure was better preserved and GFP-LC3 was observed in the vicinity of mitochondria, but not in close proximity to either PHB2 or TOMM20 (Figure 4A, right panel).
Figure 4. LC3 Localizes with the Mitochondrial Inner Membrane during Parkin-Mediated Mitophagy.
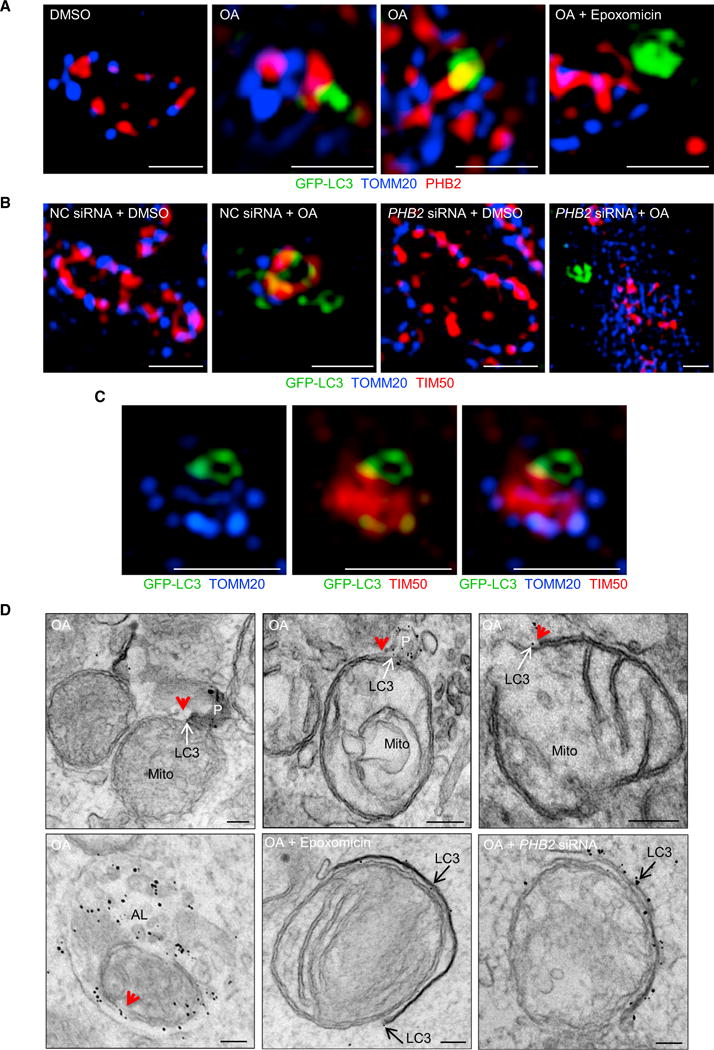
(A) Representative SIM images showing colocalization of GFP-LC3, TOMM20, and PHB2 in HeLa/Parkin/GFP-LC3 cells treated for 2 hr with DMSO, OA, or OA + epoxomicin. Scale bars, 1 μm.
(B) Representative SIM images showing colocalization of GFP-LC3, TOMM20, and TIM50 in NC or PHB2 siRNA-transfected HeLa/Parkin/GFP-LC3 cells treated with DMSO or OA for 2 hr. Scale bars, 1 μm.
(C) Representative SIM image of mitochondrion in OA-treated HeLa/Parkin/GFP-LC3 cell stained as in (B) demonstrating colocalization of GFP-LC3 with IMM marker (TIM50) at site lacking OMM marker (TOMM20). Scale bars, 1 μm.
(D) Electron micrographs of HeLa/Parkin/GFP-LC3 cells stained with immunogold-conjugated anti-GFP antibody to detect GFP-LC3. Cells were transfected with NC or PHB2 siRNA and treated for 2 hr with OA with or without epoxomicin. Red arrowheads, sites of OMM rupture. White arrows, immunogold particles labeling LC3 at sites of OMM rupture. Black arrows, immunogold particles labeling LC3 at membranes in vicinity of mitochondria. P, phagophore; Mito, mitochondrion; AL, autolysosome. Scale bars, 100 nm.
In (A)–(C), high-resolution image data acquisition was performed on seven randomly chosen cells per sample, and similar results were observed in three independent experiments. In (D), ten randomly chosen cells were imaged per sample. Two technical replicates per condition were analyzed, and similar results were observed in four independent experiments.
See also Figure S3.
We performed SIM analysis using TIM50 as an IMM marker to evaluate the effects of PHB2 knockdown on GFP-LC3 localization during mitophagy (Figures 4B and 4C). As observed for PHB2 staining, GFP-LC3 was in close proximity to TIM50 and not TOMM20 in HeLa/Parkin cells treated with OA and control siRNA (Figure 4B). For some mitochondria, the proximity of GFP-LC3 to the IMM appeared to occur sites of OMM rupture (Figure 4C). When cells were treated with OA and siRNA targeting PHB2, GFP-LC3 was observed in the vicinity of mitochondria but not in close proximity with OMM or IMM markers (Figure 4B).
We confirmed these findings using immune EM to detect GFP-LC3 in HeLa/Parkin/GFP-LC3 cells undergoing mitophagy (Figure 4D). In control OA-treated cells, we observed two patterns of GFP-LC3 immunogold staining associated with damaged mitochondria. Some mitochondria had GFP-LC3-labeled phagophores in direct contact with the mitochondria at the sites of OMM rupture (see Figure 4D, upper left and middle panels for representative examples). Other mitochondria had GFP-LC3 directly adjacent to the IMM at sites of OMM rupture in the absence of apparent LC3-associated membranous structures (see Figure 4D, upper right panel). For nearly all mitochondria, GFP-LC3 immunogold particles were absent from regions of the mitochondria with an intact OMM structure. Moreover, captured mitochondria inside autolysosomes remained decorated with LC3 at sites of OMM rupture (Figure 4D, lower left panel). As in SIM analyses, GFP-LC3-labeled membranous structures were observed in the vicinity of intact mitochondria in cells treated with OA and epoxomicin (Figure 4D, lower middle panel) and in the vicinity of damaged mitochondria in cells with PHB2 knockdown (Figure 4D, lower right panel). However, in neither condition did we observe GFP-LC3-labeled phagophores in immediate juxtaposition to the mitochondrial membrane. Consistent with a defect in autophagic targeting of damaged mitochondria, autolysosomes in epoxomicin-treated and PHB2 siRNA-treated cells formed normally, but contained contents other than mitochondria (data not shown).
Thus, GFP-LC3-associated phagophores closely associate with the IMM at sites of OMM rupture during mitophagy; proteasomal inhibition or PHB2 knockdown block this association but not the recruitment of GFP-LC3 to the vicinity of damaged mitochondria. Proteasomal inhibition or PHB2 knockdown did not block stages of mitophagy prior to recruitment of the phagophore to the region of damaged mitochondria. In biochemical studies, we did not observe alterations of ubiquitination of the OMM proteins, TOMM70 or TOMM20, with PHB2 siRNA (Figure S3A) in OA-treated HeLa/Parkin cells. Moreover, in immunofluorescence analyses, we did not observe differences in the recruitment of the autophagy adaptor protein, NBR1, or the autophagy machinery proteins, ULK1, WIPI2, HA-ATG5, or GFP-ATG16 to mitochondria after OA in the presence of proteasomal inhibition or PHB2 siRNA knockdown (Figures S3B–S3E).
The LIR Domain of PHB2 Is Essential for Interaction with LC3
We identified the LIR domain (Noda et al., 2008) of PHB2 and determined whether it is essential for mitophagy function. There are three candidate LIR domains (W/F/YxxL/I/V motifs) in PHB2, including one that is unique to PHB2 (amino acids [aa] 77–80) and two that are conserved between PHB1 and PHB2 (aa 121–124 and aa 175–178) (Figure 5A). Amino acids 77–80 are not involved in PHB2 binding to LC3 as either deletion of this region or a Y77K substitution mutation (that changes this amino acid to that found in PHB1, a protein that does not directly bind to LC3) (Figure 1D) did not block in vitro binding of purified StrepII-SUMO-PHB2 to GST-LC3 (aa 1–120) (Figure 5B). In contrast, a Y121A/L124A mutation in StrepII-SUMO- PHB2 blocked in vitro binding to GST-LC3 (Figure 5C). A mutation in the other candidate LIR domain, F175A/I178A, led to lower levels of protein expression but did not abrogate binding to GST-LC3. Thus, we conclude that amino acids 121–124 (YQRL) represent the PHB2 LIR domain. This LIR domain lies within a region of PHB2 that is predicted to be exposed within the space between the IMM and OMM (Back et al., 2002).
Figure 5. Identification of the LIR Domain of PHB2.
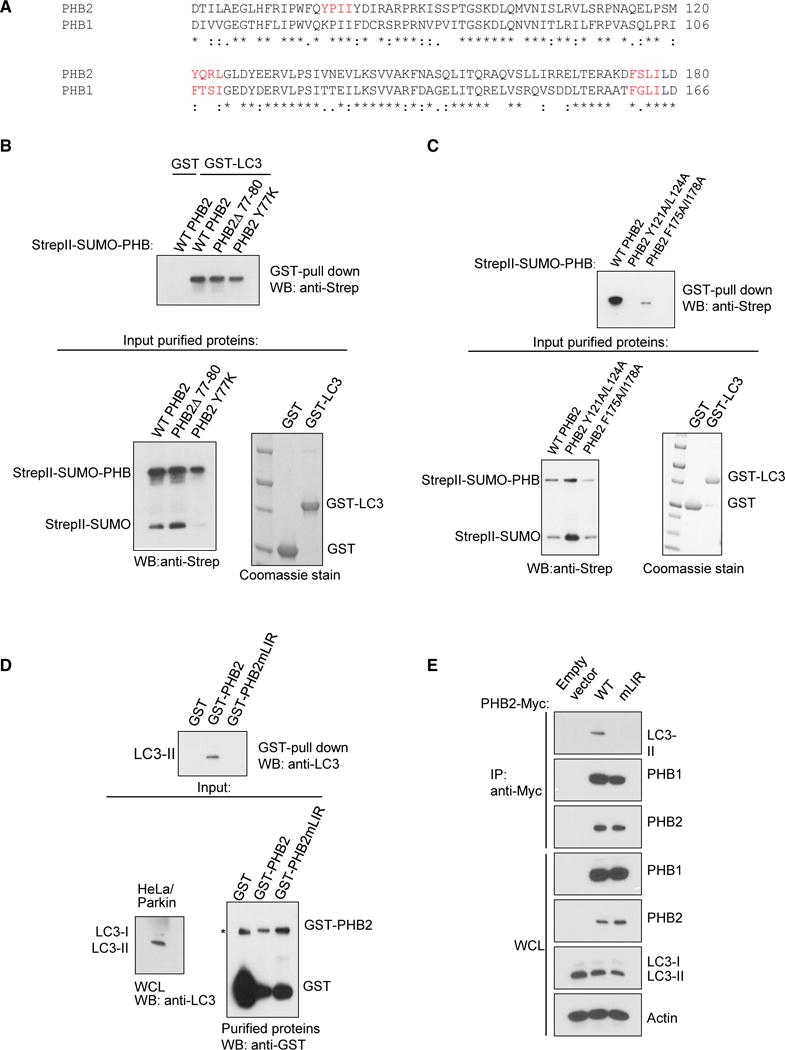
(A) Sequence alignment of PHB1 and PHB2. Candidate LIR domains (W/F/YxxL/I/V motifs) are depicted in red.
(B) In vitro interaction of purified GST-LC3 with indicated StrepII-SUMO-PHB2 constructs. Top: western blot analysis using anti-Strep after GST pull-down. Bottom left: western blot analysis using anti-Strep of purified StrepII-SUMO-PHB2 proteins used for experiment on top. Bottom right: Coomassie stain of purified GST fusion proteins used for experiment on top.
(C) In vitro interaction of purified GST-LC3 with indicated StrepII-SUMO-PHB2 constructs. Top: western blot analysis using anti-Strep after GST pull-down. Bottom left: western blot analysis using anti-Strep of purified StrepII-SUMO-PHB2 proteins used for experiment on top. Bottom right: Coomassie stain of purified GST fusion proteins used for experiment on top.
(D) Recombinant GST-PHB2 or GST-PHB2mLIR (Y121A/L124A) pull-down of LC3-II from HeLa/Parkin cell lysates treated with OA for 4 hr. Top: western blot analysis using anti-LC3 of GST-PHB pull-downs. Bottom left: western blot analysis of LC3 in whole cell lysates used for experiment on top. Bottom right: western blot analysis using anti-GST of purified recombinant GST proteins used for experiment on top. Asterisk denotes non-specific band.
(E) Co-immunoprecipitation of LC3-II with wild-type (WT) PHB2-Myc in HeLa/Parkin cells treated with OA for 4 hr. Twenty-four hours prior to experiment, endogenous PHB2 and PHB1 were depleted with PHB2 siRNA, and cells were transfected with empty vector or indicated PHB2-Myc construct and non-tagged PHB1.
We confirmed that the PHB2 LIR domain Y121A/L124A mutation blocked GST-PHB2 pull-down of LC3-II in OA-treated HeLa/Parkin cell lysates with knockdown of endogenous PHB1 and PHB2 (Figure 5D). In addition, it blocked PHB2-Myc immunoprecipitation of LC3-II in OA-treated HeLa/Parkin cells (with decreased endogenous PHB2) without affecting co-immunoprecipitation of PHB1 (Figure 5E). Thus, the LIR domain of PHB2 regulates direct binding to LC3 in vitro and binding to LC3-II in HeLa cells during mitophagy-inducing conditions.
The LIR Domain of PHB2 Is Essential for Mitophagy
To determine whether the PHB2 LIR domain is required for mitophagy, we expressed wild-type PHB2 or the PHB2 LIR domain Y121A/L124A mutant in Parkin-expressing, Cre-expressing Phb2flox/flox MEFs (Figure 5C). We also expressed exogenous PHB1 in these cells, as PHB2 is not stable in cells lacking PHB1 (Figure S1). Wild-type PHB2, but not the PHB2 LIR mutant, rescued Parkin-mediated OA-induced mitophagy, as measured by quantitative image analysis of ATP5B puncta (Figures 6A and 6B) and western blot analysis of HSP60, PHB1, and PHB2 degradation (Figures 6C and 6D). These observations are not due to aberrant localization of the PHB2 LIR domain mutant, as it colocalized with mitochondria in a manner similar to wild-type PHB2 (Figure 6E). Importantly, the PHB2 LIR domain mutant behaved similarly to wild-type PHB2 and fully rescued known mitochondrial phenotypes of PHB2 loss in Phb2 knockout MEFs (Merkwirth and Langer, 2009), including selective loss of the long isoform of the dynamin-like GTPase, OPA1 (an essential component of the mitochondrial fusion machinery) (Figure 6F), aberrant cristae morphogenesis (Figures 6G and 6H), and impaired cellular proliferation (Figure 6I). Thus, the PHB2 LIR mutation does not affect previously described mitochondrial processes controlled by prohibitins, but does abrogate the ability of PHB2 to mediate mitophagy. Together, these data indicate that PHB2 functions in mitophagy via an interaction between its LIR domain and LC3, rather than indirectly as a result of other effects on mitochondrial structure or function.
Figure 6. The LIR Domain of PHB2 Is Required for Parkin-Mediated Mitophagy.
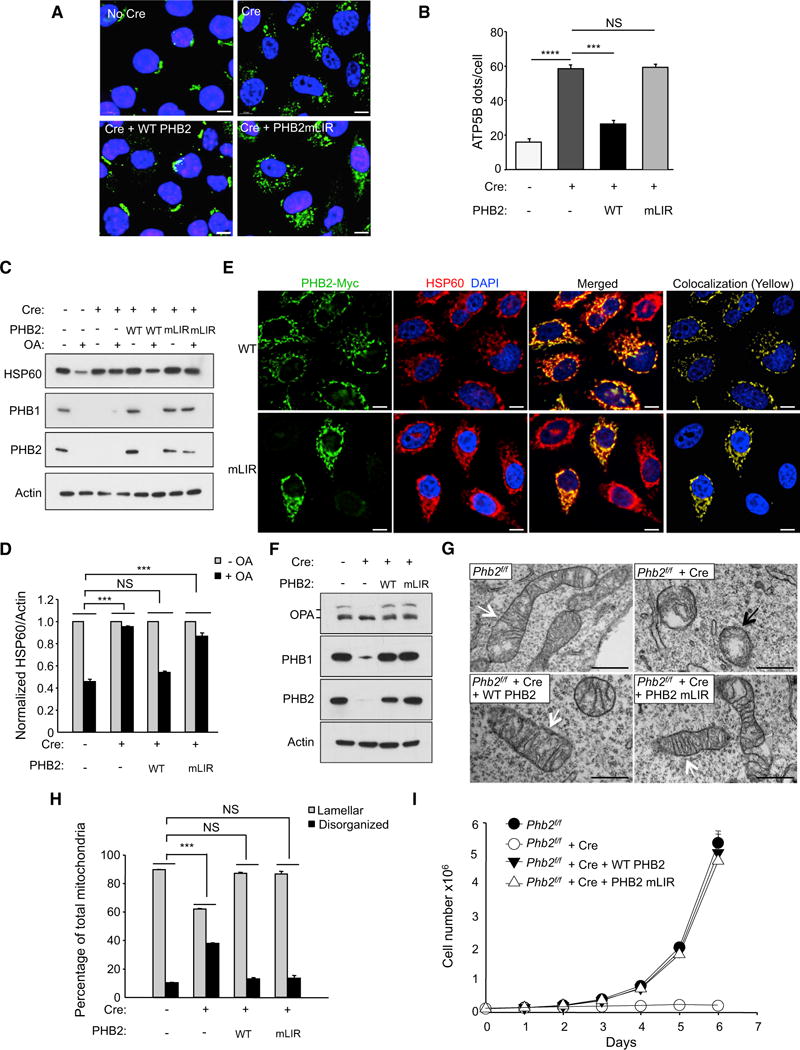
(A) Representative images of ATP5B immunofluorescence staining of Phb2flox/flox/Parkin MEFs transduced with empty vector or a Cre-expressing lentivirus and treated with OA for 16 hr prior to imaging and automated image analysis. Scale bars, 10 μm.
(B) Quantitation of cytoplasmic ATP5B puncta in Phb2flox/flox/Parkin MEFs transduced with lentiviruses expressing proteins indicated below x axis. Shown are mean ± SEM of 150 cells analyzed per condition. Similar results were observed in three independent experiments. ***p < 0.001; NS, not significant; one-way ANOVA with the Sidak test.
(C) Western blot analysis of indicated mitophagy markers in MEFs treated as shown in (A).
(D) Densitometric quantification of HSP60/Actin in (C). Shown are mean ± SEM values from western blot analyses of three independent experiments. NS, not significant; ***p < 0.001; two-way ANOVA for comparison of magnitude of change ± OA in indicated group versus Cre (−) control group.
(E) Representative images showing co-localization of PHB2-myc and HSP60 in HeLa/Parkin cells. Twenty-four hours prior to experiment, endogenous PHB2 and PHB1 were depleted with PHB2 siRNA and cells were co-transfected with indicated PHB2-myc construct and non-tagged PHB1. Scale bars, 10 μm.
(F) Western blot analysis to detect OPA1 processing Phb2flox/flox/Parkin MEFs during normal growth conditions with or without expression of Cre and WT PHB2 or PHB2 mLIR. Similar results were observed in two independent experiments.
(G) Representative images of mitochondria in MEFs shown in (F) that would be classified as lamellar (white arrows) or disorganized (black arrows).
(H) Quantification of mitochondrial cristae morphology for MEFs shown in (F) and (G). For each genotype, mitochondria were scored in 50 cell profiles. Bars are mean ± SEM for triplicate samples per genotype. NS, not significant, ***p < 0.001; two-way ANOVA for comparison of magnitude of difference between lamellar versus disorganized mitochondria in indicated group as compared to Cre (−) control group.
(I) Growth curves of MEFS of indicated genotype. Cells (2 × 104) were plated and counted at serial time points. Bars are mean ± SEM of triplicate samples for each genotype at each time point. p < 0.0001 for Phb2f/f + Cre versus all other groups; liner mixed-model effect. No differences were observed between Phb2f/f + Cre + WT PHB2 versus Phb2f/f + Cre + PHB2 mLIR.
PHB2 Is Essential for Paternal Mitochondrial Elimination in C. elegans
We sought to determine whether PHB2 is required for a well-characterized in vivo function of mitophagy, the elimination of paternal mitochondrial DNA after C. elegans oocyte fertilization (Levine and Elazar, 2011). To evaluate this, we examined the effect of paternal RNAi knockdown of phb-2 (C. elegans homolog of mammalian PHB2) on the degradation of sperm-derived mitochondria in embryos. To monitor the fate of paternal mitochondria in fertilized oocytes, males were labeled with CMXRos (MitoTracker Red), which accumulates in mitochondria in a membrane potential-dependent manner. To avoid competition from maternally derived sperm (self-fertilization), we used a strain carrying fem-3(e2006), a temperature-sensitive allele that gives rise to females at a restrictive temperature (25°C) (Hodgkin, 1986). MitoTracker-labeled males and fem-3(e2006) females were crossed, and sperm-derived mitochondria in the F1 embryos were followed microscopically. Sperm-derived Mito-Tracker signals remained strong in the 1- to 4-cell stage embryos, but gradually diminished after the 4-cell stage and almost disappeared after the 24-cell stage, consistent with previous observations (Al Rawi et al., 2011; Sato and Sato, 2011; Zhou et al., 2011) (Figure 7A). In contrast, paternal inactivation of phb-2 impaired sperm-derived mitochondrial degradation in a manner similar to maternal inactivation of atg-7, a core autophagy gene previously reported to be essential for paternal mitochondrial elimination (Al Rawi et al., 2011; Sato and Sato, 2011) (Figure 7A). By the 64- to 100-cell stage, quantitative analysis of sperm-derived MitoTracker signals (CMXRos dots/embryo) revealed almost complete clearance in control animals; in contrast, there was an accumulation of sperm-derived mitochondria after either paternal inactivation of phb-2 or maternal inactivation of atg-7 (Figure 7B). Thus, sperm-derived phb-2 is essential for the proper degradation of paternal mitochondria.
Figure 7. phb-2 Is Required for the Elimination of Paternal Mitochondria and Prevention of Heteroplasmy in C. elegans.
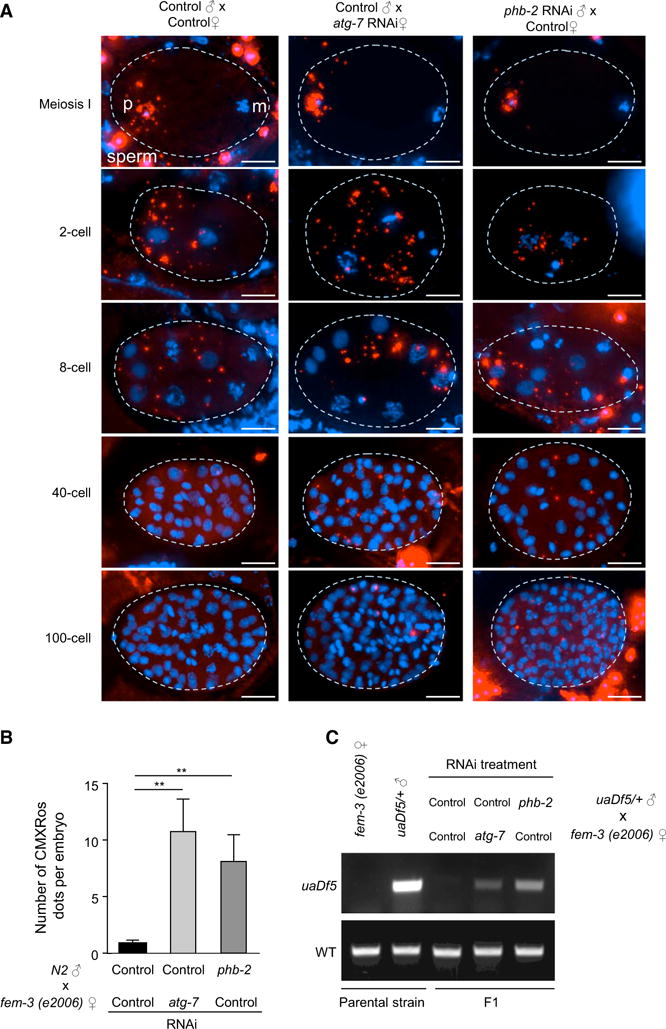
(A) Paternal inactivation of phb-2 results in the persistence of the sperm mitochondria during embryonic development. Wild-type N2 males and fem-3(e2006) females were fed on indicated RNAi bacteria. Males were labeled with CMXRos (Red) before mating with unlabeled females. F1 embryos were stained with DAPI (Blue) and examined in various stages. Projections of z stacked representative images are shown. Dotted lines indicate the outline of the embryos. Red signals outside the embryos are unfertilized sperm. p, paternal nuclear; m, maternal nuclear. Scale bars, 10 μm.
(B) Quantitative analysis of sperm-derived CMXRos signals (paternal mitochondria) in F1 embryos at 64- to 100-cell stage (mean ± SEM; **p < 0.01; one-way ANOVA with the Sidak test).
(C) Sperm-derived phb-2 is required to prevent paternal mtDNA transmission to F1 offspring. uaDf5/+ males and fem-3(e2006) females treated with the indicated RNAi bacteria were mated, and at least 30 L1 progeny were genotyped by PCR using wild-type or uaDf5 mtDNA-specific primers. Parental strains were included as controls. Similar results were observed in three independent experiments.
We investigated whether mtDNA from phb-2-deficient sperms can be passed on to F1 offspring. To do so, we utilized males from a strain carrying uaDf5, a mtDNA allele harboring a large deletion (3,054 bp) in a stable heteroplasmic state (Tsang and Lemire, 2002). The uaDf5/+ males and fem-3(e2006) females were crossed and the progeny was examined for uaDf5 mtDNA (Figure 7C). In control F1 offspring, uaDf5 mtDNA was quickly eliminated during development and could not be detected by PCR at the L1 stage (the first larva stage), consistent with prior observations (Sato and Sato, 2011). In contrast, uaDf5 mtDNA was transmitted to F1 offspring that was paternally phb-2- or maternally atg-7-deficient, indicating that sperm-derived phb-2 is required for the prevention of paternal mtDNA transmission to the next generation.
DISCUSSION
Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor
Our results identify the IMM protein PHB2 as a mitophagy receptor required for both Parkin-mediated mitophagy in mammalian cells and for paternal mitochondrial clearance in C. elegans embryos. PHB2 binds to LC3 during mitophagy through a canonical LC3-interacting region (LIR) motif and is essential for mitophagy in mammalian cells and nematode development. These data reveal a new paradigm for the cellular mechanism of mitophagy, in which IMM serves a key role in cargo recognition.
Mitophagy Involves Sequential Events at the Outer and Inner Mitochondrial Membranes
Several proteins on the OMM directly bind LC3, serving as mitophagy receptors, or indirectly bind LC3 when ubiquitinated via adaptor proteins that contain both ubiquitin binding association (UBA) domains and LIR domains (Khaminets et al., 2016). However, in Parkin-mediated mitophagy and C. elegans sperm-derived mitochondrial elimination, the OMM is ruptured (Chan et al., 2011; Yoshii et al., 2011; Wang et al., 2016), thereby exposing the IMM for potential interactions with the autophagosome. Our data show that following Parkin-mediated proteasome-dependent OMM degradation and recruitment of autophagy proteins to the OMM, the IMM serves as a direct target for autophagy via the binding of the IMM protein PHB2 to the autophagosomal membrane protein LC3-II. These observations likely explain why both the proteasome and autophagy systems are required for Parkin-mediated mitophagy. Events that occur at the OMM (including autophagy adaptor and core machinery protein recruitment) may be necessary (Lazarou et al., 2015) but not sufficient for autophagy; proteasome-dependent rupture of the OMM may be required to expose the IMM receptors for commitment to mitophagy. This dual layer of recognition of mitochondria by autophagosomes, both via OMM receptors and IMM receptors, may increase the efficiency and specificity of organelle removal.
It is not yet known how the IMM and its mitophagy receptor PHB2 become exposed during Parkin-independent mitophagy, as occurs during paternal mitochondrial elimination during C. elegans embryonic development. However, C. elegans paternal mitochondria become depolarized, lose a proportion of their cristae and undergo focal membrane rupture after their entry into the oocyte (Wang et al., 2016). Moreover, certain proteasome ubiquitin receptors are required in C. elegans oocytes for proper paternal mitochondrial clearance (Zhou et al., 2011). Although the precise mechanisms of paternal mitochondrial membrane rupture are unknown, these observations indicate that IMM proteins such as PHB2 likely become exposed to the cytoplasm during damage of sperm-derived mitochondria in oocytes.
Membrane Rupture May Represent a Common Feature of Organellar Specific Autophagy
Our data show that proteasome-dependent mitochondrial membrane rupture is necessary for Parkin-mediated mitophagy in mammalian cells in part via the cytoplasmic exposure of an IMM mitophagy receptor. Previously, membrane rupture has been shown to be required for autophagosomal sequestration of pathogen-containing endosomes and phagosomes (Shibutani and Yoshimori, 2014) and lysosomes (Maejima et al., 2013). Thus, membrane rupture may represent a common feature in selective forms of autophagy involving the sequestration of specific cellular organelles. Indeed, ubiquitination of pathogen-containing endosomal and phagosomal membranes is important in the autophagosomal response to bacterial invasion. Our findings raise the interesting question of whether the membranes of intracellular bacterial pathogens (like mitochondria) contain LC3-binding autophagy receptors that become exposed to LC3 upon rupture of the endosome or phagosome in which they reside.
Prohibitin 2 May Link Mitochondrial Dynamics and Mitophagy
The inner mitochondrial prohibitin complex, composed of large heterodimeric assemblies of PHB1 and PHB2, has a wide range of mitochondrial functions, including (1) regulating membrane protein degradation by the mitochondrial m-AAA protease, (2) acting as a chaperone in the stabilization and assembly of the oxidative phosphorylation system, (3) a role in maintaining mitochondrial genomic stability, and (4) regulating mitochondrial cristae morphogenesis via processing of OPA1, a dynamin-related GTPase essential for mitochondrial fusion (Artal-Sanz and Tavernarakis, 2009; Merkwirth and Langer, 2009; Nijtmans et al., 2002). Our data reveal another function of the mitochondrial prohibitin complex—serving as an IMM receptor for mitophagy during stress and development. Our results with the PHB2 LIR domain mutant indicate that previously described functions regulated by prohibitins, including OPA1-dependent cristae morphogenesis and cellular proliferation, are functionally distinct from its interaction with LC3 and role as a mitophagy receptor. However, prohibitins may have pleiotropic roles in mitophagy, acting simultaneously as a regulator of mitochondrial function and mitochondrial inner membrane binding to the autophagy cargo receptor LC3.
Prohibitin 2 May Play a Conserved Role in Maternal Mitochondrial DNA Inheritance
We found that prohibitin 2 plays an essential role in vivo in eliminating the transmission of paternally derived mitochondrial DNA to offspring in C. elegans. Previous studies in C. elegans and D. melanogaster have shown that mitophagy is crucial for eliminating sperm-derived mitochondria during zygotic development (Al Rawi et al., 2011; Politi et al., 2014; Sato and Sato, 2011; Zhou et al., 2011). In our study, we found similar phenotypes in F1 offspring derived from males with phb2 deletion and females with deletion of the core autophagy gene, atg7—i.e., a delay in paternal mitochondrial clearance and persistence of paternal mtDNA in the first larval stage. These results indicate that phb2 functions in mitophagy in vivo during nematode development.
The precise mechanism by which phb2 mediates mitophagy in paternal mitochondrial DNA clearance remains unknown. Unlike some species in which sperm mitochondria are ubiquitinated, ubiquitination is not believed to play a role in paternal mitochondrial clearance in C. elegans (Levine and Elazar, 2011). In mammalian cells, we did not detect PHB2 ubiquitination during Parkin-mediated mitophagy (data not shown), and we found that purified PHB2 binds LC3 in vitro, suggesting that ubiquitination and other post-translational modifications are not required for the PHB2/LC3 interaction. There may be a yet-to-be-discovered mechanism by which the OMM of paternal mitochondria become ruptured during fertilization, allowing the exposure of PHB2 to the cytoplasm and subsequent binding to the worm orthologs of LC3, LGG-1, and LGG-2, which are required for paternal mitochondrial clearance (Al Rawi et al., 2011; Sato and Sato, 2011).
Although ubiquitination does not appear to be involved in paternal mitochondrial clearance in C. elegans, ubiquitination, prohibitins, and paternal mitochondrial clearance may be linked in other species. In bull and rhesus monkey spermatozoa, prohibitin is ubiquitinated during spermatogenesis and after fertilization, suggesting that it may be involved in the selective degradation of paternal mitochondria (Sutovsky et al., 2000; Thompson et al., 2003). Moreover, similar to ubiquitin, prohibitin has also been implicated in a genetic pathway that controls mitochondrial inheritance in S. cerevisiae (Thompson et al., 2003). PHB2 ubiquitination was also noted in proteomics analyses of mammalian cells undergoing Parkin-dependent mitophagy, although no validation studies were performed to assess whether it is a bona fide Parkin substrate (Bingol et al., 2014; Sarraf et al., 2013). Thus, whether prohibitins play a conserved role in paternal mitochondrial clearance and in other mitophagy contexts, either via ubiquitin-dependent or ubiquitin-independent mechanisms, warrants further investigation.
The Mitophagy Function of Prohibitin 2 May Contribute to Its Roles in Physiology and Aging
PHB2/PHB1 are postulated to function in a broad range of cellular processes, including cell-cycle regulation, nuclear transcriptional regulation, cellular signaling, senescence, apoptosis, and mitochondrial biogenesis (Artal-Sanz and Tavernarakis, 2009; Merkwirth and Langer, 2009; Theiss and Sitaraman, 2011). Prohibitins are essential for embryonic development in C. elegans and mammals as well as lifespan regulation in yeast, plants and C. elegans (Artal-Sanz and Tavernarakis, 2009; Merkwirth and Langer, 2009). Moreover, they may regulate inflammation, obesity, and cancer in mammals (Artal-Sanz and Tavernarakis, 2009; Merkwirth and Langer, 2009; Theiss and Sitaraman, 2011), and forebrain neuron-specific prohibitin deletion in mice results in neurodegenerative disease (Korwitz et al., 2016; Merkwirth et al., 2012). As mitophagy plays an important role in extending lifespan and in protection against inflammation, metabolic diseases, cancer, and neurodegenerative diseases, the mitophagy function of the prohibitin complex may contribute to its protective effects against aging and disease.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 1.4 nm gold-conjugated fragment antigen-binding (Fab) goat antibody to mouse IgG | Nanoprobe | A24921 |
| Goat anti-mouse AlexaFluor 488 | Invitrogen | A11029 |
| Donkey anti-mouse AlexaFluor 594 | Invitrogen | A21203 |
| Goat anti-rabbit AlexaFluor 488 | Invitrogen | A11034 |
| Donkey anti-rabbit AlexaFluor 594 | Invitrogen | A21207 |
| Mouse monoclonal anti-actin-HRP | Santa Cruz | sc-47778-HRP |
| Mouse monoclonal anti-ATP5B | Santa Cruz | sc-166462 |
| Mouse monoclonal anti-ATP5B | Millipore | MAB3494 |
| Mouse monoclonal anti-FLAG M2-HRP | Sigma | A8592 |
| Mouse monoclonal anti-GFP | Abcam | AB1218 |
| Mouse monoclonal anti-Myc | Santa Cruz | sc-56634 |
| Mouse monoclonal anti-NBR1 | Santa Cruz | sc-130380 |
| Mouse monoclonal ant-OPA1 | BD Biosciences | 612606 |
| Mouse monoclonal anti-Parkin | Cell Signaling Technology | 4211 |
| Mouse monoclonal anti-PHB1 | Santa Cruz | sc-377037 |
| Mouse monoclonal anti-PHB2 | Santa Cruz | sc-133094 |
| Mouse monoclonal anti-TIM50 | Santa Cruz | sc-393678 |
| Mouse monoclonal anti-TOMM20 | Santa Cruz | sc-17764 |
| Mouse monoclonal anti-TOMM70 | Santa Cruz | sc-390545 |
| Mouse monoclonal anti-WIPI2 | Abcam | ab105459 |
| Rabbit polyclonal anti-ATG7 | Sigma | A2856 |
| Rabbit polyclonal anti-GABARAP | Novus Biologicals | NBP1-71771 |
| Rabbit polyclonal anti-GATE-16 | Millipore | ABC24 |
| Rabbit polyclonal anti-HA | Santa Cruz | sc-805 |
| Rabbit polyclonal anti-HSP60 | Santa Cruz | sc-13966 |
| Rabbit polyclonal anti-LC3B | Novus Biologicals | NB100-2220 |
| Rabbit polyclonal anti-PHB1 | ThermoFisher | PA5-19556 |
| Rabbit polyclonal anti-PHB2 | ThermoFisher | PA5-14133 |
| Rabbit polyclonal anti-TOMM20 | Santa Cruz | sc-11415 |
| Rabbit polyclonal anti-ULK1 | Novus | NBP2-29922 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Strep-Tactin Sepharose | IBA Lifesciences | 2-1201-004 |
| Anti–FLAG M2 affinity gel | Sigma | A2220 |
| MitoTracker Red (CMXRos) | Invitrogen | M7512 |
| CellMask Deep Red | ThermoFisher | C10046 |
| ProLong Diamond Antifade Mountant with DAPI | Invitrogen | P36971 |
| Oligomycin | Sigma | 75351 |
| Antimycin | Sigma | A8674 |
| Epoxomicin | Sigma | E3652 |
| Lactacystin | Sigma | L6785 |
| Critical Commercial Assays | ||
| QProteome mitochondria isolation kit | QIAGEN | 37612 |
| HQ silver enhancement kit | Nanoprobe | 2012 |
| Experimental Models: Cell Lines | ||
| Phb2flox/flox murine embryonic fibroblasts | Thomas Langer | N/A |
| Phb2+/+ murine embryonic fibroblasts | Thomas Langer | N/A |
| Phb2flox/flox/Parkin MEFs | This paper | N/A |
| HeLa/Control cells | This paper | N/A |
| HeLa/Parkin cells | This paper | N/A |
| HeLa/Parkin/Flag.StrepII-LC3 cells | This paper | N/A |
| HeLa/Parkin/GFP-LC3 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| E. coli: BL21 (DE3) | Invitrogen | C600003 |
| E. coli: OP50 | Caenorhabditis Genetics Center | RRID: WB_OP50 |
| E. coli: HT115 (DE3) | Caenorhabditis Genetics Center | RRID: WB_HT115(DE3) |
| C. elegans: wild-type Bristol strain N2 | Caenorhabditis Genetics Center | N/A |
| C. elegans: LB138: him-8(e1489) IV; uaDf5/+ | Caenorhabditis Genetics Center | RRID: WB_LB138 |
| C. elegans: CB3844: fem-3(e2006) IV | Caenorhabditis Genetics Center | RRID: WB_CB3844 |
| Recombinant DNA | ||
| pIRES-hyg3 | Clontech | 631620 |
| pIRES-hyg3-Parkin | This paper | N/A |
| pLV-Cre | Pfeifer et al., 2001 | Addgene: 38248 |
| pLenti-IRES-Neo-PHB1 | This paper | N/A |
| pLenti-IRES-Puro-PHB2 | This paper | N/A |
| pLenti-IRES-Puro-PHB2 mLIR2 | This paper | N/A |
| psPAX2 | Didier Trono | Addgene: 12260 |
| pMD2.g | Didier Trono | Addgene: 12259 |
| pGEX-4T-1 | GE healthcare | 28-9545-49 |
| ppSUMO-StrepII | This study | N/A |
| pIRES-Neo-Flag.StrepII | This study | N/A |
| L4440 | Andrew Fire | Addgene: 1654 |
| L4440-atg-7 | Julie Ahringer | WormBase: sjj_M7.5 |
| L4440-phb-2 | Julie Ahringer | WormBase: sjj_T24H7.1 |
| Sequence-Based Reagents | ||
| Dicer-substrate siRNA: NC1 negative control | Integrated DNA Technologies | 51-01-14-03 |
| 5′-CGUUAAUCGCGUAUAAUACGCGUAT-3′ | Integrated DNA Technologies | 51-01-14-03 |
| 3′-CAGCAAUUAGCGCAUAUUAUGCGCAUA-5′ | Integrated DNA Technologies | 51-01-14-03 |
| Dicer-substrate siRNA: PHB2 #1 | Integrated DNA Technologies | N/A |
| 5′-GUGAUUUCCUACAGUGUUGUUCCCT-3′; | Integrated DNA Technologies | N/A |
| 3′-GUCACTAAAGGAUGUCACAACAAGGGA-5 | Integrated DNA Technologies | N/A |
| Dicer-substrate siRNA: PHB2 #2 | Integrated DNA Technologies | N/A |
| 5′-AGAUAAACACCAACCCAGGAAUUCT-3′; | Integrated DNA Technologies | N/A |
| 3′-CCUCUAUUUGUGGUUGGGUCCUUAAGA-5′ | Integrated DNA Technologies | N/A |
| Dicer-substrate siRNA: PHB2 #3 | Integrated DNA Technologies | N/A |
| 5′-AGAUUCGAGCAGCCCAGAAUAUCTC-3′; | Integrated DNA Technologies | N/A |
| 3′-GUUCUAAGCUCGUCGGGUCUUAUAGAG-5′ | Integrated DNA Technologies | N/A |
| Primer: U1-F | Liau et al., 2007 | N/A |
| 5′-CCATCCGTGCTAGAAGACAA-3′ | Liau et al., 2007 | N/A |
| Primer: Cemt1A-R | Liau et al., 2007 | N/A |
| 5′-CTTCTACAGTGCATTGACCTAGTC-3′ | Liau et al., 2007 | N/A |
| Primer: Cemt5012-F | Liau et al., 2007 | N/A |
| 5′-TTGGTGTTACAGGGGCAACA-3′ | Liau et al., 2007 | N/A |
| Software and Algorithms | ||
| Imaris version 8.2 | Bitplane | N/A |
| Deltavision softWoRx software | GE healthcare | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact Beth Levine (beth.levine@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Cells
Phb2flox/flox and Phb2+/+ murine embryonic fibroblasts (MEFs) were gifts from Dr. Thomas Langer. Phb2flox/flox/Parkin MEFs and HeLa/Parkin cells were generated by stable transfection of a pIRES-hyg3 vector (Clontech) expressing Parkin cDNA. HeLa/Control cells were generated by stable transfection of pIRES-hyg3 vector into HeLa cells; HeLa/Parkin/Flag.StrepII-LC3 cells were generated by stable transfection of pIRES-neo- Flag.StrepII-LC3 into HeLa/Parkin cells.
Authentication of HeLa/Control, HeLa/Parkin, HeLa/Parkin/Flag.StrepII-LC3, Phb2+/+/Parkin MEF and Phbflox/flox/Parkin MEF cells was performed by the ATCC Cell Line Authentication Service.
C. elegans Genetics
The following strains and alleles were used in this study: wild-type Bristol strain N2, CB3844: fem-3(e2006) IV, LB138: him-8(e1489) IV; uaDf5/+. Animals were grown on NGM plates seeded with E.coli OP50 at 20°C unless stated otherwise. CB3844 fem-3(e2006) IV was maintained at 15°C. RNAi experiments were performed by the feeding method. Eggs or day 1 adults were placed on NGM plates containing 1 mM IPTG (isopropyl β-D-thiogalactopyranoside), 100 μg/ml carbenicillin and E. coli HT115(DE3) carrying L4440-based dsRNA-expressing constructs (obtained from RNAi library constructed by Julie Ahringer, University of Cambridge, UK) or empty vector (L4440) as a negative control.
METHOD DETAILS
Chemical Handling
Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (10 μM) oligomycin (2.5 μM) together with antimycin (250 nM) (OA), lactacystin (5 μM) and epoxomicin (100 nM) were used for all experiments. All chemicals were resuspended in DMSO and stored in small aliquots at −80°C.
Lentiviruses Generation and Transduction
The expression vectors pLV-Cre (Pfeifer et al., 2001) (Addgene #38248), pLenti-IRES-Neo-PHB1, pLenti-IRES-Puro-PHB2, and pLenti-IRES-Puro-PHB2 with mutated LIR domains were cotransfected with helper plasmids psPAX2 (Addgene #12260) and pMD2.g (Addgene #12259) into HEK293T cells. Lentiviral supernatant was filtered through a 0.45 μm membrane and then added to target cells in the presence of polybrene (8 μg/mL). After 3 hr, the virus-containing medium was removed and replaced with fresh medium. Forty-eight hr later, cells were cultured in selection medium containing 1 μg/ml puromycin and/or 500 μg/ml G418. After selection was completed, cells were maintained in 1 μg/ml puromycin and/or 100 μg/ml G418.
Dicer-Substrate siRNA Treatment
All siRNA experiments were performed using reverse transfection at a final concentration of 10 nM dicer-substrate siRNA using RNAiMAX (Invitrogen) according to the manufacturer’s instructions. At 48–72 hr after siRNA transfection, protein knockdown was assessed by western blot analysis.
LC3 Complex Tandem Affinity Purification
109 HeLa/Parkin/Flag.StrepII-LC3 cells were treated with DMSO or 10 μM CCCP for 4 hr. The cells were harvested in lysis buffer containing 25 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1× cOMPLETE protease inhibitor cocktail [Roche], and 1× HALT phosphatase inhibitor cocktail [Thermo Fisher]). The LC3 complex was purified sequentially with Strep-Tactin Superflow resin (IBA GmbH, cat. no. 2-1206-10) and anti–FLAG M2 agarose (Sigma-Aldrich) as described (Gloeckner et al., 2009). The purified LC3 complex was separated on 4%–20% SDS-PAGE denaturing gels and detected by silver staining.
Bacterial Protein Expression, Purification, and In vitro Binding Assays
LC3 (encoding amino acids 1-120), PHB1, PHB2 and PHB2 mutants, Δ77-80, Y77K, Y121A/L124A, and F175A/I178A were cloned into pGEX-4T-1 (GE Healthcare #28-9545-49) and expressed as glutathione-s-transferase (GST) fusion proteins or cloned into ppSUMO-StrepII and expressed as StrepII-SUMO fusion proteins in E. coli BL21 (DE3). The expressed GST-fusion proteins were purified by immobilized glutathione (Thermofisher #15160); StrepII-SUMO-fusion proteins were purified by Strep-Tactin-Sepharose (IBA-lifescience #2-1201-004). The purified proteins were dialyzed with a slide-A-lyzer Dialysis Kit (Thermofisher #66372).
For GST pull-down assays (Figures 1C–1E and 5D), HeLa/Control or HeLa/Parkin cells treated with DMSO or OA were collected in Buffer A, and then 10% sodium deoxycholate was added to reach a final concentration of 0.3% and samples were incubated on ice for 30 min. Lysates were centrifuged at 20,000 rcf for 10 min and the supernatants were further diluted with 2× volume of Buffer A. The diluted lysates were pre-cleared with immobilized glutathione (Thermofisher #15160) and then incubated with GST-fusion protein-loaded beads for 2 hr. Beads were washed 5 times with wash buffer (25 mM HEPES-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% Tween 20) and then boiled in Laemmli buffer + β-mercaptoethanol for 5 min.
For in vitro binding assays (Figures 1D, 5B, and 5C), purified GST-LC3 (aa 1-120) and StrepII-SUMO-PHB proteins were incubated with immobilized glutathione (Thermofisher #15160) for 2 hr, washed 5 times with wash buffer and then boiled in Laemmli buffer + β-mercaptoethanol for 5 min.
Co-Immunoprecipitations and Western Blot Analyses
For PHB2/LC3 co-immunoprecipitation, HeLa/Control or HeLa/Parkin cells treated with DMSO or OA were collected in Buffer A (25 mM HEPES-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1× cOMPLETE protease inhibitor cocktail [Roche] and 1×HALT phosphatase inhibitor cocktail [Thermo Fisher]) and 10% sodium deoxycholate was added to reach a final concentration of 0.3%. Cell lysates were incubated on ice for 30 min and centrifuged at 20,000 rcf for 10 min and the supernatants were further diluted with 2× volume of Buffer A. The diluted lysates were pre-cleared with protein G beads, and then incubated with a mouse anti-PHB1 (sc-377037, 1:100 dilution) or mouse anti-PHB2 (sc-133094, 1:100 dilution) antibody and protein G beads overnight at 4°C. Beads were washed 3 times with wash buffer (25 mM HEPES-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% Tween 20) and then boiled in Laemmli buffer + β-mercaptoethanol for 5 min.
To evaluate the ubiquitination of TOMM20 and TOMM70 (Figure S3A), HeLa/Parkin cells were co-transfected with HA-ubiquitin and control or PHB2 siRNA for 48 hr prior to treatment for 8 hr with DMSO or OA with or without epoxomicin. Cells were lysed in lysis buffer (25mM HEPES-HCL (PH7.4), 150mM NaCl, 1mM EDTA, 1% Triton X-100, 1× cOMPLETE protease inhibitor cocktail [Roche] and 1× HALT phosphatase inhibitor cocktail [Thermo Fisher]). The ubiquitinated proteins were immunoprecipitated with a polyclonal anti-HA probe antibody (Santa Cruz sc-805, 1:200 dilution).
For protease protection assays (Figure 3A), mitochondrial fractions were purified using QProteome mitochondria isolation kit (QIAGEN #37612) following the manufacturer’s instructions. For trypsin digestion, 100 μg of purified mitochondria were treated with 200 mg/ml trypsin in 200 μL of digestion buffer (10 μM sucrose, 0.1 mM EGTA/Tris and 10 mM Tris/HCl, pH 7.4) for 20 min at 4°C. The reaction was stopped by boiling in Laemmli buffer + β-mercaptoethanol for 5 min.
To assess OPA1 processing (Figure 6F), Parkin and Cre-expressing Phb2flox/flox MEFs in normal growth condition were reconstituted with wild-type PHB1 and either wild-type PHB2 or a PHB2 LIR domain (Y121A/L124A) mutant. Whole cell lysates were prepared and samples were analyzed by western blot with anti-OPA1 antibody.
For western blot analyses, cells were lysed with Laemmli buffer containing 2.5% β-mercaptoethanol, boiled for 5 min, separated on SDS-PAGE denaturing gels (Bio-Rad) and transferred to PVDF membranes (Bio-Rad). Membranes were blocked with 5% dry milk and signals were visualized with Supersignal West Pico or Femto Chemiluminescent Substrate kit (Pierce). Loading was normalized by densitometry of actin signal using ImageJ. For subcellular fractionation experiments, cells were fractionated using the QProteome mitochondria isolation kit (QIAGEN) according to the manufacturer’s instructions.
The following antibodies were used for western blot analyses: HRP-conjugated anti-actin (Santa Cruz sc-47778-HRP, 1:2000 dilution), mouse monoclonal anti-Myc (Santa Cruz sc-56634, 1:200 dilusion), rabbit anti-ATG7 (Sigma A2856, 1:500 dilution), mouse monoclonal anti-ATP5B (Santa Cruz sc-166462, 1:1000 dilution), mouse anti-TIM50 (Santa Cruz sc-393678, 1:200 dilution), rabbit anti-PHB1 (ThermoFisher PA5-19556, 1:500 dilution), rabbit anti-PHB2 (ThermoFisher PA5-14133, 1:500 dilution), HRP-conjugated anti-Flag (Sigma A8592, 1:2000 dilution), rabbit anti-LC3 (Novus NB100-2220, 1:2000 dilution), rabbit anti-HSP60 (Santa Cruz sc-13966, 1:1000 dilution), mouse monoclonal anti-Parkin (Cell Signaling Technology mAb4211, 1:1000 dilution), rabbit anti-TOMM20 (Santa Cruz sc-11415, 1:1000 dilution), rabbit anti-GABARAP (Novus NBP1-71771, 1:100 dilution), rabbit anti-GATE-16 (Millipore ABC24, 1:100 dilution), monoclonal anti-TOMM70 (Santa Cruz sc-390545, 1:200 dilution), monoclonal anti-TOMM20 antibody (Santa Cruz sc-17764, 1:500 dilution), and OPA1 antibody (BD Biosciences 612606, 1:500 dilution).
Light and Electron Microscopy
For immunofluorescence analysis of cultured cells, cells were fixed with 2% paraformaldehyde followed by 20 min methanol at −20°C. Slides were incubated for 1 hr with primary antibody and 30 min with secondary antibody and mounted with Prolong Diamond (Invitrogen). Primary antibodies were mouse monoclonal anti-ATP5B (Millipore MAB3494, 1:1000 dilution), rabbit anti-HSP60 (Santa Cruz sc-13966), rabbit anti-TOMM20 (Santa Cruz sc-11415, 1:1000 dilution), mouse monoclonal anti-Myc (Santa Cruz sc-56634, 1:200 dilution), anti-GFP (Abcam AB1218 1:500 dilution), mouse anti-NBR1 (Santa Cruz sc-130380 1:100 dilution), mouse anti-WIPI2 (Abcam ab105459 1:150), rabbit anti-ULK1 (Novus NBP2-29922 1:500 dilution) and rabbit anti-HA (Santa Cruz sc-805, 1:500 dilution). Secondary antibodies were highly cross-adsorbed goat anti-mouse AlexaFluor 488, goat anti-rabbit AlexaFluor 488, donkey anti-rabbit AlexaFluor 594, and/or donkey anti-mouse AlexaFluor 594 secondary antibodies (Invitrogen, 1:1000 dilution) with the addition of CellMask Deep Red Plasma membrane Stain (ThemoFisher #c10046, 1:1000 dilution) for experiments involving cell segmentation. Images were acquired with a Zeiss AxioImager Z2 microscope equipped with a Photometrics CoolSnap HQ2 camera using a Zeiss PLAN APOCHROMAT 20X/0.8 NA air objective using the same acquisition times for samples stained with identical primary antibody pairs. For automated image analysis, Z stacks were acquired using the same Zeiss AxioImager Z2 imaging system and deconvolved with AutoDeBlur (Bitplane). Analysis was performed using the Cell module in Imaris version 8.2 (Bitplane).
For quantitative image analyses of mitophagy, a segmentation channel was created using the Channel Arithmetic function in Imaris XT = ((5 × CellMask signal) + DAPI signal). Cell segmentation was then performed with this new channel using DAPI-positive, ~15 μm seeds. Additional filtering was applied to exclude all cells touching the edge of the image. ATP5B puncta were modeled as ~0.75 μm “vesicles.” The same creation parameters for cell segmentation and mitochondrial “vesicle” detection were then applied to all images within a given experiment using Imaris Batch. Data for numbers of cytoplasmic ATP5B “vesicles” for all cells in a given experiment were plotted in Imaris Vantage and then exported for further analysis. Thresholding for background secondary antibody staining was performed in all experiments. In some images, nonspecific nuclear staining was masked in Imaris by creating a nuclear surface using the DAPI channel and setting non-DAPI signal within the nuclear surface to zero.
For structured illumination microscopy (SIM) of mitochondria, HeLa/Parkin/GFP-LC3 cells grown on #1.5 coverslips in 6-well plates were fixed with 2% paraformaldehyde followed by 20 min methanol at −20°C. The cells were sequentially co-stained with primary antibodies (monoclonal anti-TOMM20 antibody (Santa Cruz sc-17764, 1:1000 dilution) and rabbit anti-PHB2 (ThermoFisher PA5-14133, 1:500 dilution), or rabbit anti-TOMM20 (Santa Cruz sc-11415, 1:1000 dilution) and mouse anti-TIM50 (Santa Cruz sc-393678, 1:200 dilution)) and corresponding fluorescent secondary antibodies. After washing, the coverslips were inverted onto ~50 μL Prolong Diamond mounting medium (Invitrogen) on glass slides. 3D SIM images were acquired with a Deltavision OMX SR microscope (GE Healthcare) equipped with an Olympus PLAPON 60X/1.42 NA objective and channel-dedicated pco.edge sCMOS cameras (PCO). SIM reconstructions and 3D projections were performed using Deltavision softWoRx software (GE Healthcare).
To detect GFP-LC3 labeling at the ultrastructural level by immune electron microscopy, HeLa/Parkin/GFP-LC3 cells were fixed for 15 min at room temperature with 4% paraformaldehyde (Electron Microscopy Sciences) and 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Cells were permeabilized with 0.25% saponin in PBS for 30 min and blocked with 5% goat serum in 0.01% saponin in PBS for 1 hr. Cells were then incubated with monoclonal anti-GFP (Abcam AB1218, 1:100 dilution) for 2 hr at room temperature, washed 5 times with PBS, followed by overnight incubation with 1.4 nm gold-conjugated fragment antigen-binding (Fab) goat antibodies to mouse IgG (Nanoprobes A24921, 1:100) at 4°C. After washing 5 times with PBS, cells were further fixed with 1% glutaraldehyde and washed 3 times with PBS. After washing with water, the immunogold-labeled samples were silver-enhanced for 2.5 min using the HQ silver enhancement kit (Nanoprobes) and washed again with water, followed by 0.2% OsO4 for 30 min at 4°C and en bloc staining with 1% uranyl acetate. Samples were dehydrated in graded ethanol and embedded in Epon. Thin sections were stained with uranyl acetate and lead citrate before imaging with a Tecnai Spirit Biotwin (FEI) electron microscope.
To examine the effect of a PHB2 LIR domain mutant on mitochondrial morphology in Phb2-deficient MEFs in normal growth conditions, Phb2flox/flox MEFs expressing Parkin, PHB1, and either wild-type PHB2 or the PHB LIR mutant (Y121A/L124A) were fixed and analyzed by electron microscopy. To quantify cristae morphogenesis, mitochondria were classified as either having cristae structure that is lamellar (i.e., intact) or disorganized as described (Merkwirth et al., 2008).
For TEM analyses of immunoprecipitated mitochondria, immunoprecipitation of whole mitochondria from HeLa/Parkin cells and preparation of Dynabeads for electron microscopy was performed as previously described (Sumpter et al., 2016).
In situ Proximity Ligation Assay
HeLa/Parkin/Flag-StrepII-LC3 cells were fixed with 2% paraformaldehyde followed by 20 min methanol at −20°C. The Duolink in situ proximity ligation assay was carried out according to the manufacturer’s instructions (Olink Biosciences, Sigma-Aldrich) using the following primary antibodies, mouse monoclonal anti-Flag-M2 (Sigma F3165, 1:1500 dilution) and polyclonal rabbit anti-PHB2 (ThermoFisher PA5-14133, 1:500 dilution). Images were captured with a Zeiss Axio Imager Z2 microscope.
Analysis of Paternal Mitochondrial Clearance in C. elegans
The protocols for assessing degradation of paternal mitochondria in nematodes were modified from previous studies (Al Rawi et al., 2011; Sato and Sato, 2011; Zhou et al., 2011). To avoid the deleterious effect of phb-2 knockdown during development, phb-2 RNAi was performed during adulthood. Day 1 N2 males were placed on vector control (L4440) or phb-2 RNAi plates and grown at 20°C for 36 hr. To fluorescently label paternal mitochondria, 100 μL of 200 μM MitoTracker Red (CMXRos), a red-fluorescent dye that accumulates in mitochondria dependent upon membrane potential, was added directly to the cultured nematodes and incubated in the dark at 20°C for an additional 12 hr. The fluorescently-labeled males were then transferred to fresh NGM plates seeded with OP50 and incubated for 20 min to remove excessive dye. To obtain female worms, eggs from fem-3(e2006) animals were transferred to plates seeded with either control or atg-7 RNAi bacteria and grown at 25°C for two days. Twenty-five fem-3(e2006) young females were mated with 60–80 CMXRos-labeled N2 males at 20°C for 8–12 hr. The embryos were dissected from the gravid females, treated by the freeze-crack method, and fixed by methanol for 5 min. Fixed embryos were mounted with Prolong Diamond Antifade Mountant with DAPI (Invitrogen). Z stacked images (1 μm interval) were captured by Zeiss Axio Imager Z2 system with a Zeiss PLAN APOCHROMAT 63×/1.4 oil DIC objective and a Photometrics CoolSnap HQ2 camera using the same acquisition time for all samples within each experiment. Images were deconvolved using AutoDeBlur (Bitplane) and analyzed in Imaris version 8.2 (Bitplane).
To monitor paternal mtDNA transmission in C. elegans embryos, Day 1 uaDf5/+ males were grown on vector control (L4440) or phb-2 RNAi plates at 20°C for 2 days. Eggs from fem-3(e2006) animals were transferred to control or atg-7 RNAi plates and grown at 25°C for 2 days to obtain females. Twenty-five fem-3(e2006) young females were mated with 60-80 uaDf5/+ males at 20°C for 8–12 hr. After mating, the gravid females were washed with M9 buffer supplemented with 0.01% (v/v) Triton X-100 3 times and transferred to fresh NGM plates seeded with OP50 for a 20 min recovery. Animals were then placed on new NGM plates to allow egg-laying for 1–3 hr before removal. The embryos were incubated at 20°C until hatching (about 13 hr). Genomic DNA was prepared from at least 30 L1 progeny and analyzed by genotyping PCR using primers specific to wild-type or uaDf5 mtDNA (Liau et al., 2007). U1-F and Cemt1A-R primer (see Key Resources Table for sequences) amplify the uaDf5 allele of mtDNA (299 bp) and Cemt5012-F and Cemt1A-R amplify wild-type mtDNA (518 bp) (Tsang and Lemire, 2002).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters and significance are reported in the Figures and the Figure Legends.
For comparisons of the means of two groups, one-way ANOVA was used and Sidak’s test was used for experiments with multiple comparisons. For comparison of the magnitude of changes in different conditions, two-way ANOVA was used. Chi-square analysis was used to determine difference between groups of mitochondria with unruptured or ruptured membranes. Linear-mixed effects models were used to compare the growth rate of cells with different genotypes. The model compares the differences between groups, while accounting for the correlation among samples in a given group at different time periods.
Supplementary Material
Highlights.
PHB2 is an inner mitochondrial membrane mitophagy receptor
The interaction between PHB2 and LC3 requires outer mitochondrial membrane rupture
PHB2 is required for Parkin-mediated mitophagy
PHB2 is required for paternal mitochondrial elimination in C. elegans
In Brief.
Mitophagy hinges on recognition of an inner mitochondrial protein by autophagosomes.
Acknowledgments
We thank T. Langer and A.-L. Hsu for providing critical reagents; L. Nguyen for expert technical assistance; D. Xue and M.Y. Lee for helpful discussions; X. Chen and Z. Chen for assistance with mass spectrometry; K. Luby-Phelps and A. Darehshouri for assistance with SIM imaging and immunogold EM, respectively; and H. Smith for assistance with manuscript preparation. SIM and EM studies were carried out in UT Southwestern Live Cell and Electron Microscopy Facilities. This work was supported by NIH grants, KO8AI099150 (R.S.), U19AI199725 (B.L.), and RO1CA109618 (B.L.), Cancer Prevention Research Institute of Texas (CPRIT) grant RP120718 (B.L.), and a Burroughs Wellcome Career Award for Medical Scientists (R.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.11.042.
AUTHOR CONTRIBUTIONS
Y.W., W.-C.C., P.M., and B.L. designed the study. Y.W., W.-C.C., and R.S. performed experiments. Y.W., W.-C.C., R.S., and B.L. analyzed the data. Y.W., W.-C.C., R.S., P.M., and B.L. wrote the manuscript.
References
- Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Back JW, Sanz MA, De Jong L, De Koning LJ, Nijtmans LG, De Koster CG, Grivell LA, Van Der Spek H, Muijsers AO. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002;11:2471–2478. doi: 10.1110/ps.0212602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;S0891:30033–30038. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner CJ, Boldt K, Ueffing M. Strep/FLAG tandem affinity purification (SF-TAP) to study protein interactions. Curr Protoc Protein Sci. 2009 doi: 10.1002/0471140864.ps1920s57. Chapter 19, Unit19.20. [DOI] [PubMed] [Google Scholar]
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J. Sex determination in the nematode C. elegans: analysis of tra-3 suppressors and characterization of fem genes. Genetics. 1986;114:15–52. doi: 10.1093/genetics/114.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016;26:6–16. doi: 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Korwitz A, Merkwirth C, Richter-Dennerlein R, Tröder SE, Sprenger HG, Quirós PM, López-Otín C, Rugarli EI, Langer T. Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J Cell Biol. 2016;212:157–166. doi: 10.1083/jcb.201507022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Elazar Z. Development. Inheriting maternal mtDNA. Science. 2011;334:1069–1070. doi: 10.1126/science.1215480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau WS, Gonzalez-Serricchio AS, Deshommes C, Chin K, LaMunyon CW. A persistent mitochondrial deletion reduces fitness and sperm performance in heteroplasmic populations of C. elegans. BMC Genet. 2007;8:8. doi: 10.1186/1471-2156-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32:2336–2347. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Löwer B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Martinelli P, Korwitz A, Morbin M, Brönneke HS, Jordan SD, Rugarli EI, Langer T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012;8:e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial PHB complex: roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Brandon EP, Koostra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: Efficient gene targeting in vivo. Proc Natl Acad Scie USA. 2001;98:11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E. Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev Cell. 2014;29:305–320. doi: 10.1016/j.devcel.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. 2014;53:127–133. doi: 10.1016/j.biocel.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. doi: 10.1016/j.cell.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani ST, Yoshimori T. Autophagosome formation in response to intracellular bacterial invasion. Cell Microbiol. 2014;16:1619–1626. doi: 10.1111/cmi.12357. [DOI] [PubMed] [Google Scholar]
- Sumpter R, Jr, Sirasanagandla S, Fernández AF, Wei Y, Dong X, Franco L, Zou Z, Marchal C, Lee MY, Clapp DW, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell. 2016;165:867–881. doi: 10.1016/j.cell.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol Reprod. 2000;63:582–590. doi: 10.1095/biolreprod63.2.582. [DOI] [PubMed] [Google Scholar]
- Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta. 2011;1813:1137–1143. doi: 10.1016/j.bbamcr.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WE, Ramalho-Santos J, Sutovsky P. Ubiquitination of prohibitin in mammalian sperm mitochondria: possible roles in the regulation of mitochondrial inheritance and sperm quality control. Biol Reprod. 2003;69:254–260. doi: 10.1095/biolreprod.102.010975. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD. Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem Cell Biol. 2002;80:645–654. doi: 10.1139/o02-135. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Chen L, Liang Q, Yin XM, Miao L, Kang BH, Xue D. Kinetics and specificity of paternal mitochondrial elimination in Caenorhabditis elegans. Nat Commun. 2016;7:12569. doi: 10.1038/ncomms12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi O, Murakawa T, Nishida K, Otsu K. Receptor-mediated mitophagy. J Mol Cell Cardiol. 2016;95:50–56. doi: 10.1016/j.yjmcc.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Xue D. Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans. Cell Res. 2011;21:1662–1669. doi: 10.1038/cr.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Li H, Nakagawa A, Lin JL, Lee ES, Harry BL, Skeen-Gaar RR, Suehiro Y, William D, et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science. 2016;353:394–399. doi: 10.1126/science.aaf4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.