

Summary
Type 1 diabetes is characterized by the destruction of pancreatic β cells, and generating new insulin-producing cells from other cell types is a major aim of regenerative medicine. One promising approach is transdifferentiation of developmentally related pancreatic cell types, including glucagon-producing α cells. In a genetic model, loss of the master regulatory transcription factor Arx is sufficient to induce the conversion of α cells to functional β-like cells. Here, we identify artemisinins as small molecules that functionally repress Arx by causing its translocation to the cytoplasm. We show that the protein gephyrin is the mammalian target of these antimalarial drugs and that the mechanism of action of these molecules depends on the enhancement of GABAA receptor signaling. Our results in zebrafish, rodents, and primary human pancreatic islets identify gephyrin as a druggable target for the regeneration of pancreatic β cell mass from α cells.
Keywords: artemisinins, GABA-receptor signaling, gephyrin, pancreatic endocrine transdifferentiation, ARX translocation, β cell, insulin secretion, diabetes, regenerative medicine, chemical biology
Graphical Abstract
Highlights
-
•
Artemisinins inhibit ARX function and impair α cell identity
-
•
Compounds act by stabilizing gephyrin, thus enhancing GABAA receptor signaling
-
•
Artemisinins increase β cell mass in zebrafish and rodent models
-
•
Functional and transcriptional data indicate a conserved phenotype in human islets
The anti-malarial drug Artemisinin can drive the in vivo conversion of pancreatic α cells into functional β-like cells through enhanced GABA signaling and may have potential as a therapeutic for diabetes.
Introduction
Type 1 diabetes patients often suffer from total loss of their functional β cells, as indicated by the complete absence of insulin C-peptide in their serum. Replacing β cell mass by pancreatic islet transplantation has been shown to be curative in principle but is limited by the availability of donor islets, immunological complications, and transplant survival (Shapiro et al., 2006). Therefore, attempts to regenerate patient-specific insulin-producing cells have been undertaken using different cell sources, including embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), hepatic cells, exocrine cells, and other endocrine cells (Al-Hasani et al., 2013, Chera et al., 2014, Collombat et al., 2009, Kroon et al., 2008, Pagliuca et al., 2014, Sangan et al., 2015, Talchai et al., 2012, Zhou et al., 2008). In most cases, these approaches have relied on the overexpression of master regulatory transcription factors involved in normal pancreas development, and in only a few cases, small molecules or biologicals have been used (Fomina-Yadlin et al., 2010, Kubicek et al., 2012, Pennarossa et al., 2013, Xie et al., 2013, Yi et al., 2013). α cells are particularly attractive starting points for transdifferentiation protocols, as they are developmentally closely related to β cells. These cells have been shown to replenish insulin-producing cells following extreme β cell loss (Thorel et al., 2010, Ye et al., 2015). In a genetic model, overexpression of the transcription factor Pax4 converts mouse α cells to β cells during development (Collombat et al., 2009) and when triggered in adulthood (Al-Hasani et al., 2013). Molecularly, the β cell factor Pax4 acts by directly repressing the α cell master regulatory transcription factor Arx (Collombat et al., 2003), and the loss of Arx alone is sufficient to convert α cells into β cells (Courtney et al., 2013).
So far, Pax4- and Arx-mediated α and β cell transdifferentiation has been observed in vivo in the endogenous environment of a pancreatic islet of Langerhans (Collombat et al., 2009, Courtney et al., 2013). It remains unclear whether these cell-type conversions occur by a cell-autonomous mechanism or are dependent on other islet cell types and/or signaling from remote organs, such as the liver. To discriminate the two scenarios, we generated cell-line models for inducible overexpression of pancreatic transcription factors and showed their applicability to identify small-molecule modulators of the transdifferentiation process. From a representative library of approved drugs, we identified the anti-malarial drug class of artemisinins as compounds that functionally counteract the α cell-specific transcription factor ARX, thereby overcoming a major barrier for α cell transdifferentiation. A mammalian molecular target for artemisinins has remained elusive. Here, we show that these compounds bind to gephyrin, a multifunctional protein that has primarily been studied in neurons (Tyagarajan and Fritschy, 2014). Among other functions, gephyrin is essential for active GABAA receptor signaling, a pathway that has been proposed to reverse diabetes by induction of β cell proliferation (Purwana et al., 2014, Soltani et al., 2011). We show that artemisinins increase GABA signaling and prevent glucagon secretion by α cells. Consequently, α cells acquire β cell characteristics, resulting in improved glucose homeostasis in zebrafish and rodent diabetes models.
Results
Identification of Artemisinins as Functional ARX Inhibitors in a Cell-Line Model for Transcription Factor-Mediated Transdifferentiation
To discriminate cell-autonomous effects of transcription factor-mediated transdifferentiation from phenotypes that require paracrine and endocrine signaling in an islet microenvironment, we engineered the mouse β cell line Min6 to allow the inducible overexpression of ARX. Based on the known heterogeneity of the parental cell line (Nakashima et al., 2009), we isolated clonal derivatives with integrations of constructs for the doxycycline-controlled transcriptional activation of Myc-tagged ARX (Figures S1A and S1B). While basal expression levels of α and β cell genes varied in the different subclones, we observed that induction of ARX overexpression consistently activated the transcription of α cell genes and repressed β cell-specific transcripts (Figure S1C) in a time-dependent manner (Figure S1D). These changes indicate that our cell lines faithfully model the β-to-α fate switch upon ARX overexpression and suggest that transdifferentiation can be initiated in a cell-autonomous fashion. Furthermore, the cellular system we generated is directly applicable to a high-content screen for functional repressors of ARX. In DMSO-treated control samples, we observed a >50% reduction in insulin protein levels after ARX induction compared to uninduced cells (Figures 1A and 1B). To identify small molecules that counter ARX function, we induced ARX expression and, at the same time, added compounds from a library of 280 clinically approved small molecules selected for their structural and target diversity. We then measured insulin levels after 72 hr and selected hit compounds for their ability to maintain high insulin levels in the presence of ARX while not affecting cell viability (Figure 1C). All compounds that appeared to counteract ARX in Min6 β cells were then tested for their ability to induce insulin expression in the α cell line αTC1 (Figure 1D). Two artemisinins, artemether and its active metabolite dihydroartemisinin, fully inhibited the ARX overexpression phenotype in Min6 cells while also inducing insulin expression in αTC1 cells, as predicted for functional inhibitors of ARX (Figure 1E). Importantly, artemether treatment increased insulin expression in Min6-ARX-induced cells and in αTC1 cells in a dose-dependent manner (Figures S2A and S2B). To further understand the effects of artemether on α cells, we analyzed the relative quantitative changes in the proteome of αTC1 cells treated with artemether for 72 hr. Proteins with roles in oxidative phosphorylation and mitochondrial localization were significantly altered, with some of these proteins showing increased abundance and others showing decreased abundance. In line with artemisinins acting as functional ARX antagonists, glucagon protein levels were reduced by more than 50% after artemether treatment (Figure 1F). Importantly, artemether reduced the abundance of both proglucagon and processed glucagon peptides in αTC1 cells (Figure 1G). To understand the effect of artemether on ARX, we used the Min6 overexpression model to study the cellular localization of the transcription factor. We observed that artemether induced the translocation of ARX from the nucleus to the cytoplasm (Figures 1H–1J), thereby depleting ARX from chromatin.
Figure S1.

A Min6 Cell Line for Inducible ARX Overexpression, Related to Figure 1
(A) Western blot for the Myc-tag in three clonal Min6 cell lines allowing inducible overexpression of Myc-ARX. Myc-ARX expression was induced with 1 μM doxycycline for 16 hr. Histone H2B or tubulin was used as a loading control.
(B) Western blot for the ARX as in (A).
(C) Gene expression after 24 hr ARX induction.
(D) Time course of gene expression after ARX induction for 3 and 6 days.
(E) Relative gene expression changes in ARX inducible clones after 24 hr ARX induction.
Figure 1.

A Cell-Line Model of Transcription-Factor-Mediated Transdifferentiation for Identifying Functional ARX Inhibitors
(A) Representative images of Min6-ARX cells stained for insulin and the Myc-tag present on the ARX overexpression construct with and without induction with 1 μg/ml doxycycline for 72 hr. Scale bars, 5 μm.
(B) Quantification of insulin reduction upon ARX overexpression, detected by immunofluorescence as in (A) (data indicate means and SD from 50 wells). ∗p < 0.001.
(C) High-content screen of Min6-ARX cells concomitantly treated with 1 μg/mL doxycycline and 10 μM compound for 72 hr, analyzed for cell number and insulin protein levels by immunofluorescence. Means and SD for DMSO-treated wells are indicated in red.
(D) Validation of active compounds from the primary screen in αTC1 cells, analyzed for cell number and insulin protein levels by immunofluorescence following compound treatment for 72 hr.
(E) Insulin immunofluorescence in αTC1 cells treated with 10 μM artemether or control DMSO for 72 hr. Scale bars, 5 μm.
(F) Quantitative proteome analysis of αTC1 cells treated with 10 μM artemether for 72 hr compared to control DMSO-treated cells.
(G) Western blot for glucagon in αTC1 cells treated with increasing concentrations of artemether for 72 hr.
(H) Immunofluorescence staining for the Myc-tag on overexpressed ARX in Min6-ARX cells. Cells were induced with 1 μg/mL doxycycline for 24 hr and concomitantly treated with either 10 μM artemether or control DMSO. Scale bars, 5 μm.
(I) Quantification of the cells with higher nuclear/cytoplasmic ARX staining ratio from immunofluorescence images as in (H).
(J) Western blot for nuclear ARX in cells treated as in (H).
See also Figures S1 and S2.
Figure S2.

Structure Activity Relationships of Artemisinins, Related to Figure 1
(A) Artemether dose-dependence of insulin protein maintenance quantified by immunofluorescence in induced Min6-ARX cells treated for 72 hr (Mean and SD).
(B) Artemether dose-dependence of insulin protein induction quantified by immunofluorescence in αTC1 cells treated for 72 hr.
(C) Insulin intensity in αTC1 cells following 72 hr treatment with artemisinin analogs at concentrations achieving maximum insulin induction: DMSO (0), 0.27 μM Artesunate (1), 4.5 μM Deoxyarteether (2), 2.5 μM Arteether (3), 0.17 μM N-hydroxy-11-azaartemisinin (4), 7.5 μM artemistene (5), 0.1 μM artemisone (6), 2.5 μM artemisiten (7) and 67.5 μM anhydro dihydro artemisinin (8) (Mean and SD from 3 wells).
(D) Quantification of insulin intensity in αTC1 cells following treatment with the ROS inducer elesclomol for 72 hr. (Mean and SD from 4 wells).
(E) Quantification of insulin intensity in αTC1 cells following treatment with artemether in combination with N-Acetyl-Cysteine for 72 hr (∗p = 0.034). All panels show mean and SD from three biological replicates.
Artemether Induces Insulin Expression in α Cells by Targeting Gephryin
Based on the activity of artemether, we investigated the effects of additional artemisinin analogs in α cells. The closest structural analogs, including arteether and artesunate, showed effects comparable to those of artemether. In contrast, deoxyarteether, an analog lacking the endoperoxide moiety, did not increase insulin expression in α cells (Figure S2C). To rule out the possibility that artemether, an endoperoxide, induced insulin expression due to the generation of reactive oxygen species (ROS), we applied the ROS inducer elesclomol. No significant change of insulin expression in α cells was apparent (Figure S2D). Finally, combined treatment with the anti-oxidant N-acetylcysteine, a known suppressor of the anti-malarial properties of artemisinins (Arreesrisom et al., 2007), abolished the effect of artemether on insulin expression in α cells (Figure S2E).
Artemisinins are among the most effective therapies for malaria and, based on their rapid toxicity to plasmodia, are the preferred first-line treatment for the disease (Nosten and White, 2007). Despite being widely used, the molecular mechanism of action of artemisinins is not clear. Different molecular targets have been proposed, including heme binding followed by iron oxidation in human erythrocytes (Shukla et al., 1995, Wang et al., 2015) and the inhibition of two different plasmodium-specific proteins: the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) PfATP6 (Eckstein-Ludwig et al., 2003) and phosphatidylinositol 3-kinase PfPI3K (Mbengue et al., 2015). Potent effects of artemisinins are also observed on human cells, where these compounds have been assigned anti-inflammatory and anti-cancer properties (Ho et al., 2014). There is limited evidence for possible positive effects of Artemisia extracts on animal models of type 1 diabetes (Ahmad et al., 2014) and in human patients (al-Waili, 1988), but the use of isolated artemisinins for the treatment of diabetes has never been proposed, and their effect in the pancreas has not been evaluated. To identify the molecular mechanism of artemisinins in pancreatic α cells, we first assessed whether the homologs of proposed targets of the compounds in plasmodia were involved. We used the SERCA inhibitor thapsigargin on αTC1 cells and did not observe increased insulin levels up to dose-limiting toxic concentrations (Figure S3A). Next, we tested whether artemether treatment of αTC1 cells resulted in reduced PI3P and phosphorylated (p-)Akt levels, indicative of PI3K inhibition (Figures S3B and S3C). Rather than inhibition, we observed an increased signal in both readouts, thereby excluding a conserved mechanism of action based on the proposed plasmodium targets. In order to discover potential mechanisms of action of artemisinins in mammalian endocrine cells, we undertook a chemical proteomics approach (Rix and Superti-Furga, 2009) to determine the proteins that directly interact with artemisinins in αTC1 cells. We coupled artesunate, an artemisinin active in αTC1 cells, to solid support and performed pull-down experiments in the presence and absence of competing free artemether (Figure 2A). Mass spectrometry identified gephyrin as the most significantly enriched specific interactor (Figures 2B and S3D). Artemether increased gephyrin protein levels in αTC1 cells (Figures 2C and S3E), while mRNA expression levels remained unchanged (Figure S3F), suggesting stabilization of the protein. To examine the interaction between gephyrin and artemisinins in a cellular context, we used a fluorescently labeled artelinic acid probe. The cellular localization of the compound was nearly identical with gephyrin in costaining experiments (Figure S3G). Next, we asked whether gephyrin was functionally required for the insulin increase in artemether-treated αTC1 cells. Using a specific small hairpin RNA (shRNA) targeting gephyrin, we achieved a >70% reduction of gephyrin protein in αTC1 cells (Figure S3H). In contrast to control-treated α cells, cells treated with an shRNA targeting gephyrin (shGephyrin) failed to increase intracellular insulin levels after artemether treatment (Figure 2D). Single-cell analysis of the imaging data revealed a high correlation between the increases of gephyrin and insulin in artemether-treated cells, which was largely abolished when gephyrin was knocked down (Figure 2E).
Figure S3.

Artemether Increases Gephyrin Protein Levels and GABA-Receptor Signaling, Related to Figure 2
(A) Quantification of insulin intensity in αTC1 cells following treatment with SERCA inhibitor for 72 hr (n.s.,p > 0.05). Error bars represent mean ± SD.
(B) Western blot for p-Akt and Akt of lysates from αTC1 cells treated with multiple doses of artemether for 72 hr.
(C) Measurement of PI3P in αTC1 cells treated with artemether for 72 hr (∗p = 0.007; ∗∗p = 0.002). Error bars represent mean ± SD.
(D) List of all significantly enriched proteins identified by chemical proteomics in artesunate pull-downs.
(E) Immunofluorescence staining for gephyrin in α cells treated with 10 μM artemether for 72 hr (scale bar 25 μm).
(F) Expression of GABA-receptor subunits in αTC1 cells treated with artemether for 72 hr detected by RNA-seq, measured as RPKM (Reads Per Kilobase of transcript per Million mapped reads). Error bars represent mean ± SD.
(G) Costaining of αTC1 cells treated with 10 μM fluorescently labeled artelinic acid for 2 hr for gephyrin (scale bar 10 μm).
(H) Western blot for validating gephyrin knockdown by shRNA. αTC1 cells were infected with shGephyrin, selected with 4 μg/ml puromycin and lysed after 7 days. Ponceau S staining is used as a loading control.
Figure 2.

Artemether Induces Insulin Expression in α Cells by Targeting Gephyrin
(A) Chemical proteomics to identify proteins that specifically bind to artemisinins.
(B) Volcano plot of interacting proteins identified by chemical proteomics. Both p values and fold change were calculated based on two biological replicates.
(C) Western blot for gephyrin in αTC1 cells treated with increasing doses of artemether for 72 hr.
(D) Immunofluorescence for insulin and gephyrin in αTC1 cells treated with artemether and/or shGephyrin. Scale bars, 25 μm. shRNA treatment was for 16 hr, followed by selection with 4 μg/mL puromycin for 3 days and artemether treatment at a concentration of 10 μM for 3 days.
(E) Quantification of insulin and gephyrin intensity in single cells from (D).
See also Figure S3.
Gephyrin-Mediated Increase of GABA Signaling Induces Insulin Expression in α Cells
Gephyrin exerts different functions, including enzymatic activity in the synthesis of the molybdenum cofactor Moco, regulation of mTOR signaling via direct interaction with mTOR (mechanistic target of rapamycin) (Sabatini et al., 1999), and structural roles in the transport of glycine and GABAA receptors to the membrane (Fritschy et al., 2008). Therefore, we investigated whether artemether modulated these gephyrin-mediated pathways. We observed increased Moco synthesis in artemether-treated α cell extracts, indicative of enhanced gephyrin enzymatic activity (Figure S4A). Moreover, a small fraction of endogenous gephyrin interacted with mTOR, and artemether inhibited this interaction (Figure S4B). Finally, several subunits of the GABAA-receptor complex, which physically interacts with gephyrin, were increased on both the mRNA and protein levels following artemether treatment (Figures 3A and 3B; Figure S3F). RNA sequencing (RNA-seq) experiments further underlined the effects of artemether on GABAA-receptor signaling in αTC1 cells. Gene Ontology (GO) term enrichment analysis identified synaptic transmission among the pathways significantly altered by artemether (Figure 3C), and we observed the upregulation of the genes P2rx3, Vamp1, and Nrxn3 involved in the GABA-signaling pathway (Figure S4C). Therefore, we used 3-day pre-incubation with 10 μM artemether to test whether adaptive cellular responses, such as GABAA-receptor-mediated effects, could contribute to changes in αTC1 cell excitability to modulate hormone release. Artemether-treated cells exhibited significantly lower basal Ca2+ levels (Figures 3D and 3E), including coincidently reduced nuclear (Figure S4D) and cytoplasmic (Figure S4E) Ca2+ relative to control cultures. Likewise, partial substitution of extracellular Na+ by K+ (to trigger excitation) induced differential responses in drug-exposed αTC1 cells: physiologically relevant 15 mM KCl increased the fraction of cells with Ca2+ transients, suggesting increased excitability, as compared to untreated controls (Figure 3F). In 50 mM KCl, which induces signal saturation due to maximal Ca2+ influx through voltage-gated (VG) Ca2+ channels, artemether-treated cells showed attenuated slow Ca2+ responses in the second timescale (Figure 3G), which was mimicked by pre-treatment of control cells with the GABAA-receptor antagonist picrotoxin.
Figure S4.

Artemether Effect on Gephyrin Biology, Related to Figure 3
(A) Moco synthetase activity concentration in αTC1 cell treated with artemether for 72 hr (n = 3 ∗p = 0.04). Error bars represent mean ± SD.
(B) mTOR pull-down analyzed for interaction with gephyrin by western blot in αTC1 cells treated with control DMSO or 10 μM artemether for 72 hr.
(C) Expression change of genes involved in synaptic transmission in αTC1 cells treated with 10 μM artemether for 72 hr.
(D) Basal nuclear Fura-2 ratio levels (F340/F380) in αTC1 cells treated with 10 μM artemether versus control DMSO for 72 hr (boxplots showing 10th, 25th, 50th, 75th and 90th percentiles; ∗∗∗p < 0.001, Mann-Whitney rank sum test).
(E) Basal cytoplasmic Fura-2 ratio levels (F340/F380) in αTC1 cells treated with 10 μM artemether versus control DMSO for 72 hr (boxplots showing 10th, 25th, 50th, 75th and 90th percentiles; ∗∗∗p < 0.001, Mann-Whitney rank sum test).
(F) Analysis of chloride changes upon muscimol (100 μM) application in αTC1 cells treated with 10 μM artemether versus control DMSO for 72 hr (data presented as boxplots).
(G) Voltage-clamp current recordings upon 100 ms polarization steps in control αTC1 cells in pseudophysiological conditions with K-gluconate in the patch pipette (left panel) and with CsCl-based internal solution (right panel); pink traces show the presence of voltage-gated slowly inactivated inward currents.
(H) Current clamp recordings in αTC1 cells; the representative traces demonstrate similar “firing” properties upon 5 pA current injection (1 s).
(I) Quantification of insulin intensity in αTC1 cells treated with GABA-receptor antagonists bicuculline (10 μM; ∗∗∗p = 0.0001), furosemide (12.5 μM; ∗∗p = 0.0014) or FG-7142 (50 μM; ∗p = 0.043) alone or in combination with 10 μM artemether for 72 hr (Mean and SD from 4 wells).
Figure 3.

Artemether Increases GABA-Receptor Signaling in α Cells
(A) Western blot for GABA-receptor subunits in αTC1 cells treated with 1 μM artemether for 72 hr.
(B) Representative immunofluorescence images of αTC1 cells treated with 10 μM artemether or control DMSO for 72 hr. Scale bars, 5 μm.
(C) GO term enrichment analysis of RNA-seq data from αTC1 cells treated with 10 μM artemether versus control DMSO for 72 hr.
(D) Representative calcium responses to depolarizing solutions containing 15 and 50 mM KCl of αTC1 cells pre-treated with 10 μM artemether or control DMSO for 72 hr.
(E) Basal Fura-2 ratio levels (F340/F380) in αTC1 cells treated with 10 μM artemether versus control DMSO for 72 hr (boxplots show 10th, 25th, 50th, 75th, and 90th percentiles). ∗∗∗p < 0.001, Mann-Whitney rank-sum test.
(F) Percentage of αTC1 cells generating detectable calcium responses upon 15 mM KCl containing solution (mean ± SEM). ∗p < 0.05 (n = 6 + 6).
(G) Average calcium responses to the solution containing 50 mM KCl (mean ± 99% confidence interval).
(H) Dot-density plots of mean Δratio level after stimulation by 15 mM KCl application without or with 100 μM muscimol in αTC1 cells pre-treated with 10 μM artemether alone, 6.25 μM picrotoxin alone, or the combination of the two for 72 hr. ∗∗∗p < 0.001, Wilcoxon signed-rank test.
(I) Intracellular chloride intensity in αTC1 cells treated with 10 μM artemether for 72 hr by live-cell fluorescence imaging (n = 3). ∗p < 0.001. Error bars represent mean ± SD.
(J) Quantification of insulin intensity in αTC1 cells treated with GABA-receptor antagonist gabazine (10 μM, n = 6), picrotoxin (6.25 μM, n = 4), or salicylidene salycylhydrazide (5 μM, n = 4), alone or in combination with 10 μM artemether for 72 hr (data indicate means and SD). ∗p = 0.006; ∗∗p < 0.0001.
(K) Quantification of insulin intensity in αTC1 cells treated with 10 μM artemether or GABA-receptor agonist etifoxine (12.5 μM), muscimol (50 μM), vigabatrin (100 μM), halothane (20 μM), NS11394 (10 μM), zolpidem (6.25 μM), or gaboxadol (200 μM) for 72 hr (data indicate means and SD; n = 4). ∗p < 0.05; ∗∗p < 0.005.
See also Figure S4.
Next, we tested whether acute application of muscimol, a GABAA-receptor agonist, could potentiate KCl (15 mM)-induced responsiveness and whether it exhibited an additive effect with artemether. Indeed, muscimol-exposed cells showed further increases in their intracellular Ca2+ when simultaneously exposed to KCl. This effect significantly exceeded that of control cells (Figure 3H). Pre-incubation with the GABAA-receptor antagonist picrotoxin abolished these effects (Figure 3H), confirming GABAA-receptor involvement. Based on these results, we suggest that artemether pre-treatment resets intracellular Ca2+ through GABAA-receptor-mediated mechanisms and acutely modulates αTC1 cell excitability by potentiating GABAA-receptor signaling. Moreover, the data show that GABA can act as an excitatory stimulus in αTC1 cells. Since GABAA receptors gate Cl− ions, we sought to directly monitor intracellular Cl− concentrations by using MQAE dye imaging. 3-day pre-treatment with artemether induced an increase in intracellular Cl− (Figure 3I), which was not further changed when treated with muscimol acutely (Figure S4F). Similarly, we detected muscimol-induced transmembrane currents at the low-picoampere range, if any, in both control and in artemether-treated αTC1 cells acutely using perforated patch recordings (data not shown). Even though this might be seen as contradictory to a mechanism reliant on GABAA-receptor-gated currents, we argue that the high input resistance (>1 GΩ) of all recorded cells, together with the presence of VG Ca2+ currents operating at the picoampere range (Figure S4G, pink shading), and with high-amplitude delayed-rectifier K+ channels (Figure S4G), might be sufficient to induce membrane potential oscillations because of the initial activation of VG Ca2+ channels (depolarization) followed by the activation of VG K+ channels (repolarization) (Figure S4H). Accordingly, the injection of minute currents (up to 5 pA) in current-clamp mode was sufficient to induce quasi-action potentials, which we expect to be responsible for the generation of Ca2+ transients regulating exocytosis. Thus, artemether treatment can evoke global differences in αTC1 cell excitability and hormone release through the potentiation of functional GABAA receptors. To prove that GABAA-receptor signaling plays a functional role in the mechanism of action of artemisinins in αTC1 cells, we combined artemether with structurally and mechanistically distinct GABAA-receptor antagonists (Figures 3J and S4I). While these compounds had different effects on baseline insulin expression by themselves, all compounds partially inhibited the effects of artemether on insulin expression. We next tested whether treatment with different GABAA-receptor agonists alone was sufficient to induce insulin expression in αTC1 cells (Figure 3K). Most compounds—particularly, the positive allosteric modulators etifoxine and NS11394—induced insulin expression, but all GABA-receptor agonists were less potent relative to artemether.
Hormone Secretion Controls Pancreatic Cell-type Stability
GABA is normally co-secreted with insulin by β cells, and suppressing glucagon secretion is one of the physiological functions of GABA signaling in α cells (Li et al., 2013, Rorsman et al., 1989, Wendt et al., 2004, Xu et al., 2006). In αTC1 cells, artemether treatment inhibited glucagon secretion in low-glucose conditions (Figure 4A). Interestingly, supplementing recombinant glucagon abolished the effect of artemether on insulin induction (Figure 4B). These data imply that maintaining the correct concentration of glucagon in close vicinity to α cells is critical for the stability of this cell type. In order to lower the extracellular glucagon concentration, we applied Decanoyl-RVKR-CMK (PCi), a highly potent, irreversible, cell-permeable inhibitor of proprotein convertases that inactivates prohormone convertase 2 at subnanomolar concentrations (Denault et al., 1995). Treatment of αTC1 cells with this compound resulted in a dramatic increase of intracellular glucagon staining, consistent with the accumulation of unprocessed proglucagon (Figure 4C). At the same time, these α cells stained positive for insulin protein (Figure 4C). On the transcriptional level, the decrease of glucagon secretion repressed glucagon expression and significantly upregulated the expression of β cell-specific transcription factors Pax4 and Mnx1 (Figure 4D; Table S2). These results suggest that artemether-mediated amplification of GABA-receptor signaling prevents glucagon secretion, thereby reducing extracellular glucagon and triggering the loss of α cell identity.
Figure 4.

Hormone Secretion Controls Pancreatic Cell-type Stability
(A) ELISA for secreted glucagon in αTC1 cells treated with 10 μM artemether for 72 hr (n = 4). ∗p < 0.05. Error bars represent mean ± SD.
(B) Immunofluorescence quantification of insulin intensity in αTC1 cells with treated with 10 μM artemether and 10 ng/ml glucagon for 72 hr (n = 4). ∗p < 0.001. Error bars represent mean ± SD.
(C) Representative images of insulin and glucagon immunofluorescence in αTC1 cells treated with 10 μM prohormone convertase inhibitor (PCi) for 72 hr. Scale bars, 5 μm.
(D) qRT-PCR of islet genes in αTC1 cells following treatment with 10 μM PCi for 72 hr. All data were normalized to DMSO control (n = 3). ∗p = 0.010; ∗∗p = 0.019; ∗∗∗p = 0.002; ∗∗∗∗p < 0.001. Error bars represent mean ± SD.
Artemether Increases β Cell Mass In Vivo
To characterize the physiological relevance of our findings, we applied artemether in vivo in three model organisms. For developmental effects of the compound, we studied zebrafish carrying a GFP transgene under the glucagon promoter and an mCherry construct controlled by the insulin promoter. 5-day treatment of zebrafish larvae with artemether altered islet morphology and caused a dose-dependent reduction in α cell numbers, with β cell numbers affected at higher concentrations (Figures S5A and S5B). Next, we applied the compound in a zebrafish β cell ablation model at a later developmental stage. In animals expressing an insulin-promoter-driven caspase-8 transgene that can be activated pharmacologically, β cells were completely ablated by the addition of a small-molecule dimerizer at 3 days post-fertilization (dpf) for 48 hr. The expression of a far-red fluorophore (E2crimson) under the control of the insulin promoter allowed the analysis of β cell numbers at 12 dpf (Figures 5A and 5B). Based on the known regenerative capacity of zebrafish, control ablated animals had an average of eight reestablished β cells at this time point. In animals ablated and treated with artemether for 4 days (8–12 dpf), we observed a 75% increase in β cell number to 14 cells per animal on average. Next, we analyzed the effects of β cell ablation and artemether on glucose levels in zebrafish. While β cell ablation resulted in a 34% increase in glucose levels 7 days post-ablation, artemether treatment after ablation inversed that phenotype, and animals had 42% lower glucose levels compared to unablated controls (Figure 5C). In addition to these developmental models, we also sought to study artemisinin effects on mature islets in wild-type mice. In line with in vitro data, we observed an increase in pancreatic islet size following 3-month treatment with artesunate in drinking water (Figures 5D and 5E). These changes occurred in the absence of a dramatic shift in islet subpopulations or cell proliferation (Figure S5C), thereby phenocopying Arx loss in vivo (Courtney et al., 2013). To confirm α cells as the source of new insulin-positive cells, we turned to a lineage-tracing model. We isolated islets from mice harboring a construct for Cre recombinase under the control of the glucagon promoter and a floxed red fluorescent protein (RFP) allele integrated in the Rosa26 locus. Thereby, all cells that have ever activated glucagon expression are permanently labeled RFP positive. In control DMSO-treated islets, we observed that only a background of approximately 1% cells were double positive for insulin and RFP. Treatment of these islets for 24 hr with artemether resulted in 4-fold upregulation of this cell population (Figures 5F and 5G). These data indicate that artemether induces a rapid induction of insulin expression in α cells within mouse islets, resulting in long-term expansion of the insulin-producing cell population and islet mass. Finally, to analyze the effects of artemether treatment in a rodent diabetes model, we ablated β cells in rats with a single high-dose streptozotocin injection. Animals were randomized to treatment and control groups based on matching blood glucose measurements on day 9 post-ablation and then treated orally with artemether or control vehicle for 23 days. After overnight fasting, baseline blood glucose levels in artemether-treated animals were drastically reduced compared to those of control rats (Figure 5H). These lower baseline glucose levels were also reflected in an oral glucose tolerance test performed in the two groups (Figures 5I and 5J).
Figure S5.

Effect of Artemether on Pancreatic Islets In Vivo, Related to Figure 5
(A) Islet morphology and GFP expression as marker of α cells of Tg(Gcga:GFP)ia1 zebrafish larvae treated from 26 hpf until 100 hpf with artemether.
(B) Quantification of Glucagon-GFP positive cells and Insulin-mCherry positive cells per single zebrafish islets (8-10 fish per condition; ∗p = 0.007, ∗∗p = 0.088, ∗∗∗p = 0.004, ∗∗∗∗p = 0.003). Error bars represent mean ± SD.
(C) Immunofluorescence for Sst, Pp, and BrdU co-stained with insulin or glucagon in mouse pancreas with or without artemether treatment.
Figure 5.

Artemether Increases β Cell Mass In Vivo
(A) Representative images of insulin reporter cells in zebrafish larvae. β cells in ins:caspase8;ins:E2Crimson embryos were ablated by treatment with 2 μM dimerizer AP20187 from 3 dpf to 5 dpf. Then, larvae were treated with 5 μM artemether or control DMSO for 4 days (8–12 dpf) and immunostained for dsRed and DAPI. Scale bar, 15 μm.
(B) Quantification of numbers of insulin reporter cells in zebrafish larvae described in (A). Shown are cell numbers from individual animals, and the means and SD are indicated for the three groups. ∗p = 0.0005.
(C) Glucose measurement in pooled larvae extracts, treated as in (A) and normalized to non-ablated animals (means and SD are indicated for at least four independent larvae pools for each condition).
(D) Representative staining for insulin and glucagon in mouse pancreas following a 3-month treatment with 1 mg/mL artesunate in drinking water versus control DMSO. Scale bar, 30 μm.
(E) Quantification of islets size in sections from mouse pancreata following a 3-month treatment with artesunate in drinking water (120–180 islets per animal). ∗p < 0.001. Shown are medians, 25%–75% (boxes) and 10%–90% confidence intervals.
(F) Co-staining of RFP and insulin in RFP-labeled lineage-tracing mouse islets after treatment with 10 μM artemether or control DMSO for 24 hr. Scale bar, 10 μm.
(G) Quantification of RFP/insulin double-positive cells in mouse islet from (F) (means and SD are indicated from two replicates). ∗p = 0.019.
(H) Blood glucose levels after an overnight fast in a rat β cell ablation model. β cells were ablated with 60 mg/kg streptozotocin; on day 9 post-ablation, animals were assigned to treatment and control groups based on matching fasting glucose levels and were treated with 20 mg/kg artemether p.o. for 7 days followed by 200 mg/kg artemether p.o. for 16 days. (Measured values in individual animals, means and SD, n = 10 per group, p = 0.005).
(I) Oral glucose tolerance test of animals described in (H). Means and SE are indicated for ten animals from each group.
(J) Area under the curve from the oral glucose tolerance test described in (I). Shown are measurements in individual animals, and the means and SD are indicated for the groups; p = 0.004.
See also Figure S5.
Single-Cell Transcriptomics Reveals Artemether Effects in Human α Cells
We next assessed the effects of artemether on α cells in primary human pancreatic islets (Figure 6A; Table S3). The drug caused a rapid translocation of ARX to the cytoplasm and a reduction in the staining intensity for this master regulatory transcription factor (Figure 6A). To better visualize the artemether-induced translocation of ARX to the cytoplasm, we treated intact islets and then dissociated them to single cells immediately prior to immunofluorescence staining (Figure 6B). We observed that active GABAA-receptor signaling was a requirement for the translocation of ARX to the cytoplasm following artemether treatment, since co-treatment with artemether and the GABAA-receptor antagonist picrotoxin maintained nuclear ARX localization (Figure 6C). We then studied the effect of artemether on hormone levels and gene expression in primary human islets. Compared to control DMSO-treated islets, artemether increased the number of double-positive cells expressing both insulin and glucagon (Figures S6A–S6C). The effects of artemether or GABA on global transcriptional profiles were tested in bulk samples containing 500 cells. Gene set enrichment analysis (GSEA) revealed significantly decreased expression of exocrine-specific genes (Li et al., 2016), both with artemether and GABA (Figure S7A; Table S1). In contrast, genes upregulated by both artemether and GABA significantly overlapped with an endocrine, but not an exocrine, gene set (Figure S7B). To investigate the cell-type-specific gene expression changes with artemether treatment, we performed single-cell transcriptomics on human primary islets. We treated intact islets with artemether or DMSO, dissociated the islets immediately prior to sorting single cells to individual wells of a 96-well plate containing lysis buffer, and performed single-cell RNA-seq as previously described (Li et al., 2016). We then assessed global transcriptomic alikeness between individual cells through multidimensional scaling (Figure 6D). We observed that compound-treated cells clustered with DMSO-treated controls, indicating that up to 72 hr of in vitro treatment with artemether did not induce a whole new cell type. Looking for changes in specific genes related to the compounds’ mechanism, we found that GABRB3 and GABRG2 were strongly upregulated in artemether-treated α cells (Figure 6E), consistent with the findings in the mouse cell line. An increase in gephyrin and GABA-receptor subunit protein levels on some islet cells was also evident by immunofluorescence staining (Figure 6F). To analyze transcriptome-wide changes, we used GSEA to identify differentially regulated pathways in DMSO- versus artemether-treated single-cell transcriptomes. In β cells, artemether downregulated some cell-type-specific transcripts (Figures S7C and S7D). In α cells, we observed a strong and significant downregulation of α cell-specific genes, including EIF4A1, CRYBA2, PDK4, and MUC13 following artemether treatment (Figures 6G and 6H). Key β cell-specific genes were upregulated in α cells following artemether treatment, including GNAS and ABCC8, the gene coding for the sulfonylurea receptor (Figures S7E and S7F).
Figure 6.

Artemether Affects α Cell Identity in Human α Cells
(A) Representative immunofluorescence images of sectioned human islets treated with 10 μM artemether and control DMSO for 3 hr, co-stained for nuclei (DAPI), ARX, glucagon, or C-peptide. Scale bars, 100 μm.
(B) Representative images of dissociated human islet cells stained for ARX after treatment with 10 μM artemether and/or 6.25 μM picrotoxin for 3 hr. Scale bars, 5 μm.
(C) Quantification of ARX localization from human islets cells treated as in (B) (data indicate means and SD from six imaged wells). Boxes indicate 25th and 75th percentiles, horizontal lines medians, and whiskers minima and maxima.
(D) Multidimensional scaling plot representing transcriptomes of single human islet cells treated as intact islets with either control DMSO (gray circles) or 10 μM artemether (black circles) for 24 hr or 72 hr.
(E) Expression pattern of GABA-receptor subunits in human α cells from (D).
(F) Immunofluorescence staining of dissociated human pancreatic islet cells for nuclei (DAPI), gephyrin, and GABRG2 receptor treated as intact islets with either control DMSO or 10 μM artemether for 72 hr. Scale bar, 10 μm.
(G) Gene set enrichment analysis using a gene set representing all genes significantly higher expressed in human α cells compared to β cells, comparing cumulative expression levels from single α cell transcriptomes obtained from islets treated with control DMSO or 10 μM artemether for 72 hr.
(H) Cumulative expression levels from single α or β cell transcriptomes obtained from islets treated with control DMSO or 10 μM artemether for 72 hr.
Figure S6.

Artemether-Induced Insulin/Glucagon Double-Positive Cells in Human Pancreatic Islets, Related to Figure 6
(A) Detection of insulin/glucagon double positive cells following treatment of intact human islets with 10 μM artemether for 72 hr (scale bar 10 μm).
(B) Quantification of cell fractions in human islets treated with 10 μM artemether for 72 hr by individual donor.
(C) Summary of data in B. (∗p < 0.001, n = 5). Error bars represent mean ± SD.
Figure S7.

Artemether Alters Human Pancreatic Islet Transcriptomes, Related to Figure 6
(A) GSEA in bulk human islet samples treated with 10 μM artemether or 10 mM GABA for 72 hr.
(B) Overlap between the gene set upregulated by both GABA and artemether (core-upregulation) and exocrine or endocrine gene sets. Fisher’s exact test is used for the significance test.
(C) GSEA for α cell specific genes in single human β cells from islets treated with artemether.
(D) Repression of β cell genes in artmether treated beta cells.
(E) Induction of expression of GNAS in human α cells after treatment of intact islets with 10 μM artemether for 72 hr.
(F) Induction of expression of ABCC8 in human α cells after treatment of intact islets with 10 μM artemether for 72 hr.
Artemether Improves Insulin Secretion in Human Islets
We next assessed whether the reduction in α cell character and gain of β cell characteristics following artemether treatment had functional consequences in intact human islets. First, we tested the expression of α cell factor ARX (Figure 7A). While we observed donor-to-donor variability of the effect size, ARX expression was consistently reduced. We then investigated whether the loss in α cell character corresponded to increased β cell properties. Normalized to total protein, artemether-treated islets contained more intracellular insulin compared to DMSO-treated islets from the same donor (Figure 7B). Finally, we tested whether this insulin increase was functional and performed glucose stimulated insulin secretion assays. While artemether treatment did not affect secretion in low-glucose conditions, artemether-treated islets from all donors tested secreted significantly more insulin compared to DMSO-control-treated islets when challenged with high-glucose concentrations (Figure 7C).
Figure 7.

Artemether Enhances Insulin Secretion in Human Islets
(A) RT-qPCR assay for ARX expression in human islets treated with control DMSO or 10 μM artemether for 72 hr. Error bars represent mean ± SD.
(B) Measurement of intracellular insulin content in human islets treated with control DMSO or 10 μM artemether for 72 hr. Error bars represent mean ± SD.
(C) Measurement of glucose-stimulated insulin secretion in human islets treated with control DMSO or 10 μM artemether for 72 hr (two replicates each donor; ∗∗p < 0.005; ∗p ≤ 0.05). Error bars represent mean ± SD.
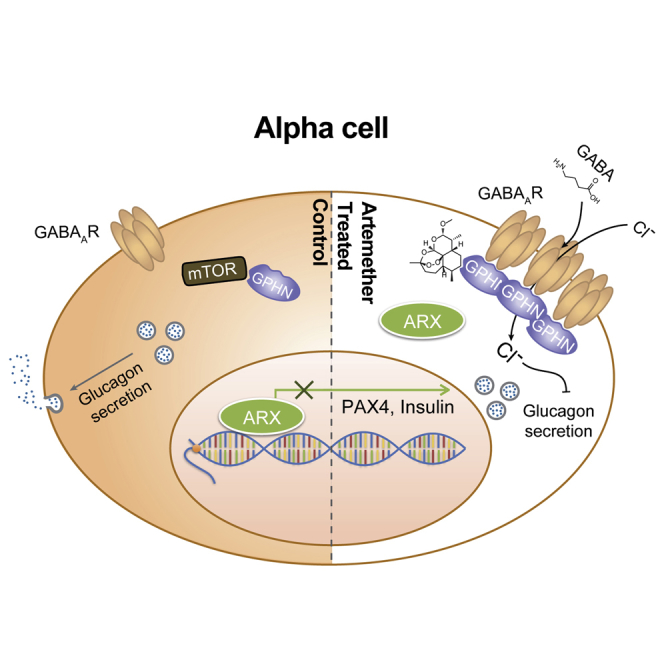
(D) Proposed mechanism of action of artemether. By stabilizing gephyrin, artemether increases GABA-receptor signaling in α cells, thereby preventing glucagon secretion. Decreased extracellular glucagon concentration induces loss of α cell identity and increase of insulin expression.
See also Figure S6.
Discussion
We identified artemisinins as approved drugs that can confer β cell characteristics to α cells. This phenotype depends on the stabilization of the protein gephyrin, which, in turn, activates GABA signaling in α cells. Active GABA signaling is required for the effects of artemisinins on the key α cell-fate determining transcription factor ARX, which translocates to the cytoplasm following drug treatment (Figure 7D). It is tempting to speculate that the changes in nuclear calcium (and/or chloride) signaling that we observed provide a direct link between extracellular glucagon, GABA-receptor signaling, and nuclear ARX localization.
Previous studies have already connected α cells’ key function of glucagon secretion to GABA-receptor signaling. GABA is primarily produced by β cells, whereas several GABAA-receptor subunits are specifically expressed on α cells (Franklin and Wollheim, 2004, Wan et al., 2015). The architecture of the vasculature in islets ensures that arterioles first contact insulin-expressing cells (Bonner-Weir and Orci, 1982, Stagner and Samols, 1992), resulting in the exposure of the downstream α cells to high local concentrations of factors secreted by β cells, including insulin and GABA. Insulin is known as a major repressor of glucagon secretion (Maruyama et al., 1984), and the action of the hormone is dependent on active Akt and GABA signaling (Xu et al., 2006). Our findings suggest that GABA signaling not only affects α cell function, as measured by glucagon secretion, but also feeds back to α cell identity. Thus, we propose an additional beneficial effect of activating GABA-receptor signaling in diabetes, beyond the modulation of the immune system and β cell proliferation previously suggested (Purwana et al., 2014, Soltani et al., 2011).
A key remaining question concerns the lack of α to β cell transdifferentiation in diabetes. Why would evolution equip α cells with a high amount of epigenetic plasticity, so that loss of a single transcription factor is sufficient for their transdifferentiation, and why would a mechanism to exclude this very transcription factor from the nucleus be maintained? However, the only conditions known, when α cells convert into β cells, are following the genetic manipulation of these master regulatory transcription factors (Collombat et al., 2009, Courtney et al., 2013) and a toxin-induced model of near-complete β cell loss (Thorel et al., 2011). Particularly in type-2-diabetic conditions, several of the factors observed to contribute to ARX repression are altered in similar ways to the changes we observe with artemisinins. For example, GABA receptors accumulate on the surface of α cells in high-glucose/high-insulin conditions (Xu et al., 2006). Moreover, long-term treatment with GABA alone is sufficient to induce α cell-mediated, β-like cell neogenesis in a mouse model (Ben-Othman et al., 2016; this issue of Cell). However, we still only have a limited understanding of the intra-islet changes associated with diabetic conditions, including local concentrations of GABA, insulin, and glucagon, as well as the composition of GABA receptors and other factors affecting the autocrine and paracrine effects of these molecules. Insufficient β cell-derived GABA levels and inappropriate glucagon secretion in diabetic high-glucose conditions could, therefore, prevent endogenous α cell transdifferentiation in diabetic conditions. In our α cell-line model, GABA-receptor agonists induced lower levels of insulin compared to artemether. Likely, the strong transcriptional induction of several GABA-receptor subunits following drug treatment induces a specific GABA-receptor subunit composition not normally present in α cells. These receptors then increase GABA responsiveness and alter baseline calcium signaling of artemether-treated α cells, and the effects could potentially synergize with other aspects of gephyrin biology and PI3K signaling.
Artemisinin combination therapy is the treatment of choice for malaria and more than 300 million doses are dispensed annually (Dondorp et al., 2010). Despite this large patient cohort, no clinical data on the effects of artemisinins on human pancreatic endocrine function are available. There are several reasons why such changes might have gone unnoticed so far. First, the acute life-threatening condition of plasmodium-infected patients, together with the known propensity of plasmodium infection to cause hypoglycemia, makes blood glucose levels highly variable in the short term. Second, in healthy individuals, a small increase in β cell number is not expected to cause a phenotype, as these cells secrete insulin in a highly glucose-regulated manner. Unfortunately, currently no imaging methods are available to directly assess human β cell mass. Third, treatment duration with artemisinins might be critical for effects on α cells. We observe rapid effects of these compounds on cell lines, isolated islets, and zebrafish larvae in the timescale of hours to days. In contrast, effects in rodent models take weeks to months, potentially because of the lower bioavailability of artemisinins in vivo. Plasma half-lives of these compounds are in the range of minutes to a few hours, and the poor solubility in aqueous solutions precludes the continuous dosing using pumps; hence, we rely on once-daily oral dosing in rats. In contrast, for malaria patients, a typical treatment cycle with artemisinins lasts only 4 days, but dosing occurs at relatively high concentrations every 8 hr. Currently, clinical studies are ongoing for the long-term effects of artemisinins in cancer patients (Ericsson et al., 2014) (NCT02263950). Therefore, investigation of the effects of long-term artemether treatment for the induction of endogenous insulin expression in C-peptide-negative type 1 diabetes patients appears clinically feasible.
Regardless of the direct clinical use of artemether in diabetes, our findings open up completely new avenues for transdifferentiating α cells by structurally different gephyrin stabilizers, GABA-receptor transcriptional activators, and additional targets in the GABA-receptor signaling pathway, toward a treatment for type 1 diabetes.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Insulin | Sigma | Cat#18510 |
| Dako | Cat#A056401; RRID: AB_2617169 | |
| Linko | Cat#4011-01 | |
| Glucagon | Abcam | Cat#ab92517; RRID: AB_10561971 |
| Arx | R&D | Cat#AF7068; RRID: AB_10973178 |
| Myc-Tag | Cell Signaling Technology | Cat#2276 |
| Histone H2B | Cell Signaling Technology | Cat#2934 |
| Gephyrin | Abcam | Cat#ab25784; RRID: AB_1209349 |
| Gephyrin (Figure S4B) | Synaptic systems | Cat#147111; RRID: AB_887719 |
| GABA-receptor subunit beta 3/4 | Millipore | Cat#MAB341; RRID: AB_2109419 |
| GABA-receptor subunit alpha 2 | Abcam | Cat#ab176170 |
| GABA-receptor subunit gamma 2 | Abcam | Cat#ab101036; RRID: AB_10672116 |
| mTOR | Cell Signaling Technology | Cat#2972 |
| p-mTOR | Cell Signaling Technology | Cat#5536 |
| Akt | Cell Signaling Technology | Cat#4691 |
| p-Akt | Cell Signaling Technology | Cat#4060P; RRID: AB_972551 |
| Actin | Sigma | Cat#A2066; RRID: AB_476693 |
| Alpha-Tubulin | Abcam | Cat#ab7291; RRID: AB_2241126 |
| dsRed | Clontech | Cat#632496; RRID: AB_10013483 |
| Goat Anti-rabbit 633 | Thermo Fischer | Cat#A-21070; RRID: AB_2535731 |
| Goat Anti-Guinea Pig 546 | Thermo Fischer | Cat#A-11074; RRID: AB_2534118 |
| Goat Anti-Guinea Pig 488 | Thermo Fischer | Cat#A-11073; RRID: AB_2534117 |
| Donkey Anti-Rabbit 488 | Thermo Fischer | Cat#A-21206; RRID: AB_2535792 |
| Donkey Anti-Mouse 488 | Thermo Fischer | Cat#A-21202; RRID: AB_2535788 |
| Donkey Anti-Sheep 546 | Thermo Fischer | Cat#A-21098; RRID: AB_2535752 |
| Donkey anti-Rabbit 546 | Thermo Fischer | Cat#A-10040/A-10036/A-10074; RRID: AB_2534016 |
| Rabbit anti-sheep IgG-HRP | Santa Cruz | Cat#SC-2770; RRID: AB_656968 |
| Donkey Anti-Rabbit HRP | Jackson ImmunoResearch | Cat#711-035-152; RRID: AB_10015282 |
| Donkey Anti-Mouse HRP | Jackson ImmunoResearch | Cat#715-035-151; RRID: AB_2340771 |
| Donkey Anti-Sheep HRP | Jackson ImmunoResearch | Cat#713-035-147; RRID: AB_2340771 |
| Donkey Anti-Goat HRP | Jackson ImmunoResearch | Cat#705-035-147; RRID: AB_2313587 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Artemether | Sigma | Cat#A9361 |
| Artemether (Figures 5H–5J) | Cayman Europe | Cat#11815 |
| Bicuculline | Tocris | Cat#0130 |
| Gabazine | Sigma | Cat#S106 |
| Picrotoxin | Sigma | Cat#P1675 |
| Salicylidene salicylhydrazide | Sigma | Cat#S8194 |
| Etifoxine | Sigma | Cat#SML0272 |
| Muscimol | Sigma | Cat#M1523 |
| Vgabatrin | Sigma | Cat#V8261 |
| Halothane | MedChemExpress | Cat#HY-B1010 |
| NS11394 | MedChemExpress | Cat#HY-11048 |
| Zolpidem | Sigma | Cat#E006 |
| Thapsigargin | Sigma | Cat#T9033 |
| Gaboxadol | Santa Cruz | Cat#sc204342 |
| Furosemide | Tocris | Cat#3109 |
| FG-7142 | Sigma | Cat#E006 |
| Elesclomol | Selleck | Cat#S1052 |
| Nacetylcysteine | Sigma | Cat#A9165 |
| Lynxtag_art | Biolynx Technologies | Cat#BL007RUO |
| PCi | Enzo Life Sciences | Cat#ALX-260-022-M001 |
| Artesunate | Sigma | Cat#A3731 |
| Artesunate (Figures 5D and E) | TCI | Cat#A2191 |
| Deoxyarteether | Santa Cruz | Cat#sc-211235 |
| Arteether | Santa Cruz | Cat#sc-217662 |
| N-hydroxy-11-azaartemisinin | TRC | Cat#H804500 |
| Artemisitene | TRC | Cat#A777550 |
| Artemisone | Alsachim | Cat#C1740 |
| Artemisiten | Alsachim | Cat#S1873 |
| Dihydro artemisinin | TRC | Cat#A637875 |
| Recombinant glucagon | Sigma | Cat#G2044 |
| Dimerizer AP20187 | Clontech | Cat#635059 |
| pE2-Crimson-N1 Vector | Clontech | Cat#632554 |
| Methylcellulose | Sigma | Cat#M6385 |
| Hoechst 33342 | Thermo Fischer | Cat#H1399 |
| Critical Commercial Assays | ||
| Xanthine Oxidase Assay Kit | Abcam | Cat#ab102522 |
| PI(3)P Mass ELISA kit | Echelon | Cat#K-3300 |
| Glucagon ELISA | Alpco | Cat#48-GLUHU-E01-AL |
| Insulin ELISA | Alpco | Cat#80-INSHU-E01.1 |
| Glucose Assay Kit | Biovision | Cat#K606-100 |
| Deposited Data | ||
| Single cell RNA-seq of the control cells | NCBI Gene Expression Omnibus | GEO: GSE73727 |
| Single cell RNA-seq of the artemether treated cells | NCBI Gene Expression Omnibus | GEO: GSE84714 |
| RNA-seq of bulk samples | NCBI Gene Expression Omnibus | GEO: GSE84592 |
| Experimental Models: Cell Lines | ||
| αTC1 | Novo Nordisk | ATCC Cat#CRL-2934 |
| αTC1-LT | This paper | N/A |
| Min6 | Novo Nordisk | Miyazaki et al., 1990 |
| Min6-ARX | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| (Gcga:GFP)ia1- (Ins:NTR-mcherry)ml10 Zebrafish | This paper | N/A |
| Ins:caspase8;ins:E2Crimson Zebrafish | This paper | N.S. and D.M., unpublished data |
| GluCre::RFP mouse | This paper | N/A |
| Recombinant DNA | ||
| TetR-CMV-ARX-MYC-FLAG | This paper | N/A |
| SV40 large T-antigen lentivirus | GenTarget | Cat#LVP016-Neo |
| Sequence-Based Reagents | ||
| Primers for Ins2, Actb, Pax4, ACTB | Fomina-Yadlin et al., 2010 | N/A |
| Primers for Mnx1, Gcg, ARX | This paper | Table S2 |
| shGephyrin | Sigma | Cat#TRCN0000098585 |
| shControl | Sigma | Cat#SHC001 |
| Software and Algorithms | ||
| Tophat v2.0.4 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| RSeQC package | Wang et al., 2012 | http://rseqc.sourceforge.net/ |
| Gene Set Enrichment Analysis | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/downloads.jsp |
| Mootha et al., 2003 | ||
| ImageJ/FIJI | Schneider et al., 2012 | https://fiji.sc/ |
| Cell Counter Plugin | Kurt De Vos, University of Sheffield, Academic Neurology | https://imagej.nih.gov/ij/plugins/cell-counter.html |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact Stefan Kubicek (skubicek@cemm.oeaw.ac.at).
Experimental Model and Subject Details
Cell Culture
αTC1 cells were purchased from ATCC (CRL-2934) and grown in low-glucose DMEM supplemented with 10% FBS, 50 U/mL penicillin and 50 μg/mL streptomycin. Min6 cells with doxycycline-inducible constructs were grown in high-glucose DMEM supplemented with 15% Tet System Approved FBS (Clonetech 631106), 71 μM 2-mercaptoethanol, 50 U/ml penicillin and 50 μg/ml streptomycin. The mouse islets from the α cell lineage-tracing animals were kept in RPMI medium supplemented with 10% FBS, 50 U/ml penicillin and 50 μg/ml streptomycin. The cell culture for human islets followed established protocols (Walpita et al., 2012).
Mice and rats
All mouse work was conducted in Patrick Collombat’s lab as previously described (Courtney et al., 2013) according to French ethical regulations under the animal license NCE/2012-85 and NCE/2014-190. Rat experiments were conducted at Physiogenex according to the standard operating procedure in place at the test facility and an agreement from the French veterinary services. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (revised 1996 and 2011, 2010/63/EU) and French laws.
Zebrafish
Transgenic (Gcga:GFP)ia1-(ins:NTR-mcherry)ml10 zebrafish were maintained according to the guidelines of the local authorities under licenses GZ:565304/2014/6 and GZ:534619/2014/4. Transgenic ins:caspase8;ins:E2Crimson (abbreviated ins:casp8) zebrafish embryos were generated under license BMWF-66.008/0017-II/3b/2013. The ages of the zebrafish are described in the method details.
Human islets
Human islets were obtained from the European Consortium for Islet Transplantation (Geneva) and the Integrated Islet Distribution Program IIDP. This study is approved by the Ethics Committee of the Medical University of Vienna (EK-Nr. 1228/2015). Detailed donor information is provided in Table S3.
Method Details
Establishment of inducible cell lines
To generate inducible Min6 cells, we first transduced cells with a lentivirus carrying TetR DNA and a neomycin resistance cassette (Gentarget, cat.# LVP017-Neo) and then isolated a single clone expressing high amounts of TetR protein, validated with the Tet01 antibody (MoBiTec). This TetR expressing cell line was then transduced with virus particles coding for Flag-Myc tagged (C-terminal) human ARX cDNA also expressing the puromycin resistance gene. The vector used for this work was pSTK007, a modified version of the pLenti4.2 (Life technologies). Then, a second round of clonal selection was performed where numerous clones were tested for maximum repression in the uninduced state and a high degree of induction upon treatment with doxycyline (1μg/ml).
All lentiviruses were produced in HEK293T cells using the Virapower plasmid mix (Life technologies), according to the manufacturer’s protocol. All virus work was done in a Class II laboratory, and before transferring the cells to a regular lab the cells were proven to be virus free using a p24 ELISA kit (Cell Biolabs, cat.#VPK-108-H).
To establish the αTC1 subclone used for all experiments described in this study, αTC1 cells were infected with a lentiviral vector causing the expression of large T antigen under the CMV promoter to make them independent of the glucagon-promoter driven large T antigen expression.
High-throughput screening and immunofluorescence staining
50 nL compound per well was transferred to black optical suitable 384-well plate (Corning 3712) from DMSO stock plates using acoustic transfer (Labcyte Echo 520). 3000 Min6 cells per well were then plated in 50 μl media containing 1 μg/ml doxycycline on top of the compounds. Three days after seeding, cells were fixed in 3.7% formaldehyde for ten minutes at room temperature. Following washing with PBS, cells were fixed with cold pure methanol at −20°C for 10 min, permeabilized with 1% Triton X-100 in PBS for 30 min and blocked with 3% BSA in PBS for 30 min. 20 μl primary anti-insulin antibody (1:2000) in 1.5% BSA/PBS, was added per well and incubated at 4°C overnight. After washing with PBS twice, 20 μl Cy-3–labeled donkey-α-guinea pig antibody (1:1000) and 10 μg/ml Hoechst 3342 in PBS was added per well and incubated for 1 hr. After two washes with PBS, plates were stored in 4°C in dark until analysis.
Images were taken by an automated microscope (Perkin Elmer Operetta) using a 20x objective. Images were exposed for 10 ms in the Hoechst channel and for 500 ms in the Alexa Fluor 546 channel. Images were analyzed using the Harmony software (Perkin Elmer). Nuclei were identified (Harmony Method C) and cytoplasm was defined based on the nuclei (Harmony Method C). In total 1152 wells were screened containing 280 compounds from the CeMM Library of Unique Drugs (CLOUD) (M. Licciardello, P. Markt, F. Klepsch, and S.K., unpublished data) in triplicates with control wells. Hits were selected based on the intensity of insulin in the Alexa Fluor 546 channel and cell numbers in the Hoechst channel. Immunofluorescence staining for all other antibodies was performed as described above, images were acquired on an automated microscope (Perkin Elmer Operetta) or a Leica DMI6000 B inverted confocal system with a 63x 1.30 ACS Apo lens. Image analysis and quantitation was performed as described above. For quantification, at least 1,000 cells were analyzed for each condition.
RNA-seq
Cells were lysed and RNA isolated using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. The amount of total RNA was quantified using a Qubit 2.0 Fluorometric Quantitation system (Life Technologies) and the RNA integrity number (RIN) was determined using an Experion Automated Electrophoresis System (Bio-Rad). The libraries for RNA-seq after 24h induction in Min6 cells were prepared with Ribo-zero and Scriptseq v2 kits (Epicenter) following the provider’s manual. Other RNA-seq libraries were prepared with TruSeq Stranded mRNA LT sample preparation kit (Illumina) using Sciclone and Zephyr liquid handling robotics (PerkinElmer). Library amount was quantified using the Qubit 2.0 Fluorometric Quantitation system (Life Technologies) and the size distribution was assessed using Experion Automated Electrophoresis System (Bio-Rad). Libraries were pooled and sequenced on the Illumina HiSeq platform using the 50 bp single-read configuration. Reads were aligned with tophat (v2.0.4) with the–no-novel-juncs–no-novel-indels options (Kim et al., 2013). Gene expression was calculated as Reads Per Kb per Millions of reads (RPKMs) using RPKM_count.py from the RSeQC package (Wang et al., 2012) and the NCBI RNA reference sequences collection (RefSeq) downloaded from UCSC. Sample preparation and data analysis for single cell RNA-seq was conducted as previously described (Li et al., 2016).
RT-qPCR
After the RNA was isolated with the RNeasy Mini Kit (QIAGEN), it was reverse transcribed with random primers using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems) on a Lightcycler 480 (Roche). Primer sequences used were published previously (Fomina-Yadlin et al., 2010), novel sequences are indicated in Table S2.
Ca2+ imaging
Ca2+ responses were measured using Fura-2AM (Invitrogen). Ratiometric imaging was performed using a VisiChrome monochromator (Visitron Systems) on a CoolSnap HQ2 camera (Photometrics) and recorded in VisiView software (Visitron Systems). Data were statistically analyzed and plotted using SigmaPlot 13.0 (Systat Software). Krebs-Ringer solution was used as basic external solution and contained: 119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 1.5 mM CaCl2, 1.5 mM MgCl2, 20 mM HEPES, 11 mM glucose (pH 7.4). A p value of < 0.05 was considered statistically significant, and calculated by the Mann-Whitney rank sum test (unpaired test) or Wilcoxon’s signed rank test (paired test), as appropriate. Experiments were performed at 25°C.
Patch-clamp electrophysiology
αTC1 cells were assayed by patch-clamp electrophysiology using the gramicidin or amphotericin perforated-patch configuration. Ionic currents and membrane potential were recorded, filtered, and analyzed using an Axopatch 200 B amplifier, DigiData1322 interface, and the pClamp8 software (Molecular Devices). The external solution contained: 119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 1.5 mM CaCl2, 1.5 mM MgSO4, 11 mM glucose and 20 mM HEPES-NaOH (pH 7.4). For the measurement of muscimol-induced currents the following intracellular solution was used: 140 mM CsCl, 4 mM NaCl, 2 mM MgCl2, 10 mM HEPES, 1 mM EGTA, 4 mM ATP-Na2, 400 mg/l amphotericin, (pH adjusted to 7.3 with NaOH). For studying excitable properties of αTC1 cells the intracellular solution contained: 114 mM K-gluconate, 6 mM KCl, 10 mM HEPES, 5 mM EGTA, 4 mM ATP-Mg, 30-50 mg/l gramicidin (pH adjusted to 7.3 with KOH). All experiments were performed at 25°C.
Chloride imaging
To assess muscimol induced Cl− changes, αTC1 cells were preincubated with MQAE for 0.5 hr (Thermo Fisher Scientific). The fluorescence was recorded with CoolSnap HQ2 camera (Photometrics) and VisiView software (Visitron Systems) using 350 nm excitation with VisiChrome monochromator (Visitron Systems) and the emission filter (445 ± 50 nm). Experiments were done at 25°C in Krebs-Ringer solution. Basal levels were measured after 1 hr incubation with MQAE and washing with PBS by live cell imaging on an automated microscope (Perkin Elmer Operetta) using a 20x objective and analyzed using the Harmony software (Perkin Elmer).
Moco synthase assay
αTC1 cells were pretreated with artemether for three days and lysed with NP-40 buffer. The measurement of Moco synthase was performed with Xanthine Oxidase Assay Kit (Abcam ab102522).
Western blotting
Whole cell extracts were generated by lysing cells in NP-40 buffer containing 150 mM sodium chloride, 1.0% NP-40 and 50 mM Tris, pH 8.0 supplemented by Protease Inhibitor Cocktail (Roche). 30 μg whole cell lysates were loaded onto a SDS–polyacrylamide gel for electrophoresis at 30 mA per gel, and then transferred by electrophoresis to a nitrocellulose membrane (GE Healthcare Life Science). All the blots were incubated with corresponding primary antibodies diluted 1:1000 in 5% milk at 4°C overnight followed by HRP-labeled secondary antibodies (1:20000) for 1 hr at room temperature. The signals were detected using ECL Prime western blotting Detection Reagent (Amersham).
Immunoprecipitation
Primary antibody was incubated with Dynabeads Protein G (Life technologies 10765583) for 15 min at room temperature and washed with PBS containing 0.02% Tween-20 (PBST). Antibody-coupled beads were then incubated with αTC1-LT cell lysates overnight at 4°C, washed with PBST for three times and eluted with SDS-loading buffer. The input, supernatant (5%) and elution were used for western blot.
PI3P assay
PI3P measurements were performed using the PI(3)P Mass ELISA kit (K-3300, Echelon). In brief, αTC1-LT cells were pretreated with artemether for three days, followed by lipid extraction and an ELISA assay according to the manufacturer’s guideline.
Glucagon secretion assays
αTC1-LT cells were pretreated with artemether for three days. Cells were first incubated in high glucose medium (5 g/L glucose in KRBS medium) for one hour followed by incubation in low glucose medium (0.5 g/L glucose in KRBS medium) for another hour. Supernatant after low glucose challenge was collected to measure glucagon content using a Glucagon ELISA (Alpco 48-GLUHU-E01-AL).
Glucagon-GFP/Insulin-mCherry zebrafish larvae assay
Transgenic zebrafish strains Tg(Gcga:GFP)ia1 and Tg(ins:NTR-mcherry)ml10 were used to demarcate α cells by GFP expression and β cells by mCherry expression. Double transgenic zebrafish were treated with indicated concentrations of artemether added to E3 medium from 26 hpf until 100 hpf. Zebrafish were embedded for confocal microscopy as described previously (Distel and Koster, 2007). Z stacks of the pancreatic islets were taken on a confocal microscope (Leica TCS SP8 X WLL) using a 25x water objective. Using the measurement counting tool of the Leica software on 3D models of the islets GFP-positive cells and mCherry-positive cells were counted.
Zebrafish Islet Ablation
Transgenic lines were generated using Tol2 mediated transgenesis (Kawakami et al., 2004). 1 kb of the insulin promoter (Huang et al., 2001) was cloned into the p5E-MCS plasmid of the Tol2 Gateway system (Kwan et al., 2007) using the BamHI and HindIII restriction sites. The FKBP-caspase8 fusion gene (Pajvani et al., 2005) (a kind gift of Philipp Scherer) was cloned downstream of the ins promoter in the p5E-ins plasmid. E2Crimson (pE2-Crimson-N1 Vector, Clontech) was cloned into the pME-MCS plasmid of the Tol2 system and expressed via a separate insulin promoter inserted via the BamHI and HindIII sites. Final constructs were generated by an LR recombination reaction as described (Kwan et al., 2007) in pDestTol2pA, and injected together with transposase mRNA into fertilized eggs to generate Tg(ins:caspase8;ins:E2Crimson), abbreviated ins:casp8, fish.
Zebrafish (Danio rerio) were maintained according to standard protocols. Transgenic lines were generated and maintained in the Mitfab692/b692/ednrb1b140/b140 background (a kind gift of Wolfgang Driever at the University of Freiburg).
3 days post fertilization (dpf) old embryos were incubated in 2 μM Dimerizer AP20187 (Dim) for 48 hr at 28°C in the dark. Dim was added to eggwater (0.3 g/l Coral Pro Salt (Red Sea) to reach the final concentration. Control embryos were exposed to corresponding amounts of ethanol. At 5 dpf (0 days post ablation, dpa), larvae were rinsed in egg water and transferred to fresh egg water at a density of 20 fish/200 ml. Starting at 3dpa, ablated larvae were treated with either 5 μM artemether or an equivalent volume of DMSO for 4 d. Compound was renewed every second day.
Larvae were harvested at 7 dpa and fixed for 1 to 2 hr at room temperature in 4% PFA in PBS, then washed 3x 5 min with 1 x PBS/0.2% Triton X-100. To improve access of antibodies to internal structures, head and tail were cut off and the gut was cut open. Larvae were incubated in blocking buffer containing 1% DMSO, 1% sheep serum, 1% BSA and 1% Triton X-100 in 1 x PBS for at least 60 min at room temperature. The embryos were then incubated overnight at 4°C with primary antibody recognizing E2Crimson (rabbit anti-dsRed, 1:100 dilution), washed and then reblocked and incubated in secondary antibody (anti-rabbit Alexa Fluor 633 conjugated, 1:1000 dilution) overnight at 4°C. Larvae were then incubated in 100 ng/ml DAPI overnight at 4°C.
Confocal images were acquired using a Zeiss LSM Exciter5 microscope with a 40x water objective.
For glucose assays, larvae were collected in pools of 4 and euthanized on ice. After removal of all liquid, larvae were resuspended in 200 μL ice-cold, autoclaved 1X PBS, then sonicated on ice with 6 pulses of 0.5 s, and centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was transferred to fresh eppendorfs and used directly for glucose measurement. The glucose assay was performed as described (Kimmel et al., 2015) on biological replicates, using 15 μL larvae extract per reaction. Sample buffer was added up to 25 μl, and combined with 25 μL reaction mix. After incubation at 37°C for 30 min, Ex/Em 535/590 was read on a microplate reader. Protein concentration (A280) was measured on a nanodrop and used to normalize glucose concentration to protein content.
Insulin-expressing (E2Crimson+/Alexa 633+) cells were counted in z stacks using the Cell Counter Plugin of ImageJ. Confocal images were processed by a median filter in ImageJ to remove speckle noise and assembled using Adobe Illustrator.
Insulin secretion assay and intracellular insulin content assay of human islet
500-1000 human islets were pretreated with artemether for three days. Cells were first incubated in low glucose medium (0.5 g/l glucose in KRBS medium) for one hour and in high glucose medium (5 g/l glucose in KRBS medium) for another hour. Supernatant was collected after low and high glucose incubations to measure insulin content using an ELISA kit (Alpco 80-INSHU-E01.1). The islet pellets after high glucose incubation were lysed in NP-40 buffer and intra-islet insulin content was measured using the same kit.
Rodent models
Two months old 129/SV mice were treated with artesunate (1mg/ml) in drinking water provided ad libitum for 3 months. Animals were sacrificed by cervical dislocation and the tissues were fixed and sectioned as previously described (Collombat et al., 2003). For the ex vivo lineage tracing experiments, islets from 2 months old GluCre::RFP animals (containing the following transgenes: the rat glucagon promoter upstream of the phage P1 Cre recombinase (Herrera, 2000) and the Rosa26 promoter upstream of a loxP-Stop-loxP-tdRFP cassette (Luche et al., 2007)) were isolated by collagenase type XI (1 mg/ml) digestion followed by a density gradient to purify the islets from the acinar tissue. Approximately 250 islets per group were handpicked under the microscope and treated with either 10 μM artemether or DMSO as control. Following 24 h of treatment, the islets were dissociated, fixed, and plated into a 384-well plate. Following washing with PBST and blocking with BSA, the cells were incubated with guinea pig polyclonal anti-insulin (1/500-Linco) for 2 h at room temperature. The cells were then washed and incubated with 488-Alexa anti-guinea pig (1/1000-Molecular probes) together with the nuclear counter-stain DAPI for 1 h. The cells were then imaged on an automated microscope (Perkin Elmer Operetta) using a 20x objective and analyzed using Harmony software (Perkin Elmer).
Rat experiments were conducted on 2 months old male SD rats which were fasted overnight and treated with 60 mg/kg streptozotocin i.p. On day 9 post ablation animals were randomized to treatment and control groups based on matching fasting glucose levels, and treated with 20 mg/kg artemether p.o. for 7 d followed by 200 mg/kg artemether p.o. for 16 d. 1% methylcellulose was used as vehicle, prepared once per week by dissolving in distilled water, heating to 40°C, then stored at 4°C. Artemether suspension in 1% methylcellulose was prepared fresh daily before treatment. Blood glucose were measured via tail nick using a handheld glucometer (Accu-Check Performa Roche Diagnostics).
Chemical proteomics
NMR spectra were recorded on a Bruker Avance III 400 (Bruker, Billerica, MA, U.S). Chemical shifts are given in ppm, and coupling constants are given in hertz. Mass spectra were recorded using a XeVo-UPLCTQ-MS system (Waters, Milford, MA, U.S.). Purification by flash column chromatography (FCC) was performed using silica gel 60 (Merck, Darmstadt, Germany), MPLC was performed on a Biotage Isolera system (Biotage, Uppsala, Sweden). The purity of the synthesized compounds was determined and confirmed by UPLC analysis. All synthesis chemicals were purchased from Sigma-Aldrich and used without further purification.
Drug-affinity matrices were prepared essentially as described previously (Fernbach et al., 2009, Huber et al., 2014). Briefly, ethylenediamine (2.7 μL, 40 μmol), ethanolamine (9.7 μL, 160 μmol), and trimethylamine (15 μL, 108 μmol) were added to 500 μL NHS-activated Sepharose 4 Fast Flow beads (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and the reaction was incubated on a rotary shaker for 24 hr. Beads were washed and resuspended in DMSO and NHS-activated artesunate (100 μL, 1.00 μmol) was added to the suspension, and the mixture was incubated on a rotary shaker for 24 hr. Unreacted beads were blocked by addition of NHS-acetate (10 μmol) and triethylamine (25 μL, 180 μmol), followed by agitation on a rotary shaker for 24 hr. After washing with DMSO and lysis buffer, beads were incubated with cell lysates.
Affinity chromatography and elution were performed in duplicate as reported previously (Fernbach et al., 2009, Huber et al., 2014), with 10 mg total cell lysate per replicate.
After elution, enriched proteins were reduced with dithiothreitol, cysteine residues alkylated by incubation with iodoacetamide and the samples digested with modified porcine trypsin (Promega, Madison, WI). Three percent (and multiples thereof) of the digested eluates were purified and concentrated by C18 reversed-phase material. Samples were analyzed at pH 2.4 on an Agilent 1200 nano-HPLC system (Agilent Biotechnologies, Palo Alto, CA) coupled to a hybrid linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Waltham, MA) utilizing the Xcalibur software version 2.1. The analysis was performed in a data-dependent acquisition mode. The 15 most intense ions were isolated and fragmented by collision-induced dissociation (CID). Dynamic exclusion for selected ions was 60 s and a single lock mass at m/z 445.120024 (Si(CH3)2O)6) was used for internal mass calibration (Olsen et al., 2005) with the target loss mass abundance of 0%. Maximal ion accumulation time allowed was 500 ms and overfilling of the C-trap was prevented by automatic gain control set to 106 ions for a full FTMS scan and 5 × 103 ions for MSn. Intact peptides were detected in the Orbitrap mass analyzer at a resolution of 60,000 with a signal threshold of 2,000 counts for triggering an MS event. Further details of the LCMS methodology are as previously described (Maurer et al., 2013).
Peak extraction and conversion of raw MS files into the mgf format was performed with msconvert (ProteoWizard Library v2.1.2708) and the peak lists searched against the mouse SwissProt database v2013.01_20130110 (24,615 sequences including isoforms obtained from varsplic.pl (Kersey et al., 2000) and appended known contaminants) with the search engines Mascot (v2.2.03, MatrixScience, London, UK) and Phenyx (v2.5.14, GeneBio, Geneva, Switzerland) (Colinge et al., 2003). Submission to the search engines was via a Perl script that performs an initial search with relatively broad mass tolerances (Mascot only) on both the precursor and fragment ions (±10 ppm and ± 0.6 Da, respectively). High-confidence peptide identifications are used to recalibrate all precursors and fragment ion masses prior to a second search with narrower mass tolerances (±4 ppm and ± 0.3 Da for CID). One missed tryptic cleavage site was allowed. Carbamidomethyl cysteine and oxidised methionine were set as fixed and variable modifications, respectively. To validate the proteins, Mascot and Phenyx output files were processed by internally developed parsers. Proteins with ≥ 2 unique peptides above a score T1, or with a single peptide above a score T2 were selected as unambiguous identifications. Additional peptides for these validated proteins with score > T3 were also accepted. The thresholds T1, T2, and T3 for each search engine were set by the target-decoy method to control the false discovery rate (FDR) of < 1% for protein identifications and < 1% for peptides (including the ones exported with lower scores). For Mascot, the following thresholds were used: T1 = 14, T2 = 40 and T3 = 10; Phenyx thresholds were set to 4.2, 4.75 and 3.5, respectively (p value < 10-3). The validated proteins retrieved by the two algorithms were merged, any spectral conflicts discarded and grouped according to shared peptides. Non-specific binders were filtered from the drug pull-downs using the SAINT software (version 2.3.4) (Choi et al., 2011). Using protein spectral counts as a measure of protein abundance and comparing the data of from the drug pull-downs with the negative control drug competition experiments, SAINT calculates the probability of a prey protein to be a real bait interactor. We also compared SAINT probabilities with the fold-reduction of spectral count upon free compound competition representing a magnitude of effect. Fold-reduction was computed as the ratio of median spectral counts observed in pull-downs with/without competition. In each condition, 4 spectral counts were available for the median (2 biological replicates and 2 technical for each).
Quantitative proteomics analysis
Quantitative proteomic analysis was performed on αTC1 cells harvested after three day compound treatment and lysed with 2% sodium dodecyl sulfate (SDS). Samples were centrifuged at 5,000 r.c.f. for 2 min and the supernatant removed. Lysis buffer (1mL) was added and the samples incubated for 20 min at RT. The samples were then heated for 5 min at 99°C and sonicated with a Covaris sonicator to reduce viscosity. After centrifugation at 16,000 r.c.f. for 15 min, the supernatant was collected. The protein amount in each sample was determined by using the bicinchoninic protein assay (BCA, Pierce Biotechnology, Thermo Scientific, IL) followed by an adaptation of the filter-aided sample preparation method (FASP) (Manza et al., 2005, Wiśniewski et al., 2009). Samples were reduced with 100 mM dithiothreitol (DTT) at 99°C for 5 min and transferred into VIVACON 500 filter units (Vivaproducts). SDS containing buffer was removed from the sample by centrifugation and exchanged with 8 M urea in 100 mM Tris-HCl buffer. Proteins were alkylated with 50 mM iodoacetamide and washed with 50 mM triethyl ammonium bicarbonate (TEAB). Porcine trypsin (Promega, Madison, WI) was used to digest the proteins in an enzyme to protein ratio of 1:100 (w/w). For relative protein quantitation, the set of six samples (three untreated and three drug-treated) were separately derivatised with TMT 6-plex reagents (ThermoFisher Scientific, Waltham, MA) according to the instructions provided by the manufacturer. The samples were labeled with TMT 126, 127, 128, 129, 130, 131, respectively. The labeled tryptic digests were pooled and concentrated by solid phase extraction (SPE) (MacroSpin columns 30-300 μg capacity, The Nest Group). Samples were basified with 20 mM ammonium formate prior to injection onto a Phenomenex column (150 × 2.0 mm Gemini®NX-C18 3 μm 110Å; Phenomenex, Torrance, CA, USA) on an Agilent 1200 series HPLC (Agilent Biotechnologies, Palo Alto, CA) with UV detection at 214 nm. HPLC solvent A consisted of 20 mM ammonium formate, pH 10 in 5% acetonitrile and solvent B consisted of 20 mM ammonium formate, pH 10 in 90% acetonitrile. Peptides were separated at a flow rate of 100 μL/min and eluted from the column with a two-step linear gradient from 0 to 70% solvent B. Seventy two time-based fractions were collected, acidified, and pooled into 50 HPLC vials based on the UV trace. After removal of organic solvent in a vacuum centrifuge, samples were reconstituted to 10 μL with 5% formic acid (Bennett et al., 2011). Individual fractions were analyzed at pH 2.4 on an Agilent 1200 nano-HPLC system (Agilent Biotechnologies, Palo Alto, CA) coupled to a hybrid linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Waltham, MA) utilizing the Xcalibur software version 2.1. for data acquisition. Single fractions were loaded onto a trap column (Zorbax 300SB-C18 5 μm, 5 × 0.3 mm, Agilent Biotechnologies, Palo Alto, CA) with a binary pump at a flow rate of 45 μL/min. Loading and washing solvents were composed of 0.1% trifluoracetic acid (TFA) in water (solvent A) and 0.1% TFA in 70% methanol and 20% isopropanol (solvent B). The peptides were eluted by back-flushing from the trap column onto a 16 cm fused silica analytical column with an inner diameter of 50 μm packed with C18 reversed-phase material (ReproSil-Pur 120 C18-AQ, 3 μm, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). LCMS solvents were composed of 0.4% formic acid (FA) in water (solvent A) and 0.4% FA in 70% methanol and 20% isopropanol (solvent B). Elution was achieved with a 27 min gradient ranging from 3 to 30% solvent B, followed by a 25 min gradient from 30 to 70% solvent B and, finally, a 7 min gradient from 70 to 100% solvent B at a constant flow rate of 100 nL/min. The analysis was performed in a data-dependent acquisition mode. The 10 most intense ions were isolated and fragmented by higher-energy collision-induced dissociation (HCD) for peptide identification and relative quantitation of TMT reporter ions. Dynamic exclusion for selected ions was 60 s and a single lock mass at m/z 445.120024 (Si(CH3)2O)6) was used for internal mass calibration (Olsen et al., 2005) with the target loss mass abundance of 0%. Maximal ion accumulation time allowed was 500 ms and overfilling of the C-trap was prevented by automatic gain control set to 106 ions for a full FTMS scan and 5 × 105 ions for MSn HCD. Intact peptides were detected in the Orbitrap mass analyzer at a resolution of 30,000 with a signal threshold of 2,000 counts for triggering an MSMS event. The maximum ion scan time was set to 200 ms for acquiring 1 microscan at a resolution of 7,500.
Peak extraction and conversion of .raw MS files into the .mgf format was performed with msconvert (ProteoWizard Library v2.1.2708) and the peak lists searched against the mouse SwissProt database v2014.03_201403131 (24,830 sequences including isoforms obtained from varsplic.pl (Kersey et al., 2000) and appended known contaminants) with the search engines Mascot (v2.2.03, MatrixScience, London, UK) and Phenyx (v2.5.14, GeneBio, Geneva, Switzerland) (Colinge et al., 2003). Submission to the search engines was via a Perl script that performs an initial search with relatively broad mass tolerances (Mascot only) on both the precursor and fragment ions (±10 ppm and ± 0.6 Da, respectively). High-confidence peptide identifications are used to recalibrate all precursor and fragment ion masses prior to a second search with narrower mass tolerances (±4 ppm and ± 0.025 Da for HCD). One missed tryptic cleavage site was allowed. Carbamidomethyl cysteine and the TMT 6-plex tag on peptide N-termini and lysine residues were set as fixed modifications. Oxidised methionine was set as a variable modification. To validate the proteins, Mascot and Phenyx output files were processed by internally developed parsers. Proteins with ≥ 2 unique peptides above a score T1, or with a single peptide above a score T2 were selected as unambiguous identifications. Additional peptides for these validated proteins with score > T3 were also accepted. The thresholds T1, T2, and T3 for each search engine were set by the target-decoy method to control the false discovery rate (FDR) of < 1% for protein identifications and < 1% for peptides (including the ones exported with lower scores). For Mascot, the following thresholds were used: T1 = 12, T2 = 45 and T3 = 10; Phenyx thresholds were set to 5.5, 9.5 and 3.5, respectively (p value < 10-3). The validated proteins retrieved by the two search engines were merged, any spectral conflicts discarded and grouped according to shared peptides. Using the isobar software (Breitwieser et al., 2011), quantitation of the relative fold change of each protein after drug treatment (Figure 1F) was calculated by determining the ratios of TMT reporter ion intensities. The TMT reporter ion channels were normalized based on the protein median intensities and only peptides unique to each protein group were used for the quantitation. The Isobar software employs statistical models to calculate the fold change of each protein and the associated p value based on the ratios and statistical confidence of each individual reporter channel.
Quantification and Statistical Analysis
Data are presented as mean ± SD unless otherwise indicated in figure legends. Sample numbers and experimental repeats are indicated in figure legends. Unless otherwise indicated, p values were calculated by Student’s t test (Graphpad Prism) and the exact values were indicated in figure legends. The Gene ontology terms enrichment was calculated using Gorilla (Eden et al., 2007, Eden et al., 2009). Gene set enrichment analysis was conducted as previously described (Mootha et al., 2003, Subramanian et al., 2005).
Data and Software Availability
Data Resources
Raw data files for the RNA sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession numbers GEO: GSE84592, GEO: GSE73727, and GEO: GSE84714.
Author Contributions
S.K., J.H.-S., and P.C. conceived the project and designed the study; T.F. and J.H.-S. developed cell lines; J.L., T.C., and S.S. conducted and analyzed cell biological experiments in rodent cell lines; J.L. and C.-H.L. performed and analyzed compound screens; K.V.M.H., P.M., A. Stukalov, M.G., K.P., J.C., K.L.B., and G.S.-F. performed and analyzed chemical proteomics experiments and global proteome studies; R.A.R., I.B., S.H., and T.H. conducted electrophysiology and Ca2+ imaging; J.L., J.K., M.F., F.M.P., T.P., and C. Bock conducted and analyzed RNA-seq experiments; A. Spittler performed fluorescence-activated cell sorting; N.S., R.A.K., C.S., M.D., and D.M. performed and analyzed zebrafish experiments; M.C., A.V., F.A., and P.C. conducted and analyzed mouse experiments; F.B. and T.S. conducted and analyzed rat experiments; C.I., M.C., J.L., P.C., and J.H.-S. conducted histology on mouse and rat pancreata; C. Barbieux and E.B. isolated human islets; J.L., T.C., and C.H. conducted functional experiments in human islets; and J.L. and S.K. wrote the manuscript, with input from all co-authors.
Acknowledgments
We thank Freya Klepsch and Patrick Markt (CeMM) for their input in the design of the drug library. Human islets were provided through the JDRF awards 31-2012-783 and 1-RSC-2014-100-I-X (ECIT Islet for Basic Research Program) and the NIDDK-funded Integrated Islet Distribution Program (IIDP) at City of Hope (NIH grant #1UC4DK098085). Next-generation sequencing was performed by the Biomedical Sequencing Facility at CeMM. This work was funded by JDRF grants 3-SRA-2015-20-Q-R, 17-2011-258 (generation of β cells from alternative pancreatic subtypes; P.C., J.H.-S., and S.K.), 17-2011-16, 2-2010-567, 26-2008-639, 17-2013-426 (P.C.), and 3-PDF-2014-206-A-N (S.S.); the European Research Council (ERC-2015-AdG-695136 Secret-Cells to T.H.; and StG-2011-281265 to P.C.); intramural funds of the Medical University of Vienna (to T.H.); an EMBO long-term research fellowship (ALTF 596-2014 to R.A.R.); the NovoNordisk Foundation (to T.H.); European Commission FP7 Marie Curie Actions (EMBOCOFUND2012 and GA-2012-600394 to R.A.R.); the Austrian Science Fund (FWF P25659-B19 to R.A.K.); a DOC Fellowship of the Austrian Academy of Sciences (to J.K.); the INSERM AVENIR program (to P.C.); the INSERM (to P.C.); the FMR (DRC20091217179 to P.C.); the ANR/BMBF (2009 GENO 105 01/01KU0906 to P.C.); the “Investments for the Future” LABEX SIGNALIFE (ANR-11-LABX-0028-01 to P.C.); the Max-Planck Society (to P.C.); Club Isatis (to P.C.); Mr. and Mrs. Dorato (to P.C.); Mr. and Mrs. Peter de Marffy-Mantuano (to P.C.); the Fondation Générale de Santé (to P.C.); and the Foundation Schlumberger pour l’Education et la Recherche (to P.C.). The single-cell sequencing infrastructure at CeMM was supported by a New Frontiers Research Infrastructure grant from the Austrian Academy of Sciences. Research in the S.K. lab is supported by the Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology, and Development. J.L. and S.K. have filed a patent application based on the findings described in this manuscript (WO/2015/155303). T.F., C.I., C.H., and J.H.-S. are employees of Novo Nordisk A/S and may also own shares in in the company. F.B. and T.S. are employees of Physiogenex S.A.S. T.H. is supported by GW Pharmaceuticals on projects unrelated in scope to the content of the present report.
Published December 1, 2016
Footnotes
Supplemental Information includes seven figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.11.010.
Supplemental Information
References
- Ahmad W., Khan I., Khan M.A., Ahmad M., Subhan F., Karim N. Evaluation of antidiabetic and antihyperlipidemic activity of Artemisia indica linn (aeriel parts) in Streptozotocin induced diabetic rats. J. Ethnopharmacol. 2014;151:618–623. doi: 10.1016/j.jep.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Al-Hasani K., Pfeifer A., Courtney M., Ben-Othman N., Gjernes E., Vieira A., Druelle N., Avolio F., Ravassard P., Leuckx G. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell. 2013;26:86–100. doi: 10.1016/j.devcel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- al-Waili N.S. Artemisia herba-alba Asso. in diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 1988;15:497. doi: 10.1111/j.1440-1681.1988.tb01106.x. [DOI] [PubMed] [Google Scholar]
- Arreesrisom P., Dondorp A.M., Looareesuwan S., Udomsangpetch R. Suppressive effects of the anti-oxidant N-acetylcysteine on the anti-malarial activity of artesunate. Parasitol. Int. 2007;56:221–226. doi: 10.1016/j.parint.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Ben-Othman N., Vieira A., Courtney M., Record F., Gjerns E., Avolio F., Hadzic B., Druelle N., Napolitano T., Navarro-Sanz S. Long-term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell. 2016;168:73–85. doi: 10.1016/j.cell.2016.11.002. this issue. [DOI] [PubMed] [Google Scholar]
- Bennett K.L., Funk M., Tschernutter M., Breitwieser F.P., Planyavsky M., Ubaida Mohien C., Müller A., Trajanoski Z., Colinge J., Superti-Furga G., Schmidt-Erfurth U. Proteomic analysis of human cataract aqueous humour: Comparison of one-dimensional gel LCMS with two-dimensional LCMS of unlabelled and iTRAQ®-labelled specimens. J. Proteomics. 2011;74:151–166. doi: 10.1016/j.jprot.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S., Orci L. New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes. 1982;31:883–889. doi: 10.2337/diab.31.10.883. [DOI] [PubMed] [Google Scholar]
- Breitwieser F.P., Müller A., Dayon L., Köcher T., Hainard A., Pichler P., Schmidt-Erfurth U., Superti-Furga G., Sanchez J.C., Mechtler K. General statistical modeling of data from protein relative expression isobaric tags. J. Proteome Res. 2011;10:2758–2766. doi: 10.1021/pr1012784. [DOI] [PubMed] [Google Scholar]
- Chera S., Baronnier D., Ghila L., Cigliola V., Jensen J.N., Gu G., Furuyama K., Thorel F., Gribble F.M., Reimann F., Herrera P.L. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature. 2014;514:503–507. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Larsen B., Lin Z.-Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.-C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinge J., Masselot A., Giron M., Dessingy T., Magnin J. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3:1454–1463. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
- Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Krull J., Gradwohl G., Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Xu X., Ravassard P., Sosa-Pineda B., Dussaud S., Billestrup N., Madsen O.D., Serup P., Heimberg H., Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M., Gjernes E., Druelle N., Ravaud C., Vieira A., Ben-Othman N., Pfeifer A., Avolio F., Leuckx G., Lacas-Gervais S. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denault J.B., D’Orléans-Juste P., Masaki T., Leduc R. Inhibition of convertase-related processing of proendothelin-1. J. Cardiovasc. Pharmacol. 1995;26(Suppl 3):S47–S50. [PubMed] [Google Scholar]
- Distel M., Koster R.W. In vivo time-lapse imaging of zebrafish embryonic development. CSH Protoc. 2007;2007 doi: 10.1101/pdb.prot4816. pdb prot4816. [DOI] [PubMed] [Google Scholar]
- Dondorp A.M., Fanello C.I., Hendriksen I.C., Gomes E., Seni A., Chhaganlal K.D., Bojang K., Olaosebikan R., Anunobi N., Maitland K., AQUAMAT group Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein-Ludwig U., Webb R.J., Van Goethem I.D., East J.M., Lee A.G., Kimura M., O’Neill P.M., Bray P.G., Ward S.A., Krishna S. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- Eden E., Lipson D., Yogev S., Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson T., Blank A., von Hagens C., Ashton M., Äbelö A. Population pharmacokinetics of artesunate and dihydroartemisinin during long-term oral administration of artesunate to patients with metastatic breast cancer. Eur. J. Clin. Pharmacol. 2014;70:1453–1463. doi: 10.1007/s00228-014-1754-2. [DOI] [PubMed] [Google Scholar]
- Fernbach N.V., Planyavsky M., Müller A., Breitwieser F.P., Colinge J., Rix U., Bennett K.L. Acid elution and one-dimensional shotgun analysis on an Orbitrap mass spectrometer: an application to drug affinity chromatography. J. Proteome Res. 2009;8:4753–4765. doi: 10.1021/pr900455x. [DOI] [PubMed] [Google Scholar]
- Fomina-Yadlin D., Kubicek S., Walpita D., Dancik V., Hecksher-Sørensen J., Bittker J.A., Sharifnia T., Shamji A., Clemons P.A., Wagner B.K., Schreiber S.L. Small-molecule inducers of insulin expression in pancreatic alpha-cells. Proc. Natl. Acad. Sci. USA. 2010;107:15099–15104. doi: 10.1073/pnas.1010018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin I.K., Wollheim C.B. GABA in the endocrine pancreas: its putative role as an islet cell paracrine-signalling molecule. J. Gen. Physiol. 2004;123:185–190. doi: 10.1085/jgp.200409016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy J.M., Harvey R.J., Schwarz G. Gephyrin: where do we stand, where do we go? Trends Neurosci. 2008;31:257–264. doi: 10.1016/j.tins.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Herrera P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Ho W.E., Peh H.Y., Chan T.K., Wong W.S. Artemisinins: pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014;142:126–139. doi: 10.1016/j.pharmthera.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Huang H., Vogel S.S., Liu N., Melton D.A., Lin S. Analysis of pancreatic development in living transgenic zebrafish embryos. Mol Cell Endocrinol. 2001;177:117–124. doi: 10.1016/s0303-7207(01)00408-7. [DOI] [PubMed] [Google Scholar]
- Huber K.V., Salah E., Radic B., Gridling M., Elkins J.M., Stukalov A., Jemth A.S., Göktürk C., Sanjiv K., Strömberg K. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014;508:222–227. doi: 10.1038/nature13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell. 2004;7:133–144. doi: 10.1016/j.devcel.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Kersey P., Hermjakob H., Apweiler R. VARSPLIC: alternatively-spliced protein sequences derived from SWISS-PROT and TrEMBL. Bioinformatics. 2000;16:1048–1049. doi: 10.1093/bioinformatics/16.11.1048. [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel R.A., Dobler S., Schmitner N., Walsen T., Freudenblum J., Meyer D. Diabetic pdx1-mutant zebrafish show conserved responses to nutrient overload and anti-glycemic treatment. Sci. Rep. 2015;5:14241. doi: 10.1038/srep14241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E., Martinson L.A., Kadoya K., Bang A.G., Kelly O.G., Eliazer S., Young H., Richardson M., Smart N.G., Cunningham J. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Kubicek S., Gilbert J.C., Fomina-Yadlin D., Gitlin A.D., Yuan Y., Wagner F.F., Holson E.B., Luo T., Lewis T.A., Taylor B. Chromatin-targeting small molecules cause class-specific transcriptional changes in pancreatic endocrine cells. Proc. Natl. Acad. Sci. USA. 2012;109:5364–5369. doi: 10.1073/pnas.1201079109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C.B. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Li C., Liu C., Nissim I., Chen J., Chen P., Doliba N., Zhang T., Nissim I., Daikhin Y., Stokes D. Regulation of glucagon secretion in normal and diabetic human islets by γ-hydroxybutyrate and glycine. J. Biol. Chem. 2013;288:3938–3951. doi: 10.1074/jbc.M112.385682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Klughammer J., Farlik M., Penz T., Spittler A., Barbieux C., Berishvili E., Bock C., Kubicek S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016;17:178–187. doi: 10.15252/embr.201540946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luche H., Weber O., Nageswara Rao T., Blum C., Fehling H.J. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- Manza L.L., Stamer S.L., Ham A.J., Codreanu S.G., Liebler D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics. 2005;5:1742–1745. doi: 10.1002/pmic.200401063. [DOI] [PubMed] [Google Scholar]
- Maruyama H., Hisatomi A., Orci L., Grodsky G.M., Unger R.H. Insulin within islets is a physiologic glucagon release inhibitor. J. Clin. Invest. 1984;74:2296–2299. doi: 10.1172/JCI111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M., Müller A.C., Wagner C., Huber M.L., Rudashevskaya E.L., Wagner S.N., Bennett K.L. Combining filter-aided sample preparation and pseudoshotgun technology to profile the proteome of a low number of early passage human melanoma cells. J. Proteome Res. 2013;12:1040–1048. doi: 10.1021/pr301009u. [DOI] [PubMed] [Google Scholar]
- Mbengue A., Bhattacharjee S., Pandharkar T., Liu H., Estiu G., Stahelin R.V., Rizk S.S., Njimoh D.L., Ryan Y., Chotivanich K. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 2015;520:683–687. doi: 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Kanda Y., Hirokawa Y., Kawasaki F., Matsuki M., Kaku K. MIN6 is not a pure beta cell line but a mixed cell line with other pancreatic endocrine hormones. Endocr. J. 2009;56:45–53. doi: 10.1507/endocrj.k08e-172. [DOI] [PubMed] [Google Scholar]
- Nosten F., White N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007;77(6, Suppl):181–192. [PubMed] [Google Scholar]
- Olsen J.V., de Godoy L.M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Pagliuca F.W., Millman J.R., Gürtler M., Segel M., Van Dervort A., Ryu J.H., Peterson Q.P., Greiner D., Melton D.A. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani U.B., Trujillo M.E., Combs T.P., Iyengar P., Jelicks L., Roth K.A., Kitsis R.N., Scherer P.E. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat. Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- Pennarossa G., Maffei S., Campagnol M., Tarantini L., Gandolfi F., Brevini T.A. Brief demethylation step allows the conversion of adult human skin fibroblasts into insulin-secreting cells. Proc. Natl. Acad. Sci. USA. 2013;110:8948–8953. doi: 10.1073/pnas.1220637110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwana I., Zheng J., Li X., Deurloo M., Son D.O., Zhang Z., Liang C., Shen E., Tadkase A., Feng Z.P. GABA promotes human β-cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63:4197–4205. doi: 10.2337/db14-0153. [DOI] [PubMed] [Google Scholar]
- Rix U., Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Rorsman P., Berggren P.O., Bokvist K., Ericson H., Möhler H., Ostenson C.G., Smith P.A. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature. 1989;341:233–236. doi: 10.1038/341233a0. [DOI] [PubMed] [Google Scholar]
- Sabatini D.M., Barrow R.K., Blackshaw S., Burnett P.E., Lai M.M., Field M.E., Bahr B.A., Kirsch J., Betz H., Snyder S.H. Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science. 1999;284:1161–1164. doi: 10.1126/science.284.5417.1161. [DOI] [PubMed] [Google Scholar]
- Sangan C.B., Jover R., Heimberg H., Tosh D. In vitro reprogramming of pancreatic alpha cells towards a beta cell phenotype following ectopic HNF4α expression. Mol. Cell. Endocrinol. 2015;399:50–59. doi: 10.1016/j.mce.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A.M., Ricordi C., Hering B.J., Auchincloss H., Lindblad R., Robertson R.P., Secchi A., Brendel M.D., Berney T., Brennan D.C. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006;355:1318–1330. doi: 10.1056/NEJMoa061267. [DOI] [PubMed] [Google Scholar]
- Shukla K.L., Gund T.M., Meshnick S.R. Molecular modeling studies of the artemisinin (qinghaosu)-hemin interaction: docking between the antimalarial agent and its putative receptor. J. Mol. Graph. 1995;13:215–222. doi: 10.1016/0263-7855(94)00001-9. [DOI] [PubMed] [Google Scholar]
- Soltani N., Qiu H., Aleksic M., Glinka Y., Zhao F., Liu R., Li Y., Zhang N., Chakrabarti R., Ng T. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. USA. 2011;108:11692–11697. doi: 10.1073/pnas.1102715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagner J.I., Samols E. The vascular order of islet cellular perfusion in the human pancreas. Diabetes. 1992;41:93–97. doi: 10.2337/diab.41.1.93. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C., Xuan S., Kitamura T., DePinho R.A., Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat. Genet. 2012;44:406–412. doi: 10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F., Népote V., Avril I., Kohno K., Desgraz R., Chera S., Herrera P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F., Damond N., Chera S., Wiederkehr A., Thorens B., Meda P., Wollheim C.B., Herrera P.L. Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes. 2011;60:2872–2882. doi: 10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan S.K., Fritschy J.M. Gephyrin: a master regulator of neuronal function? Nat. Rev. Neurosci. 2014;15:141–156. doi: 10.1038/nrn3670. [DOI] [PubMed] [Google Scholar]
- Walpita D., Hasaka T., Spoonamore J., Vetere A., Takane K.K., Fomina-Yadlin D., Fiaschi-Taesch N., Shamji A., Clemons P.A., Stewart A.F. A human islet cell culture system for high-throughput screening. J. Biomol. Screen. 2012;17:509–518. doi: 10.1177/1087057111430253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Wang Q., Prud’homme G.J. GABAergic system in the endocrine pancreas: a new target for diabetes treatment. Diabetes Metab. Syndr. Obes. 2015;8:79–87. doi: 10.2147/DMSO.S50642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wang J., Zhang C.J., Chia W.N., Loh C.C., Li Z., Lee Y.M., He Y., Yuan L.X., Lim T.K., Liu M. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015;6:10111. doi: 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt A., Birnir B., Buschard K., Gromada J., Salehi A., Sewing S., Rorsman P., Braun M. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes. 2004;53:1038–1045. doi: 10.2337/diabetes.53.4.1038. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Xie S., Sinha R.A., Singh B.K., Li G.D., Han W., Yen P.M. Resveratrol induces insulin gene expression in mouse pancreatic α-cells. Cell Biosci. 2013;3:47. doi: 10.1186/2045-3701-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E., Kumar M., Zhang Y., Ju W., Obata T., Zhang N., Liu S., Wendt A., Deng S., Ebina Y. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 2006;3:47–58. doi: 10.1016/j.cmet.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Ye L., Robertson M.A., Hesselson D., Stainier D.Y., Anderson R.M. Glucagon is essential for alpha cell transdifferentiation and beta cell neogenesis. Development. 2015;142:1407–1417. doi: 10.1242/dev.117911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P., Park J.S., Melton D.A. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell. 2013;153:747–758. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhou Q., Brown J., Kanarek A., Rajagopal J., Melton D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.