

Abstract
Deer mice in the genus Peromyscus occupy nearly every terrestrial habitat in North America, and have a long history as subjects of behavioral, ecological, evolutionary, and physiological study. Recent advances in transcriptomics, the study of the complete set of RNA transcripts produced by certain cell types or under certain conditions, have contributed to the development of Peromyscus as a model system. We review the recent use of transcriptomics to investigate how natural selection and gene expression plasticity contribute to the existence of deer mice in challenging environments such as highlands, deserts, and cities across North America. Transcriptomics also holds great promise for elucidating the genetic basis of mating systems and other behaviors in Peromyscus, but has to date been underutilized for developmental biology and disease studies. Future Peromyscus studies should apply robust comparative frameworks to analyze the transcriptomics of multiple populations of the same species across varying environmental conditions, as well as multiple species that vary in traits of interest.
Keywords: deer mice, ecological genomics, gene expression, Peromyscus, RNA-Seq, transcriptome
1. Introduction
The ability to identify the specific genes underlying phenotypic variation has long been the holy grail of studies investigating adaptive evolution [1,2]. While phenotypes are the grist through which evolution occurs via natural selection, variation in nucleotide sequence or expression levels of the relevant genes provides an alternative, and often more sensitive, way of detecting a response to environmental conditions [3]. This is particularly true in non-model species that may not have high-quality reference genomes and may be difficult to raise in laboratory settings to F1 or F2 generations [4,5].
With the recent proliferation of high-throughput sequencing, the ability to sequence transcribed RNA (i.e. transcriptomes) has made it feasible for scientists to measure the relative expression levels of different coding regions of the genome. Such approaches are particularly useful when studying species that do not have sequenced genomes or annotated transcriptomes where gene function is well characterized [6]. RNA sequencing (RNA-Seq) has been used effectively to identify gene expression differences between individuals from different natural environments and individuals exposed to different experimental conditions (Figure 1). RNA-Seq is now widely recognized as a powerful tool allowing researchers to expand transcriptomics beyond clinical settings and a handful of model species [3].
Figure 1.
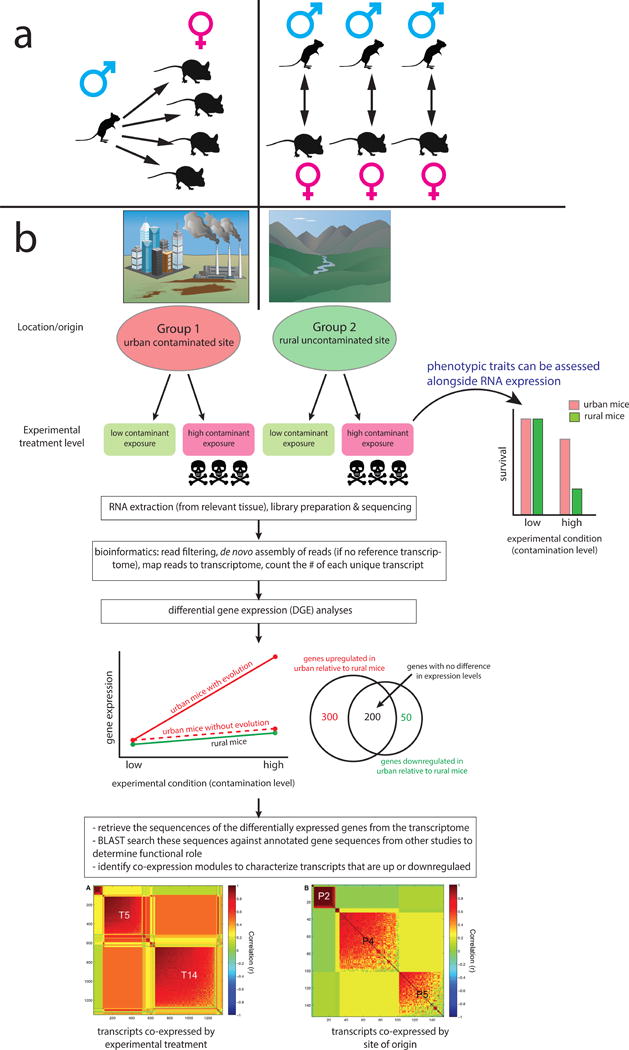
Schematic illustration of the potential for transcriptomic research in Peromyscus. Panel A demonstrates how transcriptomics could used to identify gene expression differences between different species of Peromyscus with distinct mating behavior and parental care (e.g. P. californicus and P. maniculatus). Panel B illustrates the utility of an experimental transcriptomics approach in identifying the genes under selection in divergent habitats. In this example, individuals of the same species are obtained from contrasting habitats (urban vs. rural areas) with divergent natural selection regimes (contaminated vs. uncontaminated soils). Common-garden experiments are performed on individuals acclimated to the common environment, or their F1 offspring. These experiments manipulate environmental variables hypothesized to be agents of natural selection in their natural habitat. Experimental animals are then evaluated for phenotype, tissue is preserved from the tissue where gene expression is hypothesized to be under selection, and RNA is extracted. After sequencing and bioinformatics is performed, differential gene expression (DGE) analyses are performed. The resulting patterns of gene expression can highlight the effects of both experimental treatment and evolutionary history of the population of origin. Finally, the sequences of the up- or down-regulated genes are searched against a database of gene sequences with annotated functions to identify the likely role of differentially expressed genes. Co-expression modules can also be characterized to look for suites of genes with common functions and shared regulatory pathways. Peromyscus vector image from [7]; co-expression module images from [11].
Peromyscus (AKA deer mice) are quickly becoming models for studies on evolutionary ecology, physiology and behavior [7]. Peromyscus have been the focus of ecological, evolutionary and epidemiological research for more than a century, and there is now abundant information on the natural history of this taxon [8]. In addition, several Peromyscus species have continental-wide ranges across North and Central America. These species occur across a wide range of habitat types, and a correspondingly diverse array of natural selection regimes that would promote adaptive divergence between populations experiencing different environmental conditions. In addition to their wild counterparts, deer mice have also been amenable to laboratory rearing and experimentation. As a result, abundant information exists on the basic biology of lab-raised Peromyscus [9,10].
Below we detail the important role that RNA sequencing and transcriptomics have played in developing Peromyscus as a new model system in biology. We highlight seminal studies on the genes underpinning Peromyscus adaptation, including adaptation to high-elevation hypoxia [11], osmotic balance in deserts [12], and tolerance to urbanization [13]. We also detail the design of experimental transcriptomic studies that can be used to evaluate the genes underpinning divergent phenotypes and evolution in Peromyscus.
2. Adaptive evolution and gene expression in challenging environments
One or more species of deer mice are present in nearly every habitat in North America, and Peromyscus is the most speciose genus of North American mammals with 56 currently recognized species [7]. Given their radiation across an impressive range of environments, deer mice have recently become one of the preferred mammalian systems for understanding adaptive responses of wild populations to environmental challenges. Adaptive evolution occurs when advantageous traits increase in population frequency from one generation to the next. The most widespread species, P. maniculatus and to a lesser extent, P. leucopus, occur nearly continent-wide, and thus are particularly useful for intraspecific studies of adaptive evolution and gene expression between populations that occupy highly contrasting environments [13,14]. Habitat specialists, such as P. eremicus and other species that are limited to arid regions, are excellent candidates for both intra- and interspecific studies of gene expression differences between habitats [12].
Transcriptomics provides a two-fold value for studies of deer mice in challenging environments because a single, well-designed RNA-Seq experiment can discover 1) both single nucleotide polymorphisms (SNPs) within protein-coding genes that are functionally connected to adaptive phenotypic changes, and 2) changes in gene expression that are associated with physiological or behavioral responses to challenging environments. Variation in gene expression in wild populations will likely be high due to variable environmental conditions and plasticity, and thus large sample sizes will be necessary to categorize the variation of interest. Guidelines for designing robust RNA-Seq experiments in natural habitats are emerging [3]. Once identified in the field, these SNPs or gene expression changes can be functionally validated using captive Peromyscus populations that are subjected to a common environment and experimental treatments related to hypotheses about selection pressures in the wild (Figure 1). Below we review studies on adaptive evolution and gene expression in Peromyscus that occupy distinct habitats with strong natural selection differentials: high elevation, light-colored substrates, deserts, and cities.
2.1 High altitude: hypoxia and cold
P. maniculatus is the most widespread deer mouse species, including montane habitats at > 4,300 m above sea level in the Rocky Mountains [7]. Mammals at such elevations must deal with substantially less oxygen available for respiration, as well as extreme cold during winter months when non-hibernatory species such as deer mice are still active. Directional selection for adaptive phenotypes or transcriptional plasticity are possible explanations for the enhanced respiration and metabolic heat production that would be necessary for mammalian existence under hypoxia. Several studies have now confirmed that both of these phenomena contribute to the existence of deer mice at such high altitudes.
It has been known for decades that P. maniculatus exhibit diverse hemoglobin phenotypes due to multiple, polymorphic hemoglobin loci that resulted from a complex history of gene duplications. Furthermore, some of these genotypes are associated with high-altitude populations and relevant physiological traits [15,16]. In particular, one highland haplotype is associated with higher blood oxygen affinity and oxygen consumption when mice are exposed to cold or aerobic activity [17]. Intense directional selection on the maximal rate of oxygen consumption was documented for wild, highland P. maniculatus, thus confirming the adaptive value of the highland haplotype [18]. Storz et al. [19] subsequently identified five specific amino acid substitutions in two closely linked, α-globin gene duplicates that underlie adaptive increase in oxygen-binding affinity. Two β-globin genes were also later identified as targets of divergent selection between low- and high-elevation populations [20]. Having identified the relevant sets of paralogs, Storz et al. [21] then characterized the isoform diversity produced by allelic diversity between two altitudinal zones, and recently reported that substantial epistasis between amino acid mutations contributes to increased blood oxygen affinity [22]. These mutations also occur in P. maniculatus populations from lower elevations and with other closely-related species, suggesting that variation in blood oxygen affinity is influenced by many genes of small effect [23]. Presence of adaptive alleles of small effect in lowland populations also suggests that adaptation to high altitude proceeded from standing variation in P. maniculatus.
The functional relationship between hemoglobin mutations and oxygen-binding affinity in hypoxic environments is one of the best examples of an adaptive phenotype linked to genotype in Peromyscus, or perhaps any free-living mammal. However, other candidate loci and plasticity in gene expression are likely to play roles in adaptive physiological responses to high-altitude environments. Several transcriptomic studies have now employed experimental approaches to examine regulatory plasticity in P. maniculatus. Cheviron et al. [14] used RNA-Seq to identify gene expression differences between highland and lowland P. maniculatus, as well as interspecific differences between P. maniculatus and lowland P. leucopus. Genes involved in metabolic pathways were significantly overrepresented compared to the total set of genes that were differentially expressed between P. maniculatus populations, as well as between P. maniculatus and P. leucopus. In particular, dozens of genes in the fatty acid β-oxidation and oxidative phosphorylation (OXPHOS) pathways were differentially expressed between highland P. maniculatus and lowland P. maniculatus/leucopus. More than 75% of these genes were upregulated in highland P. maniculatus compared to the other two groups, indicating that enhanced expression in these pathways is associated with adaptive high-altitude phenotypes in deer mice. Cheviron et al. [14] further found that genes involved in glycolysis and carbohydrate metabolism were not overrepresented among genes exhibiting differential expression. They interpreted these patterns as evidence that highland deer mice rely on lipid metabolism to generate heat through shivering. These findings are in contrast to hypotheses for humans that stress the important role of carbohydrate metabolism in acclimation to high altitude [24], indicating that highland deer mice have evolved at least partially along a different physiological trajectory.
To examine the role of transcriptomic plasticity in thermogenesis at high altitude, Cheviron et al. [11] followed up on the work above with a large-scale RNA-Seq experiment examining expression differences between low- (430 m) and high-altitude (4,350 m) deer mice within 1–2 days of capture in their native habitat, after 6 weeks in common-garden conditions (360 m), and on F1 progeny reared in common-garden conditions. In common-garden experiments, organisms are transplanted from their native environment to an experimental environment with the same conditions for all groups. These experiments allow researchers to identify differences between groups that are due to heritable rather than environmental factors. Cheviron et al. identified 1,353 genes that exhibited expression differences due to experimental treatment, and found that these genes existed in five transcriptional modules (i.e., transcripts co-expressed together with related functions) related to protein metabolism, regulation of skeletal muscle cell differentiation, regulation of erythrocyte differentiation, water transport, carbohydrate and lipid metabolism, muscle and blood vessel development, brown fat cell differentiation, and other more general regulatory categories. Comparing highland and lowland mice revealed several transcriptional modules associated with thermogenic capacity and endurance in the experimental common garden and F1 mice. As reported previously [14], genes in lipid oxidation and OXPHOS pathways were upregulated in highland mice, but glycolysis and carbohydrate metabolism were not [11]. These results indicate that plasticity in gene expression plays an important role in adaptive phenotypic differences between highland and lowland deer mice, and that flexible regulation of lipid metabolism is a significant functional component of the shivering thermogenesis phenotype in high altitude populations.
Along with thermogenic capacity and oxidation pathways, Scott et al. [25] hypothesized that highland mice exhibit an adaptive skeletal muscle phenotype characterized by greater capillary density and oxidative capacity. To test this prediction, they measured muscle fiber type and surface capillary density of muscle from highland and lowland deer mice, as well as from laboratory F1 progeny as above. They then used RNA-Seq to identify gene expression associated with muscle phenotypes, and add to the broader sets of genes functionally related to adaptation to hypoxia. As predicted, skeletal muscle in highland deer mice had greater capillary density and a more oxidative phenotype. The RNA-Seq analysis identified two transcriptional modules of 68 genes that differed in expression levels between highland and lowland populations, and five modules with 589 genes that differed between wild and lab-reared F1 mice. Population differences in expression were associated with muscle phenotypes and pathways that influence metabolism, muscle fiber composition, and vascularization [25]. Increase in capillary density and oxidative capacity of muscle may enhance oxygen retention and thermogenesis through shivering as well as mitochondrial respiration of muscle. These differences between groups likely resulted from both divergent natural selection and plasticity as previously reported for highland deer mice [11,14].
The same experimental design used to examine muscle phenotypes was employed by Velotta et al. [26] to examine the role of brown adipose tissue in non-shivering thermogenesis in deer mice. Heat is generated by brown adipose tissue through well-characterized pathways involving the uncoupling of oxidative phosphorylation from ATP synthesis. Nonshivering thermogenesis was estimated in deer mice by measuring oxygen consumption after stimulation of brown adipose tissue by injection of an agonist of β3-adrenergic receptors. As with the skeletal muscle/shivering phenotypes, highland deer mice exhibited increased nonshivering thermogenesis. RNA-Seq identified transcriptional differences in several co-regulated genes influencing traits such as brown adipose proliferation, oxygen supply to brown adipose tissue, and the heat generation pathway in brown adipose tissue [26]. Adaptive divergence in hemoglobin genes and oxygen binding affinity, and both adaptive divergence and gene expression plasticity in shivering and non-shivering thermogenesis, all explain how deer mouse populations exist in hypoxic conditions over 4,000 m above sea level.
2.2 Deserts, renal physiology, and water stress
A number of Peromyscus species (e.g. P. eremicus, P. crinitus) occupy the deserts of the SW United States and northern Mexico, which are among the harshest environments on Earth. These desert ecosystems are characterized by the highest daytime maximum temperatures in North America, minimal precipitation, and almost no standing water sources, thus posing a severe challenge to maintaining water balance in desert-dwelling organisms. Deserts do not exhibit high mammalian biodiversity, but rodents are relatively common in deserts and thus have likely evolved many adaptations to water stress. Recently, full transcriptomes of the kidney were generated for three non-Peromyscine rodents that inhabit deserts [27,28]. Adaptive divergence and differential expression in genes related to osmoregulation were identified in two of these species [27]. Desert Peromyscus offer several advantages over these other taxa because they are relatively easy to keep in captivity, other closely-related non-desert species are available for interspecific comparisons (e.g. P. californicus), and genomic resources for Peromyscus are under active development to serve the community studying this emerging model group [7].
Recently, a research program to develop P. eremicus as a model system for examining desert adaptations was initiated by characterizing the transcriptome of multiple tissue types (kidney, liver, brain, testes) for one individual using RNA-Seq [12]. A subsequent study added three male reproductive transcriptomes (epididymis, testes, and vas deferens) to transcriptome resources for this species [29]. Nearly four times more unique transcripts were identified in the kidney transcriptome compared to the next highest tissue type (brain), although it is hard to rule out that more rare transcripts were detected by chance in the kidney. Male reproductive transcriptomes were not compared to non-reproductive tissues, but the testes transcriptome exhibited a greater number of unique transcripts than epididymis or vas deferens. Peromyscus genomic resources still lag far behind other model rodents, but these transcriptomes were successfully annotated using Mus musculus resources and the P. maniculatus reference transcriptome. All of the P. eremicus resources generated in these studies are freely available to the Peromyscus research community (Table 1).
Table 1.
Peromyscus transcriptome datasets generated using high-throughput sequencing and currently available on GenBank. na = not applicable
| Species | GenBank Accession | Cell types | Experimental Treatment | Reference(s) |
|---|---|---|---|---|
| P. eremicus | PRJNA242486 | brain, liver, kidney, testes | na | 12 |
| PRJEB13364 | Epididymis, testes, vas deferens | na | 29 | |
| P. leucopus | PRJNA192723 | brain, liver, ovary, testes | urban vs. rural | 13, 42, 48 |
| PRJNA184055 | brain, liver, kidney | na | 82 | |
| P. maniculatus | PRJNA240902 | liver | na | unpub. |
| PRJNA217920 | skeletal muscle | highland vs. lowland, common garden, F1 hybrids | 11, 25 | |
| SRP073101 | Brown adipose tissue | highland vs. lowland, common garden | 26 |
To further explore potential desert adaptations, Macmanes & Eisen [12] generated kidney RNA-Seq data for an additional 15 P. eremicus individuals from Palm Desert, California. The top ten values of Tajima’s D calculated for each transcript in this dataset were for genes related to water balance that may be experiencing balancing selection, although the influence of historical demography on these values has not yet been examined. Branch site tests and Tajima’s D for six candidate genes identified in desert heteromyid rodents by Marra et al. [27] revealed one significant transcript in P. eremicus: Slc2a9, a gene involved in solute balance. The other five genes, mostly in the aquaporin gene families, exhibited Tajima’s D values consistent with selection.
As with high-altitude P. maniculatus above, RNA-Seq comparisons of wild-caught P. eremicus from the extreme portions of its range (i.e. most arid and most humid locations) are feasible in this study system. Experimental comparisons of lab-reared P. eremicus and other Peromyscus exposed to simulated desert conditions in the lab will also be productive research areas moving forward. Given the preliminary work at generating high-quality reference transcriptomes, it is likely that such experiments would very quickly improve our knowledge of desert adaptation in Peromsycus and develop one or more species as powerful models for functional genomic studies of osmoregulation in mammals. To this end, Kordonowy et al. [30] recently developed an environmental chamber that simulates desert conditions for P. eremicus. Preliminary experiments established that P. eremicus drink very little water (0.11 mL/day when provided ad libitum). Experimental dehydration in the chamber revealed that dehydrated mice lose a substantial percentage of their body mass (nearly 25% in three days), and their serum electrolyte profile changes. Despite these effects, there were no obvious ill effects of water deprivation.
2.3 Cities: diet, pollution, immunology
Given the broad geographic range and habitat tolerance of P. maniculatus and P. leucopus, it seems inevitable in hindsight that these species would become common inhabitants of landscapes constructed by humans. P. leucopus, for example, may occur at higher densities near roads than in contiguous forests [31], and small forest fragments may even act as source populations for larger fragments because of ecological release from predators and competitors [32]. Populations fragmented by agriculture maintain high levels of heterozygosity and become only weakly differentiated [33], and white-footed mice have recently expanded their range northward across agricultural areas into Quebec in response to milder and shorter winters [34][35]. P. leucopus also occurs in highly-disturbed secondary forests within cities across eastern North America, even in the most densely populated areas of the country such as Central Park in New York City (NYC) [36]. Unlike agricultural landscapes, however, urbanization results in genetic differentiation between populations separated by a few km or less [37]; only narrow, vegetated corridors comprised of rivers, parkways, cemeteries and other “green” infrastructure still maintain any genetic connectivity between NYC populations of white-footed mice [38].
These findings that NYC populations are genetically isolated but still maintain high levels of neutral genetic variation [36–38] indicated that the conditions for local adaptation to urban conditions may be widespread for white-footed mice and other urban Peromyscus [39]. Indeed, temporal replacement of mitochondrial haplotypes had previously been reported for Chicago-area P. leucopus, suggesting rapid evolution in urban populations [40,41]. However, these earlier studies on urban P. leucopus relied on relatively small numbers of microsatellite markers and mitochondrial haplotypes to investigate evolutionary change. In 2013, Harris et al. [13] used RNA-Seq to generate transcriptome data from liver, brain, and gonads from both urban and rural populations around NYC. They identified over 7K gene candidates with functional annotations from Mus and Rattus reference genomes, as well as hundreds of over-represented gene ontology (GO) terms for individual tissue types. Across the dataset, they also reported over 30K SNPs and 11 candidate genes with elevated ratios of nonsynonymous to synonymous substitutions in urban vs. rural or urban vs. urban comparisons. These candidates had GO terms associated with immune responses, metabolism of xenobiotic compounds, and other functions that could be associated with a priori hypotheses about local adaptation to polluted, densely populated habitat patches in cities.
Harris et al. [42] conducted a much larger RNA-Seq study using liver tissue from urban and rural white-footed mice to build transcriptome resources for this species and identify additional candidate genes under selection in urban environments. This effort produced over 30 Mb of sequence, 40K contigs, 104K SNPs derived from the transcriptome, and 10 genes that were differentially expressed between urban and rural populations. Similar to the candidates identified previously [13], these expression outliers were related to immune responses and transcriptional/translational processes [42]. Scans for selection are often subject to false positive outliers because the influence of historical demography (e.g. population bottlenecks or expansions) may create genomic signatures that mimic the actions of selection [43]. To account for historical demography in this system, Munshi-South et al. [44] generated more than 10K SNPs using double-digest restriction site-associated DNA sequencing (ddRAD-Seq) [45] among 23 white-footed mouse populations spanning an urban-to-rural gradient in the NYC area. They found that levels of genome-wide variation are inversely related to degree of urbanization of populations, and populations within urban areas are more differentiated from each other than populations in rural areas. They concluded that standing genetic variation is lower in urban white-footed mice, but not so low nor genetic drift so strong as to preclude local adaptation [44]. Harris et al. [46] then built a full demographic model for these 23 populations using a recently developed coalescent approach to estimate parameters [47]. This model revealed that a group of mainland populations diverged from Long Island populations after post-glacial colonization events several thousand years ago. However, several contemporary urban populations also diverged from other populations in the last few hundred generations and experienced recent bottlenecks, indicating that recent urbanization influenced genomic patterns in NYC’s white-footed mice [46].
Having inferred the demographic history of urban P. leucopus in NYC, Harris and Munshi-South [48] then conducted genome scans using methods that incorporate the demographic history in the null model. From their transcriptome data for 48 individuals from 3 urban and 3 rural populations [42], they identified overlapping subsets of outlier genes using scans based on patterns of differentiation (i.e. FST calibrated using inferred demographic parameters), linkage disequilibrium, and genotype-by-environment associations. The majority of these outlier genes have GO terms associated with metabolic functions, particularly dietary specialization. Many of these genes are in pathways involved in either carbohydrate or lipid metabolism, suggesting that urban white-footed mice in NYC have experienced directional selection for novel diets [48]. These findings will be assessed in the near future by field investigations of diet, a dataset of SNP genotypes from many more than 3 urban and 3 rural populations, and comparisons of SNP allele frequencies between contemporary populations and museum specimens collected over 100 years ago [49].
3. Behavior
Closely-related species of Peromyscus may exhibit markedly different behaviors that evolved due to natural selection in contrasting social and environmental contexts [7]. If highly heritable, then uncovering the genetic architecture of these behaviors is tractable using laboratory crosses when species are interfertile. Burrow building [50] and mating systems [51,52] are two such complex behaviors that vary between closely-related species and have recently been the subject of transcriptomic studies. Peromyscus have also attracted interest as potential alternatives to Mus and Rattus as laboratory rodent models for human behavior [53]. Behavioral transcriptomics is a young field with high growth potential that is likely to benefit from methodological advances in genomics for years to come [54].
3.1 Burrow construction
One of the best examples of studies linking genotypes to behavioral phenotypes that are relevant to wild organisms is Weber et al.’s [55] analysis of burrow construction in two species of Peromyscus. One species, P. polionotus, construct long burrows with both entrance and escape tunnels, whereas its sister species, P. maniculatus, exhibits a more ancestral, relatively short burrow without escape tunnels [56]. Both taxa exhibit their characteristic burrow-building behavior after several generations in captivity, and F1 hybrids exhibit the P. polionotus burrow phenotype because the P. polionotus trait is dominant [50]. Weber et al. [55] extended this work by creating first-generation backcross mice that exhibited a wide range of entrance tunnel lengths and intermediate frequencies of escape tunnels, suggesting that these two traits are influenced by separate genes. A quantitative trait locus (QTL) analysis with hundreds of SNPs genotyped using ddRAD-Seq identified one locus associated with escape tunnels, and three loci associated with entrance tunnel length. They argued that the two burrow traits are influenced by discrete genetic modules, and evolved through the accumulation of alleles that combined in novel ways.
A new Ph.D. dissertation by H. Metz [57] from this same research group further explored the proximate causes of burrow variation in Peromyscus using approaches from developmental biology and transcriptomics. She reports that a locus associated with tunnel length predicts precocious burrowing behavior (typical of P. polionotus) in recombinant F2 backcross hybrids, and that escape tunnels are likely a threshold trait. Using a transcriptomic experiment, it was found that F1 hybrids exhibit brain gene expression that is biased towards P. polionotus alleles. Allele-specific expression also characterized several candidate genes in QTLs that are associated with burrow building behavior [57]. GO terms related to behavior and activity patterns, likely established in functional studies of lab rodents, characterized these candidate genes. Given that several QTLs, candidate genes, and expression differences between burrow phenotypes are now known, the P. polionotus/P. maniculatus species pair would be an ideal system for functional studies using CRISPR-Cas9 [58] or other genomics manipulations. The success of these approaches at elucidating the genomics of a complex behavior in Peromyscus will rightfully inspire behavioral transcriptomic studies in many other wild-living organisms.
3.2 Social behavior and mating systems
A substantial majority of mammals, including most Peromyscus, exhibit polygynous or promiscuous mating systems [59–61]. However, at least two deer mice, P. californicus [51] and P. polionotus [52], have evolved genetic monogamy and biparental care [62] (although to a more modest degree in the latter) [63]. This variation in mating behavior allows for robust genomic comparisons between closely-related taxa that exhibit interesting behavioral differences. Comparison of vaginal bacterial communities from promiscuous P. maniculatus and monogamous P. californicus captured from the same site revealed that the promiscuous species has greater microbial diversity [64]. A prediction that could then be tested with transcriptomic comparisons is that promiscuous species may exhibit upregulated gene expression, or signatures of selection, in genes associated with the immune system. Indeed, MacManes and Lacey [65] reported higher levels of diversity and enhanced selection in the major histocompatibility complex of P. maniculatus vs. P. californicus, but many other genes are likely to exhibit differences related to mating system variation. Gene expression in the brain is also likely to vary in relation to social behavior and mating system [66]. For example, estrogen receptor alpha (ERα) is expressed at higher levels in male brains of monogamous P californicus and P. polionotus compared to polygynous P. maniculatus and P. leucopus [67]. ERα was already known to be associated with social monogamy in some voles (Microtus spp.) and dwarf hamsters (Phopodus spp.). There are many other candidate genes that have been identified after decades of research in monogamous Microtus [68,69] that could be examined in Peromyscus [70]. This candidate gene strategy would not identify genes that are important in Peromyscus but not Microtus, and thus transcriptomic studies could develop deer mice as an alternative model system for the genetic basis of mating system variation. For example, ERα expression is not sexually dimorphic in all studied regions of the Peromyscus brain whereas it is in murid rodents, voles, and dwarf hamsters [67].
Morphological, physiological, and life history traits associated with mating behavior are also likely to exhibit genetic differences between monogamous and promiscuous species. For example, sperm competition has driven the evolution of cooperative sperm aggregation in P. maniculatus, but not in the monogamous P. polionotus [71]. This trait would be beneficial for post-copulatory sexual selection when a female has serially mated with multiple males in a short period of time. Faster reproductive development is also likely to be favored in promiscuous vs. monogamous species [72], especially in short-lived taxa where the temporal window of reproductive opportunities may be small. Jacobs-Palmer [73] found that the onset of spermatogenesis occurs earlier in male P. maniculatus than in male P. polionotus, in line with predictions based on their respective mating systems. To examine the influence of gene expression on sexual development, she then created F1 hybrids of the two species and conducted RNA-Seq on testis tissues collected at 10 post-natal periods from 16 to 64 days. These sequence data were then filtered for genes that fit experimental prerequisites (e.g. fixed SNPs between the two species) and had adequate read counts for examining allele-specific expression. She reported that 702/4,337 (16.2%) loci exhibited two-fold or greater differences in expression between P. maniculatus and P. polionotus alleles for at least one time point. These differentially expressed genes are related to spermatogenesis, testis development, and sperm phenotype, suggesting that accelerated onset of spermatogenesis in P. maniculatus is influenced by cis-regulatory changes [73].
4. Development
Transcriptomics has been particularly transformational for developmental biology, given the crucial role of gene expression regulation in tissue differentiation. Laboratory Mus have been the workhorse of mammalian developmental biology; the mouse genome [74], encyclopedia of mouse DNA elements [75,76], and other resources [77] for studying gene function and regulation are unparalleled among non-human mammals. However, gene expression patterns are not very similar between humans and Mus [78], prompting calls for efforts to develop nonmodel organisms for developmental study [79]. Such efforts could examine developmental phenomena in broader ecological and evolutionary contexts, and these goals are achievable using RNA-Seq for even highly complex developmental phenotypes.
Vrana et al. [80] recently published a thorough review of the use of Peromyscus as developmental models. In brief, they outlined the availability of wild-derived populations available through the Peromyscus Genetic Stock Center (http://stkctr.biol.sc.edu/), which includes five species and F1 hybrids of P. maniculatus × P. polionotus at the time of writing this review. Transcriptomic studies of contrasting populations of many of these species and stocks are detailed above; P. leucopus have also been proposed as a useful model for understanding the evolution and development of long lifespan relative to other small mammals [81,82]. Several mutant strains of P. maniculatus are also available for examining the development of pelage color, hairlessness, syndactyly, tremors, epilepsy, and juvenile ataxia. P. maniculatus × P. polionotus hybrids also exhibit parent-of-origin effects on growth and development, with the male P. maniculatus × female P. polionotus cross producing typically inviable offspring. Thus, these two species have been extremely useful for studying hybrid dysgenesis [83,84]. Transcriptomic experiments to examine intra- and interspecific differences in development have not yet been conducted using these Peromyscus resources. The availability of wild-derived and mutant strains, as well as emerging resources such as descriptions of embryonic stages in Peromyscus [85], have set the stage for a productive research program of great value to evolutionary developmental biology.
5. Pathogens and Disease
Transcriptomics also holds great promise for understanding the role of Peromyscus in the etiology of several human diseases. P. maniculatus is the primary reservoir host of Sin Nombre hantavirus [86], and P. leucopus is an important reservoir of several tick-borne pathogens including Borellia [87,88], Anaplasma [89,90], and Babesia [91,92]. Lyme disease from Borellia infection is the most common tick-borne disease in humans. Recent findings indicate that both tick vectors and P. leucopus are often co-infected with two or more of these pathogens [93,94], enhancing the risk that humans will be infected by more than one pathogen at a time by a single tick bite. However, the host factors that promote co-infection and disease severity in humans are poorly understood. P. leucopus exhibit a broad range of responses to experimental infections, and transcriptomic studies could shed light on the genetic architecture and gene expression factors related to their immune traits [95]. Candidate gene approaches have been used to examine immunology in wild populations [96], with a particular focus on Toll-like receptors in rodents [97–99]. Wild bank voles (Myodes glareolous) in Europe with certain Toll-like receptor 2 haplotypes are much less likely to be infected with Borellia [100], but the influence of this gene in P. leucopus has not yet been established. It is likely that many pathways of the innate and adaptive immune responses are involved in variability in host infection with tick-borne pathogens. A recent transcriptomic analysis in humans infected with Borellia found many differentially expressed genes that are unique to this infection [101]; similar studies in Peromyscus are needed to fully understand the ecology of Borellia and its primary host.
6. Conclusions & Future Directions
Transcriptomic and RNA-Seq approaches have demonstrated their value in revealing how natural selection and gene expression plasticity contribute to the existence of deer mice in challenging environments across North America. Transcriptomics holds great promise for elucidating the genetic basis of mating systems and other behaviors in Peromyscus, but has been underutilized for developmental biology and disease studies.
In 2001 Dewey and Dawson [102] argued for Peromyscus could be the “Drosophila of North American Mammalogy”, and began their review with reference to the success of the human and laboratory mouse genome projects. The development of much greater genomic resources, particularly deeply sequenced, assembled, and annotated reference genomes and transcriptomes for multiple species, will be necessary to move Peromyscus transcriptomics forward. Currently only a single Peromyscus genome assembly is available on GenBank (P. maniculatus, GCA_000500345.1 Pman_1.0), along with genome sequencing reads for P. leucopus, P. polionotus, and P. californicus (GenBank BioProject Accession PRJNA170428). Transcriptomes are currently available from only three species (Table 1). Bedford and Hoekstra [7] also identified the need for improved genomic resources, as well as phylogenetic revision, natural history of poorly known Peromyscus species, and use of deer mice in biomedical studies as priorities for research. RNA-Seq applied to many more Peromyscus taxa could be very useful for identifying genes for use in phylogenetic analyses of the group. Range-wide surveys of genetic, phenotypic, and ecological parameters, especially for the widespread species, would likely uncover as yet unknown diversity. The power of the Peromyscus system will not be fully realized until more studies apply robust comparative frameworks to analyze populations in multiple habitats/study sites, multiple populations of the same species, and/or multiple species.
Acknowledgments
Work on this publication was supported by the National Science Foundation under award number DEB 1457523 and the National Institute of General Medical Sciences of the National Institutes of Health under award number R15GM099055 to Jason Munshi-South. The content is solely the responsibility of the authors and does not represent the official views of the National Science Foundation or National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wright S. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. Proc Sixth Int Congr Genet. 1932;1:356–366. [Google Scholar]
- 2.Stinchcombe JR, Hoekstra HE. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity. 2007;100:158–170. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- 3.Todd EV, Black MA, Gemmell NJ. The power and promise of RNA-seq in ecology and evolution. Mol Ecol. 2016;25:1224–1241. doi: 10.1111/mec.13526. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez M, Schrey AW, Richards CL. Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution? Mol Ecol. 2015;24:710–725. doi: 10.1111/mec.13055. [DOI] [PubMed] [Google Scholar]
- 5.Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 2014;29:51–63. doi: 10.1016/j.tree.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Gleason LU, Burton RS. RNA-seq reveals regional differences in transcriptome response to heat stress in the marine snail Chlorostoma funebralis. Mol Ecol. 2015;24:610–627. doi: 10.1111/mec.13047. [DOI] [PubMed] [Google Scholar]
- 7.Bedford NL, Hoekstra HE. Peromyscus mice as a model for studying natural variation. eLife. 2015;4:e06813. doi: 10.7554/eLife.06813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sumner FB. The Role of Isolation in the Formation of a Narrowly Localized Race of Deer-Mice (Peromyscus) Am Nat. 1917;51:173–185. [Google Scholar]
- 9.King JA. Biology of Peromyscus (Rodentia) The American Society of Mammalogists; Stillwater, OK: 1968. (Special Publication No. 2). [Google Scholar]
- 10.Kirkland GL, Layne JN. Advances in the Study of Peromyscus (Rodentia) Texas Tech University Press; Lubbock, TX: 1989. [Google Scholar]
- 11.Cheviron ZA, Connaty AD, McClelland GB, Storz JF. Functional genomics of adaptation to hypoxic cold-stress in high-altitude deer mice: transcriptomic plasticity and thermogenic performance. Evolution. 2014;68:48–62. doi: 10.1111/evo.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacManes MD, Eisen MB. Characterization of the transcriptome, nucleotide sequence polymorphism, and natural selection in the desert adapted mouse Peromyscus eremicus. PeerJ. 2014;2:e642. doi: 10.7717/peerj.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris SE, Munshi-South J, Obergfell C, O’Neill R. Signatures of Rapid Evolution in Urban and Rural Transcriptomes of White-Footed Mice (Peromyscus leucopus) in the New York Metropolitan Area. PLoS ONE. 2013;8:e74938. doi: 10.1371/journal.pone.0074938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheviron ZA, Bachman GC, Connaty AD, McClelland GB, Storz JF. Regulatory changes contribute to the adaptive enhancement of thermogenic capacity in high-altitude deer mice. Proc Natl Acad Sci. 2012;109:8635–8640. doi: 10.1073/pnas.1120523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder LRG. Closely Linked Alpha-Chain Hemoglobin Loci in Peromyscus and Other Animals: Speculations on the Evolution of Duplicate Loci. Evolution. 1980;34:1077–1098. doi: 10.2307/2408289. [DOI] [PubMed] [Google Scholar]
- 16.Snyder LRG. Deer Mouse Hemoglobins: Is There Genetic Adaptation to High Altitude? BioScience. 1981;31:299–304. doi: 10.2307/1308147. [DOI] [Google Scholar]
- 17.Chappell MA, Snyder LR. Biochemical and physiological correlates of deer mouse alpha-chain hemoglobin polymorphisms. Proc Natl Acad Sci. 1984;81:5484–5488. doi: 10.1073/pnas.81.17.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes JP, O’Connor CS. Natural Selection on Thermogenic Capacity of High-Altitude Deer Mice. Evolution. 1999;53:1280–1287. doi: 10.2307/2640830. [DOI] [PubMed] [Google Scholar]
- 19.Storz JF, Sabatino SJ, Hoffmann FG, Gering EJ, Moriyama H, Ferrand N, Monteiro B, Nachman MW. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 2007;3:e45. doi: 10.1371/journal.pgen.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storz JF, Runck AM, Sabatino SJ, Kelly JK, Ferrand N, Moriyama H, Weber RE, Fago A. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proc Natl Acad Sci. 2009;106:14450–14455. doi: 10.1073/pnas.0905224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storz JF, Runck AM, Moriyama H, Weber RE, Fago A. Genetic differences in hemoglobin function between highland and lowland deer mice. J Exp Biol. 2010;213:2565–2574. doi: 10.1242/jeb.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis among adaptive mutations in deer mouse hemoglobin. Science. 2013;340:1324–1327. doi: 10.1126/science.1236862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natarajan C, Hoffmann FG, Lanier HC, Wolf CJ, Cheviron ZA, Spangler ML, Weber RE, Fago A, Storz JF. Intraspecific polymorphism, interspecific divergence, and the origins of function-altering mutations in deer mouse hemoglobin. Mol Biol Evol. 2015;32:978–997. doi: 10.1093/molbev/msu403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holden JE, Stone CK, Clark CM, Brown WD, Nickles RJ, Stanley C, Hochachka PW. Enhanced cardiac metabolism of plasma glucose in high-altitude natives: adaptation against chronic hypoxia. J Appl Physiol. 1995;79:222–228. doi: 10.1152/jappl.1995.79.1.222. [DOI] [PubMed] [Google Scholar]
- 25.Scott GR, Elogio TS, Lui MA, Storz JF, Cheviron ZA. Adaptive modifications of muscle phenotype in high-altitude deer mice are associated with evolved changes in gene regulation. Mol Biol Evol. 2015 doi: 10.1093/molbev/msv076. msv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velotta JP, Jones J, Wolf CJ, Cheviron ZA. Transcriptomic plasticity in brown adipose tissue contributes to an enhanced capacity for non-shivering thermogenesis in deer mice. Mol Ecol. 2016;25:2870–2886. doi: 10.1111/mec.13661. [DOI] [PubMed] [Google Scholar]
- 27.Marra NJ, Romero A, DeWoody JA. Natural selection and the genetic basis of osmoregulation in heteromyid rodents as revealed by RNA-seq. Mol Ecol. 2014;23:2699–2711. doi: 10.1111/mec.12764. [DOI] [PubMed] [Google Scholar]
- 28.Giorello FM, Feijoo M, D’Elía G, Valdez L, Opazo JC, Varas V, Naya DE, Lessa EP. Characterization of the kidney transcriptome of the South American olive mouse Abrothrix olivacea. BMC Genomics. 2014;15:446. doi: 10.1186/1471-2164-15-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kordonowy LL, MacManes MD. Characterization of a Male Reproductive Transcriptome for Peromyscus eremicus (Cactus mouse) bioRxiv. 2016:048348. doi: 10.1101/048348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kordonowy L, Lombardo K, Green H, LaCourse S, Bolton E, MacManes M. Physiological and biochemical changes associated with experimental dehydration in the desert adapted cactus mouse, Peromyscus eremicus. bioRxiv. 2016:047704. doi: 10.1101/047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rytwinski T, Fahrig L. Effect of road density on abundance of white-footed mice. Landsc Ecol. 2007;22:1501–1512. doi: 10.1007/s10980-007-9134-2. [DOI] [Google Scholar]
- 32.Anderson CS, Meikle DB. Genetic estimates of immigration and emigration rates in relation to population density and forest patch area in Peromyscus leucopus. Conserv Genet. 2010;11:1593–1605. [Google Scholar]
- 33.Mossman CA, Waser PM. Effects of habitat fragmentation on population genetic structure in the white-footed mouse (Peromyscus leucopus) Can J Zool. 2001;79:285–295. [Google Scholar]
- 34.Roy-Dufresne E, Logan T, Simon JA, Chmura GL, Millien V. Poleward Expansion of the White-Footed Mouse (Peromyscus leucopus) under Climate Change: Implications for the Spread of Lyme Disease. PloS One. 2013;8:e80724. doi: 10.1371/journal.pone.0080724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogic A, Tessier N, Legendre P, Lapointe FJ, Millien V. Genetic structure of the white-footed mouse in the context of the emergence of Lyme disease in southern Québec. Ecol Evol. 2013;3:2075–2088. doi: 10.1002/ece3.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munshi-South J, Nagy C. Urban park characteristics, genetic variation, and historical demography of white-footed mouse (Peromyscus leucopus) populations in New York City. PeerJ. 2014;2:e310. doi: 10.7717/peerj.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munshi-South J, Kharchenko K. Rapid, pervasive genetic differentiation of urban white-footed mouse (Peromyscus leucopus) populations in New York City. Mol Ecol. 2010;19:4242–4254. doi: 10.1111/j.1365-294X.2010.04816.x. [DOI] [PubMed] [Google Scholar]
- 38.Munshi-South J. Urban landscape genetics: canopy cover predicts gene flow between white-footed mouse (Peromyscus leucopus) populations in New York City. Mol Ecol. 2012;21:1360–1378. doi: 10.1111/j.1365-294X.2012.05476.x. [DOI] [PubMed] [Google Scholar]
- 39.Tanner CJ, Adler FR, Grimm NB, Groffman PM, Levin SA, Munshi-South J, Pataki DE, Pavao-Zuckerman M, Wilson WG. Urban ecology: advancing science and society. Front Ecol Environ. 2014;12:574–581. doi: 10.1890/140019. [DOI] [Google Scholar]
- 40.Pergams ORW, Lacy RC. Rapid morphological and genetic change in Chicago-area Peromyscus. Mol Ecol. 2008;17:450–463. doi: 10.1111/j.1365-294X.2007.03517.x. [DOI] [PubMed] [Google Scholar]
- 41.Pergams ORW, Barnes WM, Nyberg D. Rapid change in mouse mitochondrial DNA. Nature. 2003;423:397. doi: 10.1038/423397a. [DOI] [PubMed] [Google Scholar]
- 42.Harris SE, O’Neill RJ, Munshi-South J. Transcriptome resources for the white-footed mouse (Peromyscus leucopus): new genomic tools for investigating ecologically divergent urban and rural populations. Mol Ecol Resour. 2015;15:382–394. doi: 10.1111/1755-0998.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen R, Williamson S, Kim Y, Hubisz M, Clark A, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munshi-South J, Zolnik CP, Harris SE. Population genomics of the Anthropocene: urbanization is negatively associated with genome-wide variation in white-footed mouse populations. Evol Appl. 2016;9:546–564. doi: 10.1111/eva.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris SE, Xue AT, Alvarado-Serrano D, Boehm JT, Joseph T, Hickerson MJ, Munshi-South J. Urbanization shapes the demographic history of a native rodent (the white-footed mouse, Peromyscus leucopus) in New York City. Biol Lett. 2016;12:20150983. doi: 10.1098/rsbl.2015.0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust Demographic Inference from Genomic and SNP Data. PLOS Genet. 2013;9:e1003905. doi: 10.1371/journal.pgen.1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris SE, Munshi-South J. Scans for positive selection reveal candidate genes and local adaptation of Peromyscus leucopus populations to urbanization. bioRxiv. 2016:038141. [Google Scholar]
- 49.Bi K, Linderoth T, Vanderpool D, Good JM, Nielsen R, Moritz C. Unlocking the vault: next-generation museum population genomics. Mol Ecol. 2013;22:6018–6032. doi: 10.1111/mec.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dawson WD, Lake CE, Schumpert SS. Inheritance of burrow building in Peromyscus. Behav Genet. 1988;18:371–382. doi: 10.1007/BF01260937. [DOI] [PubMed] [Google Scholar]
- 51.Ribble DO. The monogamous mating system of Peromyscus californicus as revealed by DNA fingerprinting. Behav Ecol Sociobiol. 1991;29:161–166. [Google Scholar]
- 52.Foltz DW. Genetic evidence for long-term monogamy in a small rodent, Peromyscus polionotus. Am Nat. 1981:665–675. [Google Scholar]
- 53.Shorter KR, Owen A, Anderson V, Hall-South AC, Hayford S, Cakora P, Crossland JP, Georgi VR, Perkins A, Kelly SJ, et al. Natural genetic variation underlying differences in Peromyscus repetitive and social/aggressive behaviors. Behav Genet. 2014;44:126–135. doi: 10.1007/s10519-013-9640-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calisi RM, MacManes MD. RNAseq-ing a more integrative understanding of animal behavior. Curr Opin Behav Sci. 2015;6:65–68. doi: 10.1016/j.cobeha.2015.09.007. [DOI] [Google Scholar]
- 55.Weber JN, Peterson BK, Hoekstra HE. Discrete genetic modules are responsible for complex burrow evolution in Peromyscus mice. Nature. 2013;493:402–405. doi: 10.1038/nature11816. [DOI] [PubMed] [Google Scholar]
- 56.Weber JN, Hoekstra HE. The evolution of burrowing behaviour in deer mice (genus Peromyscus) Anim Behav. 2009;77:603–609. [Google Scholar]
- 57.Metz H. PhD Dissertation. Harvard University; 2015. The Genetic Basis of Behavior: Burrow Construction in Deer Mice (Genus Peromyscus) https://dash.harvard.edu/handle/1/17467514 (accessed March 23, 2016) [Google Scholar]
- 58.Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 59.Orians GH. On the evolution of mating systems in birds and mammals. Am Nat. 1969:589–603. [Google Scholar]
- 60.Clutton-Brock TH. Review lecture: mammalian mating systems. Proc R Soc Lond B Biol Sci. 1989;236:339–372. doi: 10.1098/rspb.1989.0027. [DOI] [PubMed] [Google Scholar]
- 61.Emlen ST, Oring LW. Ecology, sexual selection, and the evolution of mating systems. Science. 1977;197:215–223. doi: 10.1126/science.327542. [DOI] [PubMed] [Google Scholar]
- 62.Gubernick DJ, Alberts JR. The biparental care system of the California mouse, Peromyscus californicus. J Comp Psychol. 1987;101:169. [PubMed] [Google Scholar]
- 63.Margulis SW, Nabong M, Alaks G, Walsh A, Lacy RC. Effects of early experience on subsequent parental behaviour and reproductive success in oldfield mice, Peromyscus polionotus. Anim Behav. 2005;69:627–634. doi: 10.1016/j.anbehav.2004.04.021. [DOI] [Google Scholar]
- 64.MacManes MD. Promiscuity in mice is associated with increased vaginal bacterial diversity. Naturwissenschaften. 2011;98:951. doi: 10.1007/s00114-011-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.MacManes MD, Lacey EA. Is Promiscuity Associated with Enhanced Selection on MHC-DQα in Mice (genus Peromyscus)? PLOS ONE. 2012;7:e37562. doi: 10.1371/journal.pone.0037562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacManes MD, Lacey EA. The Social Brain: Transcriptome Assembly and Characterization of the Hippocampus from a Social Subterranean Rodent, the Colonial Tuco-Tuco (Ctenomys sociabilis) PLOS ONE. 2012;7:e45524. doi: 10.1371/journal.pone.0045524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cushing BS. Estrogen Receptor Alpha Distribution and Expression in the Social Neural Network of Monogamous and Polygynous Peromyscus. PLOS ONE. 2016;11:e0150373. doi: 10.1371/journal.pone.0150373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson GE. Dissecting diversity in the social brain. Science. 2015;350:1310–1312. doi: 10.1126/science.aad8071. [DOI] [PubMed] [Google Scholar]
- 69.Okhovat M, Berrio A, Wallace G, Ophir AG, Phelps SM. Sexual fidelity trade-offs promote regulatory variation in the prairie vole brain. Science. 2015;350:1371–1374. doi: 10.1126/science.aac5791. [DOI] [PubMed] [Google Scholar]
- 70.Bester-Meredith JK, Young LJ, Marler CA. Species differences in paternal behavior and aggression in Peromyscus and their associations with vasopressin immunoreactivity and receptors. Horm Behav. 1999;36:25–38. doi: 10.1006/hbeh.1999.1522. [DOI] [PubMed] [Google Scholar]
- 71.Fisher H, Hoekstra H. Competition drives cooperation among closely related sperm of deer mice. Nature. 2010;463:801–803. doi: 10.1038/nature08736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleiman DG. Monogamy in Mammals. Q Rev Biol. 1977;52:39–69. doi: 10.1086/409721. [DOI] [PubMed] [Google Scholar]
- 73.Jacobs-Palmer E. PhD Dissertation. Harvard University; 2015. The Genetics of Sexually Selected Male Reproductive Traits in Mice (Mus and Peromyscus Species) https://dash.harvard.edu/handle/1/17463151 (accessed March 23, 2016) [Google Scholar]
- 74.Chinwalla AT, Cook LL, Delehaunty KD, Fewell GA, Fulton LA, Fulton RS, Graves TA, Hillier LW, Mardis ER, McPherson JD, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 75.Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:1. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xue Z, Huang K, Cai C, Cai L, Jiang C, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, Liu J, Horvath S, Fan G. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin S, Lin Y, Nery JR, Urich MA, Breschi A, Davis CA, Dobin A, Zaleski C, Beer MA, Chapman WC, et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci. 2014;111:17224–17229. doi: 10.1073/pnas.1413624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rowan BA, Weigel D, Koenig D. Developmental genetics and new sequencing technologies: the rise of nonmodel organisms. Dev Cell. 2011;21:65–76. doi: 10.1016/j.devcel.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 80.Vrana PB, Shorter KR, Szalai G, Felder MR, Crossland JP, Veres M, Allen JE, Wiley CD, Duselis AR, Dewey MJ, et al. Peromyscus (deer mice) as developmental models. Wiley Interdiscip Rev Dev Biol. 2014;3:211–230. doi: 10.1002/wdev.132. [DOI] [PubMed] [Google Scholar]
- 81.Shi Y, Pulliam DA, Liu Y, Hamilton RT, Jernigan AL, Bhattacharya A, Sloane LB, Qi W, Chaudhuri A, Buffenstein R, Ungvari Z, Austad SN, Remmen HV. Reduced mitochondrial ROS, enhanced antioxidant defense, and distinct age-related changes in oxidative damage in muscles of long-lived Peromyscus leucopus. Am J Physiol - Regul Integr Comp Physiol. 2013;304:R343–R355. doi: 10.1152/ajpregu.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fushan AA, Turanov AA, Lee SG, Kim EB, Lobanov AV, Yim SH, Buffenstein R, Lee SR, Chang KT, Rhee H, Kim JS, Yang KS, Gladyshev VN. Gene expression defines natural changes in mammalian lifespan. Aging Cell. 2015;14:352–365. doi: 10.1111/acel.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vrana PB, Fossella JA, Matteson P, del Rio T, O’Neill MJ, Tilghman SM. Genetic and epigenetic incompatibilities underlie hybrid dysgenesis in Peromyscus. Nat Genet. 2000;25:120–124. doi: 10.1038/75518. [DOI] [PubMed] [Google Scholar]
- 84.Duselis AR, Vrana PB. Aberrant growth and pattern formation in Peromyscus hybrid placental development. Biol Reprod. 2010;83:988–996. doi: 10.1095/biolreprod.110.085654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davis SW, Keisler JL. Embryonic Development of the Deer Mouse, Peromyscus maniculatus. PloS One. 2016;11:e0150598. doi: 10.1371/journal.pone.0150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Netski D, Thran BH, St Jeor SC. Sin Nombre Virus Pathogenesis in Peromyscus maniculatus. J Virol. 1999;73:585–591. doi: 10.1128/jvi.73.1.585-591.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levine JF, Wilson ML, Spielman A. Mice as reservoirs of the Lyme disease spirochete. Am J Trop Med Hyg. 1985;34:355–360. doi: 10.4269/ajtmh.1985.34.355. [DOI] [PubMed] [Google Scholar]
- 88.Donahue JG, Piesman J, Spielman A. Reservoir competence of white-footed mice for Lyme disease spirochetes. Am J Trop Med Hyg. 1987;36:92–96. doi: 10.4269/ajtmh.1987.36.92. [DOI] [PubMed] [Google Scholar]
- 89.Levin ML, Nicholson WL, Massung RF, Sumner JW, Fish D. Comparison of the Reservoir Competence of Medium-Sized Mammals and Peromyscus leucopus for Anaplasma phagocytophilum in Connecticut. Vector-Borne Zoonotic Dis. 2002;2:125–136. doi: 10.1089/15303660260613693. [DOI] [PubMed] [Google Scholar]
- 90.Keesing F, Hersh MH, Tibbetts M, McHenry DJ, Duerr S, Brunner J, Killilea M, LoGiudice K, Schmidt KA, Ostfeld RS. Reservoir competence of vertebrate hosts for Anaplasma phagocytophilum. Emerg Infect Dis. 2012;18:2013. doi: 10.3201/eid1812.120919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spielman A, Etkind P, Piesman J, Ruebush TK, 2nd, Juranek DD, Jacobs MS. Reservoir hosts of human babesiosis on Nantucket Island. Am J Trop Med Hyg. 1981;30:560–565. doi: 10.4269/ajtmh.1981.30.560. [DOI] [PubMed] [Google Scholar]
- 92.Hersh MH, Tibbetts M, Strauss M, Ostfeld RS, Keesing F. Reservoir competence of wildlife host species for Babesia microti. Emerg Infect Dis. 2012;18:1951. doi: 10.3201/eid1812.111392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hersh MH, Ostfeld RS, McHenry DJ, Tibbetts M, Brunner JL, Killilea ME, LoGiudice K, Schmidt KA, Keesing F. Co-Infection of Blacklegged Ticks with Babesia microti and Borrelia burgdorferi Is Higher than Expected and Acquired from Small Mammal Hosts. PLOS ONE. 2014;9:e99348. doi: 10.1371/journal.pone.0099348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Diuk-Wasser MA, Vannier E, Krause PJ. Coinfection by Ixodes Tick-Borne Pathogens: Ecological, Epidemiological, and Clinical Consequences. Trends Parasitol. 2016;32:30–42. doi: 10.1016/j.pt.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook V, Barbour AG. Broad diversity of host responses of the white-footed mouse Peromyscus leucopus to Borrelia infection and antigens. Ticks Tick-Borne Dis. 2015;6:549–558. doi: 10.1016/j.ttbdis.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brown EA, Pilkington JG, Nussey DH, Watt KA, Hayward AD, Tucker R, Graham AL, Paterson S, Beraldi D, Pemberton JM, Slate J. Detecting genes for variation in parasite burden and immunological traits in a wild population: testing the candidate gene approach. Mol Ecol. 2013;22:757–773. doi: 10.1111/j.1365-294X.2012.05757.x. [DOI] [PubMed] [Google Scholar]
- 97.Areal H, Abrantes J, Esteves PJ. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol. 2011;11:1. doi: 10.1186/1471-2148-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tschirren B, Råberg L, Westerdahl H. Signatures of selection acting on the innate immunity gene Toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J Evol Biol. 2011;24:1232–1240. doi: 10.1111/j.1420-9101.2011.02254.x. [DOI] [PubMed] [Google Scholar]
- 99.Tschirren B, Andersson M, Scherman K, Westerdahl H, Råberg L. Contrasting patterns of diversity and population differentiation at the innate immunity gene toll-like receptor 2 (TLR2) in two sympatric rodent species. Evolution. 2012;66:720–731. doi: 10.1111/j.1558-5646.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- 100.Tschirren B, Andersson M, Scherman K, Westerdahl H, Mittl PRE, Råberg L. Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc R Soc Lond B Biol Sci. 2013;280:20130364. doi: 10.1098/rspb.2013.0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bouquet J, Soloski MJ, Swei A, Cheadle C, Federman S, Billaud JN, Rebman AW, Kabre B, Halpert R, Boorgula M, Aucott JN, Chiu CY. Longitudinal Transcriptome Analysis Reveals a Sustained Differential Gene Expression Signature in Patients Treated for Acute Lyme Disease. mBio. 2016;7:e00100–16. doi: 10.1128/mBio.00100-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dewey MJ, Dawson WD. Deer mice: “the Drosophila of North American mammalogy”. Genesis. 2001;29:105–109. doi: 10.1002/gene.1011. [DOI] [PubMed] [Google Scholar]