

Abstract
Purpose
Predicting target site drug concentration in the brain is of key importance for the successful development of drugs acting on the central nervous system. We propose a generic mathematical model to describe the pharmacokinetics in brain compartments, and apply this model to predict human brain disposition.
Methods
A mathematical model consisting of several physiological brain compartments in the rat was developed using rich concentration-time profiles from nine structurally diverse drugs in plasma, brain extracellular fluid, and two cerebrospinal fluid compartments. The effect of active drug transporters was also accounted for. Subsequently, the model was translated to predict human concentration-time profiles for acetaminophen and morphine, by scaling or replacing system- and drug-specific parameters in the model.
Results
A common model structure was identified that adequately described the rat pharmacokinetic profiles for each of the nine drugs across brain compartments, with good precision of structural model parameters (relative standard error <37.5%). The model predicted the human concentration-time profiles in different brain compartments well (symmetric mean absolute percentage error <90%).
Conclusions
A multi-compartmental brain pharmacokinetic model was developed and its structure could adequately describe data across nine different drugs. The model could be successfully translated to predict human brain concentrations.
Electronic supplementary material
The online version of this article (doi:10.1007/s11095-016-2065-3) contains supplementary material, which is available to authorized users.
KEY WORDS: blood-brain barrier, central nervous system (CNS), human prediction, pharmacokinetics, translational model
Introduction
Central nervous system (CNS) drug development suffers from 91% attrition rate and especially the success rate in phase II is very low (1,2). The primary reasons for attrition are safety issues (3). Although the underlying physiological and pharmacological reasons for such failures are often not fully known they are likely related to a lack of knowledge or failure to account for a combination of on- and off-target site concentrations, target interaction and downstream signal processing. The first step in this cascade, obtaining quantitative insight into CNS target site concentration kinetics, is already a major challenge, and has been suggested as a major factor contributing to failure of novel drug candidates (4). During clinical drug development, typically only drug plasma concentrations are considered as marker for drug exposure, because quantifying drug concentrations in the brain is challenging. Hence, the ability to predict brain concentrations based on plasma data is highly relevant to further optimize CNS drug development.
The prediction of brain target site concentrations is controlled by several factors. First, the poorly penetrable blood-brain barrier (BBB) and the blood-cerebrospinal barrier (BCSFB) (5) limit passage of drugs from the systemic circulation into the brain. These barriers are associated with limited passive diffusion, and in addition various active transport and drug metabolism processes that systematically administered drugs need to pass. Second, the brain can be further subdivided into several distinct physiological compartments, including the brain extracellular fluid (ECF), brain intracellular fluid (ICF), and multiple cerebrospinal fluid (CSF) compartments. The specific disposition characteristics across these specific compartments further determines drug target site concentrations. Third, CNS drug target site concentrations are mediated by physiological flows including the microvascular blood flow, and brain ECF and CSF flows. Lastly, drug protein binding and the localized pH in specific sub-compartments further affect ultimate brain target site concentrations.
Passive drug transport processes are mediated through a combination of drug permeability properties, trans-membrane transport routes, and the surface areas of the BBB (SABBB) and BCSFB (SABCSFB) (5). Active drug transport is mediated by transport proteins such as P-glycoprotein (P-gp), multidrug resistance-associated protein (MRPs), organic anion transporters (OATs), and organic anion transporting polypeptides (OATPs). Even though the function and localization of these transporters has been extensively investigated in in-vitro and in-vivo studies, their precise functions is in some cases not fully understood (6).
Several experimental preclinical models have been developed to assess drug distribution to brain compartments. These models differ in terms of temporal and spatial resolution, and in their consideration of drug protein binding (7–10). For example, the combinatorial mapping approach has been recently introduced using unbound drug concentration with the brain slice technique (10,11). This approach can predict unbound drug CNS exposure at steady state in multiple brain compartments, but does not allow temporal characterization of drug concentration changes. Positron emission tomography (PET) is sometimes used also clinically, as a non-invasive imaging method to visualize spatiotemporal drug distribution in the brain. However, PET scan signals cannot distinguish parent compounds from their metabolites, or bound and unbound drug compounds in the brain (12). Finally, microdialysis allows serial sampling in multiple physiological compartments of unbound drug concentrations, hence is suited to characterize the time profile of drug concentrations in the brain (13).
In order to capture the time profile and complexity of interacting factors governing drug distribution across brain compartments as determined by microdialysis methods, mathematical modeling represents an indispensable tool. Specifically, physiologically based pharmacokinetic (PBPK) models are of interest, as these models aim to distinguish between system- and drug-specific parameters, allowing for translational predictions by scaling or replacing system- or drug-specific parameters from the rat to man (14). Several (semi-) PBPK models for CNS drug distribution have been published, with different levels of complexity (15–20). However, these models did not yet include validations of predicted human CNS concentrations (21). Recently, Gaohoa et al published a CNS PBPK model, which consists of four compartments such as brain blood volume, brain mass, cranial CSF and spinal CSF. This model was validated with human acetaminophen and phenytoin data. However, a limitation of this model is the lack of consideration of a brain extracellular fluid compartment (brainECF), which is of critical importance for prediction of receptor binding kinetics for drugs acting on membrane bound receptors and ultimately drug efficacy (22).
Previously we have developed separate semi-physiological CNS PBPK models for three drugs based on microdialysis experiments in rats, which included unbound drug concentration-time profiles across multiple brain compartments (23–25). These models described the data well, but resulted in different individual model structures for each of these drugs.
The purpose of the current work was to develop a more generally applicable model structure that can be used to predict drug target site concentration-time profiles in human brain compartments based on rat pharmacokinetic (PK) studies. To this aim, we used published and newly generated datasets for a larger number of drugs, and we performed rigorous model validation on external datasets. Furthermore, the impact of key drug transporters was also included in our model. Finally, we investigated the performance of the developed model structure to predict human brain concentration-time profiles for acetaminophen and morphine.
Materials and Methods
Data for Model Development
An overview of experimental data for nine compounds with different physicochemical characteristics used for model development is provided in Table I. The physicochemical characteristics of the nine compounds are provided in Table SI. Data on 6 compounds were previously published, as indicated in Table I. For three compounds (paliperidone, phenytoin and risperidone), data were newly produced after single intravenous administration, as described below.
Table I.
Summary of the Rat Brain Distribution Data for Model Development and External Validation
| Study design | Model development | External validation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Published data | Newly produced data | Published data | Newly produced data | ||||||||||
| Acetaminophen | Atenolol | Methotrexate | Morphine | Morphine | Quinidine | Remoxipride | Paliperidone | Phenytoin | Risperidone | Acetaminophen | Remoxipride | ||
| Species | rat | rat | rat | rat | rat | rat | rat | rat | rat | rat | rat | rat | |
| Nr of animals | 16 | 5 | 23 | 65 | 18 | 41 | 29 | 21 | 14 | 16 | 8 | 65 | |
| Dosage, mg/kg (infusion time, min) | 16 (10) | 10 (1) | 40, 80 (10) | 4, 10, 40 (10) | 10, 40 (10) | 10, 20 (10) | 4, 8, 16 (30) | 0.5 (20) | 20, 30, 40 (10) | 2 (20) | 200a (1) | 0.7, 5.2, 14 (10) | |
| Nr of samples (sampling times, min) | plasma | 67 (0–240) | 32 (0–120) | 186 (0–300) | 825 (0–360) | 306 (0–190) | 313 (0–360) | 189 (0–240) | 182 (0–360) | 109 (0–480) | 124 (0–360) | 67 (0–180) | 290 (0–240) |
| dialysate | 592 (0–240) | 106 (0–120) | 1065 (0–300) | 238 (0–360) | 299 (0–180) | 1678 (0–360) | 125 (0–240) | 660 (0–240) | 152 (0–480) | 436 (0–240) | 72 (0–180) | 489 (0–240) | |
| Active transport inhibitor | – | – | probenecidb | GF120918c | – | tariquidarc | – | tariquidarc | tariquidarc, probenecidb | tariquidarc | – | – | |
| Dosage of active transport inhibitor, mg/kg (infusion time, min) | – | – | 150 (10) | 6 (cont)d | – | 15 (10) | – | 15 (10) | 15 (10) 150 (10) | 15 (10) | – | – | |
| Data | |||||||||||||
| plasma | X | X | X | X | X | X | X | X | X | X | X | X | |
| brainECF | X | X | X | X | X | X | X | X | X | X | X | X | |
| CSFLV | X | X | X | X | |||||||||
| CSFCM | X | X | X | X | X | X | |||||||
| References | (6) | (69) | (25) | (70) | (71) | (24) | (26) | (72) | |||||
brain ECF a brain extracellular fluid compartment, CSF LV a compartment of cerebrospinal fluid in lateral ventricle, CSF CM a compartment of cerebrospinal fluid in cisterna magna
a; mg, b; inhibitor of MRPs, OATs and OATPs, c; inhibitor of P-gp,d; continuous infusion
For some of the drugs, active transport inhibitors were co-administered intravenously to characterize the effect of P-gp, MRP, OATs and OATPs, as indicated in Table I. The transport inhibitors included were probenecid as an inhibitor of MRPs, OATs and OATPs, and GF120918 or tariquidar as inhibitor of P-gp.
Data for External Model Validation
For an external validation of the model, we used two separate rat datasets for acetaminophen and remoxipride, as indicated in Table I. The acetaminophen data was previously published, the remoxipride data was newly generated as described in the experimental section. For acetaminophen and remoxipride, two separate experimental datasets were available. For each drug, one of these datasets was used for model development, whilst the second dataset was used for external validation. The external validation with these second sets of data allows assessment of the robustness of our model predictions with respect to a different experiment and variation in experimental design.
Animals
Animal study protocols were approved by the Animal Ethics Committee of Leiden University and all animal experiments were performed in accordance with the Dutch Law of Animal Experimentation (for approval numbers see Table SII). Male Wistar rats (225–275 g, Charles River, The Netherlands) were housed in groups for a few days (5–13 days) under standard environmental conditions with ad libitum access to food (Laboratory chow, Hope Farms, Woerden, The Netherlands) and acidified water. Between surgery and experiments, the animals were kept individually in Makrolon type three cages for 7 days to recover from surgical procedures.
Surgery
Rats were anesthetized (5% isoflurane for induction, 1–2% as maintenance), and subsequently received cannulas in the femoral artery for serial blood sampling, and in the femoral vein for drug administration, respectively. Subsequently, microdialysis guides were inserted into different brain locations. The animals were allowed to recover for 1 week before the experiments were performed. One day before the experiment, the microdialysis dummies were replaced by microdialysis probes. For details on guides, probes and locations see Table SII.
Microdialysis and Drug Administration
Experiments generally started at 9:00 a.m. to minimize the influence of circadian rhythms. Microdialysis probes were continuously flushed with microdialysis perfusion fluid (PF) until equilibration before the start of drug administration. Drugs were administered at t = 0 h by intravenous infusion through the cannula implanted in the femoral vein. For the quantification of active drug transport, the active transport inhibitor was administered before the drug’s administration. The general procedure of microdialysis is depicted in Fig. 1. Dosage and infusion time for each drug and the active transport inhibitor were summarized in Table I, and the composition of microdialysis PF and flow rate of microdialysis PF are summarized in Table SII.
Fig. 1.

Microdialysis procedures for the compounds used for the development of the multi-compartmental brain PK model.
Bioanalytical methods
The developed analytical methods for risperidone, paliperidone, phenytoin and remoxipride are described below.
Chemicals and Reagents
For all procedures, nanopure lab water (18.2 MΩ cm) was used. All chemicals used were obtained from Sigma Aldrich (Zwijndrecht, The Netherlands), and analytical grade unless stated otherwise. The internal standards risperidone-D4 and paliperidone-D4 were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). Remoxipride. HCl was obtained from TOCRIS (Bristol, United Kingdom). Tariquidar (TQD, XR9576) was obtained from Xenova group PLC (Cambridge, United Kingdom). Ammonium formate, ammonium bicarbonate (ULC/MS grade), acetonitrile (LC-MS grade), methanol, isopropanol, and formic acid (ULC/MS grade) were obtained from Biosolve B.V. (Valkenswaard, The Netherlands). Sodium hydroxide was obtained from Baker (Deventer, The Netherlands).
Sample Preparation of Plasma
Risperidone and Paliperidone
The calibration curve was in a range of 5 to 1000 ng/ml. Quality controls (QC’s) were prepared in blank rat plasma at three different concentration levels and stored at −20°C. The lower limit of quantification (LLOQ) for both risperidone and paliperidone was 5 ng/ml. To 20 μl of plasma, 20 μl of internal standard solution (risperidone-D4 and paliperidone-D4) and 20 μl water (or 20 μl calibration solution in the case of the calibration curve) were added. After brief vortexing, 1 ml of acetonitrile was added. Brief vortexing and subsequent centrifugation at 10,000 g led to a clear supernatant, which was transferred to a glass tube and evaporated in the vortex evaporator (Labconco, Beun de Ronde, Breda, The Netherlands). The residue was redissolved in 200 μl of 2% methanol, 10 mM ammonium formate, pH 4.1 and processed in according to the solid phase extraction (SPE)- liquid chromatography (LC) method.
Phenytoin
20 μl of plasma sample was mixed with 20 μl of water in an Eppendorf vial. An aliquot of 40 μl acetonitrile was added for protein precipitation. After centrifugation at 11,000 g for 10 min, 40 μl of supernatant was mixed with 40 μl ammonium acetate buffer (pH 5.0). Calibration was performed by adding 20 μl of calibration solution to 20 μl of blank plasma, using the same clean-up procedure. The calibration solutions ranged from 0.2 to 100 μg/ml. 30 μl was injected into the high-performance liquid chromatography (HPLC) system. The LLOQ was 250 ng/ml.
Remoxipride
Sample preparation was performed according to Stevens et al (26). Briefly, 20 μl of sample was mixed with 20 μl of water and 20 μl internal standard (raclopride). Proteins were precipitated with 6% perchloric acid and centrifugation. After addition of sodium carbonate, 10 μl was injected into the SPE-LC system.
Sample Preparation for Microdialysates
Risperidone and Paliperidone
The calibration curve for the microdialysis samples was prepared in buffered PF (composition in Table SII). The concentrations were in the range of 0.1 to 20 ng/ml. QC’s were prepared using a different batch of buffered PF. Before injection of 10 μl into the LC system, the microdialysate samples were diluted with internal standard solution in a ratio of 1:1 v/v. The internal standard solution consisted of 100 ng/ml risperidone-D4 and paliperidone-D4 in nanopure water. The LLOQs for risperidone and paliperidone were 0.4 and 0.2 ng/ml, respectively.
Phenytoin
Calibration curves were made in minimal PF at a concentration range of 25 to 5000 ng/ml. QC’s were prepared using a different batch of buffered PF. Of a typical sample that consisted of 40 μl of microdialysate, 30 μl was injected into the HPLC system. The LLOQ was 25 ng/ml.
Remoxipride
Calibration curves were prepared in buffered PF. The calibration range was from 1 to 200 ng/ml. QC’s were prepared using a different batch of buffered PF. Samples were mixed in a 1:1 v/v ratio with the internal standard raclopride (100 ng/ml) before injection of 5 μl into the LC system. The LLOQ was 0.5 ng/ml.
Chromatography
Paliperidone and Risperidone
SPE-LC Method
For plasma samples the SPE-method was applied. The SPE system consisted of a Hyphere C8 HD, SE column (10 × 2 mm) (Spark Holland, Emmen, The Netherlands) in a cartridge holder and served for the clean-up of the sample. The cartridge holder was connected to a Gynkotek gradient pump (Thermo Scientific, Breda, The Netherlands) and a Waters 717 autosampler (Waters, Etten-Leur, The Netherlands). The MS Surveyor pump from Thermo Scientific (Breda, The Netherlands) provided the flow for the LC column, which was the same type as in the LC-method. The sample was injected onto the SPE, which was preconditioned with 2% methanol (pH 4.1). After 1 min of flushing, the SPE was switched into the LC system. After 4 min, the SPE was cleaned with 98% methanol (pH 4.1) for 2 min and reconditioned with 2% methanol (pH 4.1). The flow of the SPE pump was 0.75 ml/min. The flow of the LC system was 0.25 ml/min. The gradient was from 10 to 90% methanol (1–8.5 min after injection). The SPE column was used for a maximum of 240 injections.
LC-Method
For microdialysates, LC-Method was applied. The separation of the active compounds was possible using Hyper Clone HPLC column (3 μm BDS C18 130 Å) from Phenomenex (Utrecht, The Netherlands) placed at 40°C. The LC system was used at a flow of 0.25 ml/min using a linear gradient from 20 to 74% methanol (1–6 min after injection). Before the next injection, the column was re-equilibrated with 20% methanol for 2 min.
Phenytoin
HPLC Method and Detection
For both plasma and microdialysates samples an HPLC method was used. The mobile phase consisted of 15 mM ammonium acetate adjusted to pH 5.0 with acetic acid and acetonitrile in a 2:1 ratio (v/v). Separation was achieved using an Altima HP C18-Amide HPLC column (5 μm, 150 × 4.6 mm) from Grace Alltech (Breda, The Netherlands). The injector was from Waters (Etten-Leur, The Netherlands). The LC pump (LC-10 ADVP) was obtained from Shimadzu (‘s-Hertogenbosch, The Netherlands). The ultraviolet (UV) detector (Spectroflow 757) was obtained from Applied Biosystems (Waltham, Massachusetts) and was used at a wavelength of 210 nm. Data acquisition was achieved using Empower software from Waters (Etten-Leur, The Netherlands).
Remoxipride
SPE-LC Method
For the precipitated plasma samples, on-line SPE was combined with HPLC and mass spectrometry according to Stevens et al (26). Briefly, a pretreated sample was loaded into a Hysphere GP resin cartridge column (10 × 2 mm) from Spark Holland (Emmen, The Netherlands) at pH 8.3 and flushed for 1 min. Elution was performed using a low pH and an Altima HP C18 column (150 × 1.0 mm, 5 μm).
LC-Method
For microdialysates, a Kinetex 2.6 μm column (50 × 2.0 mm, XB-C-18) from Phenomenex (Utrecht, The Netherlands) was used at a flow of 0.6 ml/min and placed at 40 oC. The system was a Nexera-X2 UHPLC system, consisting of two ultra high performance liquid chromatography (UHPLC) pumps delivering the high pressure gradient. A SIL-30 AC auto sampler was used to inject 5 μl of the microdialysis sample. The flow was diverted for the first 0.5 min, while a gradient from 10 to 90% methanol in 1.5 min served to elute both remoxipride and raclopride to the mass spectrometer.
Mass Spectrometry
For risperidone, paliperidone and remoxipride, mass spectrometry was used to measure the concentrations. The mass spectrometer was a TSQ Quantum Ultra from Thermo Fisher Scientific (Breda, the Netherlands) and was used in MS/MS mode. Electrospray was used for ionization in the positive mode, nitrogen served as the desolvation gas and argon was used as collision gas. Data acquisition for both remoxipride and risperidone and paliperidone was performed using LCQuan 2.5 software from Thermo Scientific (Breda, The Netherlands).
Risperidone and paliperidone had the following transitions (m/z): 411.2→191.1 (risperidone), 415.2 →195.1 (paliperidone), 415.2 →195.1 (risperidone-D4), 431.2 →211.1 (paliperidone-D4). The scan width was set at 0.2 m/z, the scan time was 0.05 s. Collision was performed at fixed voltages between 27 and 38 V, using a skimmer offset of 2 V.
The transitions (m/z) were 371→242.8 for remoxipride and 247.0→84.0, 112, 218.8 for raclopride. The skimmer offset was 18 and collision was performed between at fixed voltages between 24 and 45 V. Scan width and scan time were the same as above.
Determination of Fraction Unbound in Plasma
To determine the free fraction of paliperidone and risperidone in plasma samples, Centrifree Ultrafiltration Devices from Merck Millipore (Amsterdam, The Netherlands) were used to separate the free from the protein bound risperidone and paliperidone in pooled plasma samples. Both the ultrafiltrate and the original pooled plasma sample (without ultrafiltration step) were measured. The free fraction was calculated according to the following Eq. 1:
| 1 |
For phenytoin and remoxipride, the free fraction in plasma was calculated using a protein binding constant of 91 and 26% respectively which were obtained from literature (27,28).
Determination of In-Vivo Recovery (retro dialysis) (29)
The in-vivo recovery of paliperidone, risperidone phenytoin and remoxipride was calculated using the compound concentration in the dialysate (Cdial) and in PF (Cin) according to the following Eq. 2:
| 2 |
Brain microdialysis data of paliperidone, risperidone, phenytoin and remoxipride were corrected for in-vivo recovery to obtain brainECF and CSF data.
The in-vivo recovery and free fraction for the nine compounds are summarized in Table SII.
Human Data
Table II summarizes the clinical concentration data for acetaminophen and morphine used to assess model performance to predict human concentrations. These data consisted of two clinical studies for acetaminophen and two studies for morphine. All studies were published, except for study 1 for acetaminophen that consists of newly generated data (see in Table II).
Table II.
Summary of the Human Acetaminophen and Morphine Data
| Study design | Acetaminophen | Morphine | |||
|---|---|---|---|---|---|
| Study 1 | Study 2 | Study 1 | Study 2 | ||
| Condition of patients | human with traumatic brain injury | human with nerve-root compression pain | human with traumatic brain injury | human with traumatic brain injury | |
| Nr of patients | 7 | 1 (mean values) | 2 | 1 | |
| Dosage | 1 g, 30 min infusion | 2 g (propacetamol), short infusion | 10 mg, 10 min infusion | 10 mg, 10 min infusion | |
| Nr of samples (sampling time, h) | plasma | 38 (0–6 h) | 11 (0–12 h) | 23 (0–3 h) | 11 (0–3 h) |
| brain ECF or CSF | 54 (0–5.5 h) | 11 (0–13 h) | 74 (0–3 h) | 37 (0–3 h) | |
| data references | Newly generated | (31) | (34) | (33) | |
| Data | |||||
| plasma | X | X | X | X | |
| brainECF | X (“normal” and “injured” brain tissue) | X (“normal” and “injured” brain tissue) | |||
| CSFEVD | X | ||||
| CSFSAS | X | ||||
| fp a | 85% | 85% | – | – | |
| fp references | (32) | (32) | (34) | (33) | |
brain ECF a brain extracellular fluid compartment, CSF EVD a compartment of cerebrospinal fluid in EVD, CSF SAS a compartment of cerebrospinal fluid in subarachnoid space
a free fraction in plasma
Acetaminophen
Acetaminophen human plasma samples and CSF samples were obtained at Poitier University Hospital. Seven patients who had a traumatic brain injury (TBI) were enrolled in the clinical study. They were treated with a 30 min intravenous infusion of 1 g of acetaminophen. CSF samples were collected from a compartment of cerebrospinal fluid in the lateral ventricle (CSFLV) by external-ventricular drainage (EVD) to control the intra-cranial overpressure (named CSFEVD) (30). All clinical studies were conducted according to the Declaration of Helsinki, and written informed consent was obtained from each subject after the approval of the institutional review board at the medical institute. The demographic data is summarized in Table SIII. Acetaminophen concentrations at the start of the study (some patients already received acetaminophen before) were used as an initial value in the plasma compartment. The volume of EVD samples and EVD flow rate during a certain time interval were experimentally determined (Table SIV).
A second human acetaminophen PK dataset (study 2) in plasma and in CSF subarachnoid space (CSFSAS) was obtained from the literature, and was based on patients with nerve-root compression pain (31).
For both datasets, total plasma concentrations for acetaminophen were converted to free plasma concentrations using the free fraction obtained from literature (32).
Morphine
Morphine human concentration-time profiles in plasma and in brainECF were obtained from the physiologically “normal” side of the brain and also from the “injured” side of the brain of TBI patients (33,34). For both datasets, the unbound plasma concentrations were already reported in the original publications (33,34).
Software
The PK analysis was performed using NONMEM version 7.3 (ICON Development Solutions, Hanover, MD, USA) (35). For the brain PK modeling of rat data, the extended least squares estimation method was applied. Other analyses were performed by using the first-order conditional estimation method with interaction (FOCE-I). The compartmental models were defined using the ADVAN6 differential equation solver in NONMEM (35). The plots and the statistical analysis were conducted using R (Version 3.2.5; R Foundation for Statistical Computing, Vienna, Austria) (36).
Model Development
Separate models describing plasma and brain concentration-time profiles for all nine compounds were developed whereby plasma- and brain-related parameters were estimated simultaneously. A naïve pooling approach was used (37), i.e. inter-individual variability in each compound’s data was not quantified, because of the highly standardized experimental settings combined with the homogeneous nature of the animals within each study.
The structural model that was used as a starting point was based on our previously developed models (23–25). To develop a more generally applicable model structure with parameters that can be precisely estimated across drugs, we systematically assessed the following two model structure characteristics.
First, a combined drug dispersion parameter was estimated to capture the CSF and ECF flow and turbulence flow of the drug molecules (38,39).
Second, drug transfer across the BCSFB was excluded. SABCSFB is 2–15 times smaller than SABBB (40–42), suggesting that drug exchange at BCSFB can be ignored from the model.
We evaluated for each drug the validity of the changes to the basic model with regard to a single or two different flow rates for drug dispersion and drug transport at the BCSFB.
Quantification of Active Drug Transport
For the 6 compounds, data were obtained using co-administration of inhibitors of active transport. For all these compounds, the effect of the active transport inhibitors was tested on drug exchange at the BBB (QPL_ECF) and plasma clearance (CLPL), and in combination, as a categorical covariate. (Eq.3)
| 3 |
where PPAT represents the parameter including passive and active transport (net transport), P represents the parameter which takes into account the active transport inhibitors if there is any such effect, Cov is the value of the covariate (0: without an active transport inhibitor, 1: with an active transport inhibitor), θcov represents the effect of the active transport inhibitor.
Model Evaluation
The systematic inclusion of aforementioned factors was guided by a likelihood ratio test, by an adequate parameter estimation precision, by assessment of the parameter correlation matrix to ensure parameter identifiability, and by the graphical evaluation of plots for observations versus predictions and weighted residuals versus time and versus predictions. The likelihood ratio test is based on the assumption that changes in the NONMEM objective function values (OFV, -2 log likelihood) are asymptotically chi-square distributed. A decrease of OFV ≥ 3.84 was considered statistically significant (p < 0.05). For a clear assessment of model predictions and observations we also computed the following metrics (Eq.4 and 5).
| 4 |
| 5 |
where PE is a prediction error, and SMAPE is symmetric mean absolute percentage error (43). YOBS,ij is the jth observation of the ith subject, YPRED,ij is the jth prediction of the ith subject. N is number of observations. In the cases where we did not estimate inter-individual variability, e.g. for all brain PK data, YPRED,ij equals the mean population prediction YPRED,j.
External Model Validation
Validation of the brain PK model was performed by investigating the quality of the prediction of external rat data. The prediction was done as follows, 1) estimating plasma-related parameters (CLPL, QPL-PER1 VPL and VPL_PER1) using the external rat plasma data, 2) fixing the brain-related parameters (QPL_ECF, QDIFF, VECF, VLV, VTFV, VCM, and VSAS) to the values which were estimated from the brain PK model and 3) predicting the brainECF or CSF concentrations using estimated rat plasma-related parameters and fixed brain-related parameters.
Plasma PK Analysis of External Rat Data
The plasma-related parameters including inter-individual variability on these parameters and residual errors were estimated using the external rat plasma data. We used a mixed effects modeling approach to investigate the predictability of the brain concentration based on each plasma concentration. The same plasma model structure, which was obtained from the brain PK model was applied for each compound. Inter-individual variability were tested on each PK parameter using an exponential model (Eq. 6).
| 6 |
where θi represents the parameters of the ith subject, θ represents the population mean value of the parameter, and ηi is the random effect of the ith subject under the assumption of a normal distribution with a mean value of 0 and variance of ω2.
A proportional error model and the mixed error model (Eq. 7-8) were tested for the residual errors:
| 7 |
| 8 |
where Cij represents the jth observed concentration of the ith subject, YPRED,ij represents the jth predicted concentration of the ith subject, and εij is the random effect of the jth observed concentration of the ith subject under the assumption of a normal distribution with a mean value of 0 and variance of σ2.
Model selection was guided by a likelihood ratio test with p < 0.05 and by the precision of the parameter estimates.
Handling of the Brain-Related Parameter Values
For QPL_ECF, QDIFF, the same values, which were estimated from the brain PK model, were used for acetaminophen and remoxipride, respectively. VECF, VLV, VTFV, VCM, and VSAS are system-specific parameters, therefore, the same rat physiological values were used, indicated in Table III.
Table III.
Parameter Estimates for the Nine Compounds in Rat
| Parameter estimates (RSE, %) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Acetaminophen | Atenolol | Methotrexate | Morphine | Paliperidone | Phenytoin | Quinidine | Remoxipride | Risperidone | ||
| CLPL | mL/min | 15.9 (4.80) | 6.09 (6.80) | 8.12 (3.90) | 21.6 (2.60) | 192 (7.80) | 44.7 (5.40) | 152 (2.40) | 114 (2.70) | 465 (13.0) |
| QPL_PER1 | mL/min | 29.2 (19.9) | 6.55 (12.2) | 28.1 (18.0) | 8.72 (3.80) | NA | 133 (18.2) | 1070 (5.80) | 105 (10.7) | NA |
| QPL_PER2 | mL/min | NA | NA | 1.50 (14.1) | 53.3 (5.80) | NA | NA | NA | NA | NA |
| QPL_ECF | mL/min | 0.0281 (12.1) | 0.00749 (15.6) | 0.00109 (10.6) | 0.00458 (7.40) 0.00750 (8.90)d | 0.0123 (12.8) | 0.00340 (13.7) | 0.0354 (3.90) | 0.0141 (8.90) | 0.0247 (18.6) |
| QLV_PL | mL/min | NA | NA | 0.105 (10.6) | NA | NA | NA | NA | NA | NA |
| QECF_ICF | mL/min | NA | NA | NA | NA | 0.0126 (21.0) | NA | 0.0250 (6.70) | NA | NA |
| QDIFF | mL/min | 0.0556 (10.3) | 0.0205 (11.7) | 0.0598 (9.30) | 0.0200 (4.30) | 0.0248 (10.7) | 0.0133 (13.0) | 0.0237 (2.40) | 0.0176 (8.20) | 0.0254 (15.0) |
| VPL | mL | 65.7 (25.4) | 115 (12.3) | 51.2 (20.3) | 118 (6.40) | 28400 (8.00) | 2890 (7.90) | 194 (37.5) | 286 (16.6) | 60000 (13.0) |
| VPER1 | mL | 219 (8.90) | 280 (19.0) | 210 (7.20) | 1210 (7.80) | NA | 5320 (7.80) | 13300 (3.00) | 2310 (6.40) | NA |
| VPER2 | mL | NA | NA | 114 (6.60) | 570 (4.40) | NA | NA | NA | NA | NA |
| VECF e (53) | mL | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX | 0.29 FIX |
| VICF e (73) | mL | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX | 1.44 FIX |
| VLV e (45,46) | mL | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX |
| VTFV e (74) | mL | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX | 0.05 FIX |
| VCM e (48,49) | mL | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX | 0.017 FIX |
| VSAS e (74,75) | mL | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX | 0.18 FIX |
| fraction | % | 93.3(1.20) | NA | NA | NA | NA | NA | NA | NA | NA |
| Impact of active transport on | ||||||||||
| QPL_ECF | NA | NA | 4.09 (3.7)a | 1.62 (11.4)b | 0.434 (11.4)b | 0.355 (25.1)b | 4.43 (2.80)b | NA | 1.24 (16.3)b | |
| QLV_PL | NA | NA | 0.410 (16.0)a | NA | NA | NA | NA | NA | NA | |
| Standard deviations of residual error | ||||||||||
| σ _plasma | 0.341 (8.60) | 0.218 (17.9) | 0.522 (7.00) | 0.647 (4.00) | 0.631 (12.1) | 0.444 (8.40) | 0.418 (4.90) | 0.348 (6.80) | 1.44 (5.90) | |
| σ _brainECF | 1.88 (6.70) | 0.480 (17.8) | 0.529 (6.20) | 0.779 (7.40) | 0.946 (6.80) | 0.415 (7.00) | 0.628 (3.30) | 0.673 (6.80) | 0.911 (6.80) | |
| σ _CSFLV | 0.607 (6.40) | NA | 0.663 (5.70) | NA | NA | NA | 0.629 (4.10) | NA | NA | |
| σ _CSFCM | 0.640 (8.20) | NA | 1.00 (9.30) | NA | 0.770 (7.60) | NA | 0.466 (4.10) | NA | 0.827 (9.40) | |
CL PL clearance from the central compartment, Q PL_PER1 inter-compartmental clearance between the central compartment and the peripheral compartment 1, Q PL_PER2 inter-compartmental clearance between the central compartment and the peripheral compartment 2, Q PL_ECF clearance from the central compartment to brainECF, Q LV_PL clearance from CSFLV to the central compartment, Q ECF_ICF inter-compartmental clearance between brainECF and brainICF, Q DIFF drug dispersion rate in brain and CSF, V PL distribution volume of the central compartment, V PER1 distribution volume of the peripheral compartment 1, V PER2 distribution volume of the peripheral compartment 2,V ECF distribution volume of brainECF, V ICF distribution volume of brainICF, V LV distribution volume of CSFLV, V TFV distribution volume of CSFTFV, V CM distribution volume of CSFCM, V SAS distribution volume of CSFSAS, fraction; percentage of the drug which is reabsorbed by enterohepatic circulation, RSE relative standard error.
a; probenecid, b; GF120918 or tariquidar, c; dosage of 10 and 40 mg/kg, d; dosage of 4 mg/kg, e; physiological values
Prediction of brainECF and CSF Concentrations of External Data
Simulations were performed 200 times for each compound. The 95% prediction interval (using the calculated 2.5% tile and 97.5% tile) and the median of the simulated concentrations were plotted together with the external data. Accuracy of the mean population prediction for brain PK data was evaluated with SMAPE mentioned above (Eq. 5).
Translation of the Model to Humans
The translational prediction was performed by the following steps, 1) estimating plasma-related parameters (CLPL, QPL-PER1 VPL and VPL_PER1) using human plasma data, 2) replacing brain-related system-specific parameters (VECF, VLV, VTFV, VCM and VSAS) by human values, 3) applying allometric scaling to the brain-related drug-specific parameters which were estimated with the rat in-vivo data (QPL_ECF and QDIFF), 4) adding clinical sampling procedure related fixed parameters which were obtained from the EVD into the model (QLV_EVD and VEVD) and 5) predicting the brainECF and CSF concentrations using estimated human plasma PK parameters, replacing system-specific parameters, scaling drug-specific parameters and using clinical sampling procedure related fixed parameters. The details of the translational methods for each parameter are explained in Fig. 2.
Fig. 2.

The brain PK model structure and translational methods for each parameter. The brain PK model consists of plasma, brainECF, brainICF, CSFLV, CSFTFV, CSFCM and CSFSAS, which consists of 4 different categories parameters (colors). The scaling method on each parameter is indicated with color coding.
Human Plasma PK Analysis
Plasma-related parameters including inter-individual variability and residual errors were estimated using the human data using the Eqs. 6–8. A 1-compartment, 2-compartment and 3-compartment model were tested. Model selection was guided by a likelihood ratio test with p < 0.05, by the precision and correlation between parameter estimates and by the graphical evaluation of plots for observations versus predictions and weighted residuals versus time and versus predictions.
Replacement of the System-Specific Parameters
System-specific parameters in the brain distribution rat model (VECF, VLV, VTFV, VCM and VSAS) were replaced with the human physiological values, which are available from literature (44–50) (see Table IV).
Table IV.
Parameter Values used for the Translational Prediction to Humans
| Translational methods | Unit | Parameter estimates (RSE, %) | ||
|---|---|---|---|---|
| Acetaminophen | Morphine | |||
| Plasma-related parameters | ||||
| CLPL | estimation from human PK data | mL/min | 562 (20.1) | 3070 (15.8) |
| QPL_PER1 | estimation from human PK data | mL/min | 2060 (31.1) | 3030 (0.60) |
| VPL | estimation from human PK data | mL | 9880 (41.1) | 16000 (35.3) |
| VPER1 | estimation from human PK data | mL | 51900 (18.3) | 95400 (2.50) |
| Brain-related parameters | ||||
| Drug-specific parameters | ||||
| QPL_ECF | allometric scaling | mL/min | 1.92 FIX | 0.513 FIX |
| QDIFF | allometric scaling | mL/min | 3.81 FIX | 1.37 FIX |
| System-specific parameters | ||||
| VECF a (44) | replacement | mL | 240 FIX | 240 FIX |
| VLV a (45–47) | replacement | mL | 22.5 FIX | 22.5 FIX |
| VTFV a (45–47) | replacement | mL | 22.5 FIX | 22.5 FIX |
| VCM a (48,49) | replacement | mL | 7.5 FIX | 7.5 FIX |
| VSAS a (50) | replacement | mL | 90 FIX | 90 FIX |
| Clinical sampling procedure related fixed parameters | ||||
| QLV_EVD | use the fixed parameter | mL/min | values are in supplemental Table IV | |
| VEVD | use the fixed parameter | mL | ||
| Standard deviations of inter-individual variability (estimated from human PK data) | ||||
| ω_CLPL | 0.490 (30.2) | 0.271 (19.9) | ||
| ω_QPL_PER1 | NA | NA | ||
| ω_VPL | NA | 0.596 (20.0) | ||
| ω_VPER1 | 0.235 (22.5) | NA | ||
| Standard deviations of residual error (estimated from human PK data) | ||||
| σ_plasma | 0.250 (8.20) | 0.0960 (22.9) | ||
CL PL clearance from the central compartment, Q PL_PER1 inter-compartmental clearance between the central compartment and the peripheral compartment 1, V PL distribution volume of the central compartment, V PER1 distribution volume of the peripheral compartment 1, Q PL_ECF clearance from the central compartment to brainECF, Q DIFF drug diffusion rate in brain and CSF, V ECF distribution volume of brainECF, V LV distribution volume of CSFLV, V TFV distribution volume of CSFTFV, V CM distribution volume of CSFCM, V SAS distribution volume of CSFSAS, Q LV_EVD flow from CSFLV to CSFEVD, V EVD volume of CSFEVD
a; physiological values
Scaling of the Drug-Specific Parameters
Drug-specific parameters (CLPL_ECF and QDIFF) were scaled to human values using allometric principles following Eq. 9 (18).
| 9 |
where Phuman is the scaled human parameter, Prat is the estimated rat parameter from the model, BWhuman is the average human body weight (70 kg), and BWrat is the average rat body weight (250 g).
Adding Clinical Sampling Procedure Related Fixed Parameters
In addition to those parameters which were used in the rat brain PK model, we have data obtained from the EVD approach, therefore the EVD compartment was added into the translated brain distribution model (see Fig. 2). To describe the PK of acetaminophen in the EVD compartment, the values of flow rate from CSFLV to CSFEVD (QLV_EVD) and the volume of EVD compartment (VEVD) were added into the model. The values of QLV_EVD and VEVD for each patient are obtained from EVD approach and available in Table SIV.
Prediction of Human brainECF and CSF Concentrations
Simulations were performed using the same methods as we mentioned for the external model validation.
Results
The analysis work flow is depicted in Fig. 3. The developed multi-compartmental brain PK model adequately described the data for the nine compounds, as can be observed from the selected observed and predicted concentration-time profiles (Fig. 4a) and the prediction error plots for all of the nine compounds (Fig. 4b). The prediction errors were mostly within two standard deviations of zero, i.e. no systematic differences between observations and predictions were found. No specific trend across time, also with respect to the presence or absence of active transport inhibitors, were observed. More extensive plots for individual observations versus predictions and weighted residuals versus time across drugs, dose levels and active transport inhibitors, are provided in the supplemental material (Figure S1 and S2).
Fig. 3.
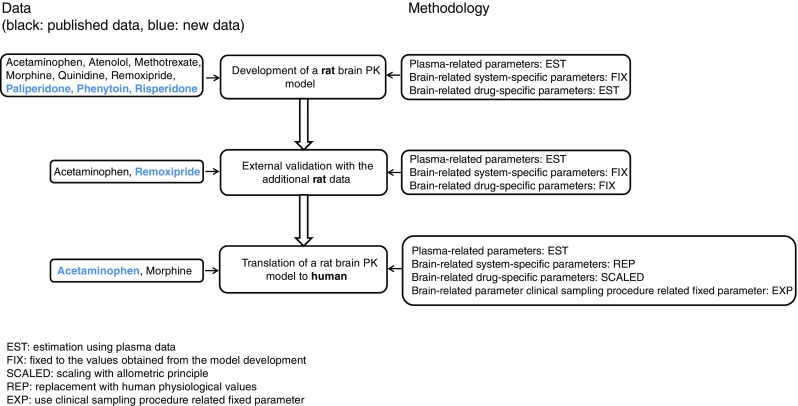
Schematic flow chart of the analysis.
Fig. 4.

Prediction of the multi-compartmental brain PK model. (a) Individual observed drug concentrations (lines and circles) and mean model prediction (solid lines). Unbound concentration (ng/mL) versus time (min) profiles for acetaminophen and morphine. (b) Box-whisker plots for the prediction errors across all nine drugs evaluated. The plots were stratified by brain compartments (panels) and by active transport blockers (colors).
We identified a generally applicable model structure (Fig. 2) with physiologically relevant compartments. The final model consists of plasma, brainECF, brain intracellular fluid compartment (brainICF), CSFLV, compartment of CSF in third and fourth ventricle (CSFTFV), compartment of CSF in cisterna magna (CSFCM) and CSFSAS, which included processes for drug exchange at the BBB (QPL_ECF) and drug dispersion through brainECF and CSF compartments (QDIFF). The parameter estimates were obtained with good precision, and are summarized in Table III.
A single drug dispersion rate (QDIFF) was shown to be sufficient for describing the sum of the drug distribution in the brainECF and CSF for the nine compounds. QDIFF was comparable among the compounds, and ranged between 0.0598 mL/min for methotrexate to 0.0133 mL/min for phenytoin, and could be precisely identified (RSE < 15.0%), suggesting this parameter could be potentially considered to represent a system-specific parameter.
The parameter representing drug transfer at the BBB (QPL_ECF) was critical to quantify drug exchange between blood and brain. QPL_ECF was substantially different between drugs, ranging from 0.0354 mL/min for quinidine to 0.00109 mL/min for methotrexate.
On the other hand, drug exchange at BCSFB was identified only for methotrexate, and could not be identified for the other 8 compounds. For methotrexate, the efflux transport at BCSFB (QLV_PL) was 0.105 mL/min.
Among the nine compounds, clearance between brainECF and brainICF (QECF_ICF) could be estimated for paliperidone and quinidine: QECF_ICF is 0.0250 mL/min for quinidine, and 0.0126 mL/min for paliperidone, implying for quinidine a slightly faster uptake into brainICF after crossing the BBB (Table III).
For morphine, brainECF concentration displayed a nonlinear relationship with dose and plasma concentrations. A categorical dose effect was therefore estimated. Continuous linear or nonlinear concentration-dependent effects to account for this effect were not supported by the data.
No statistically significant impact of P-gp and the combination of MRPs, OATs and OATPs on CLPL could be identified, whereas those transporters were identified to act as efflux transporters at the BBB for our compounds. The P-gp function was quantified on the data of morphine, paliperidone, phenytoin, quinidine, and risperidone, and the impact of the combination of MRPs, OATs and OATPs was quantified on the data of methotrexate, as a categorical covariate on QPL_ECF. The presence of P-gp inhibitors increased the QPL_ECF values of morphine, paliperidone, phenytoin, quinidine, and risperidone by 162, 43.4, 35.5, 443 and 124% respectively. The presence of the inhibitor of MRPs, OATs and OATPs increased the QPL_ECF values of methotrexate by 409%.
The developed model adequately predicted the external rat acetaminophen and remoxipride data. Figure 5 presents the prediction results for the external rat data of acetaminophen and remoxipride using the developed multi-compartmental brain PK model. Prediction of the acetaminophen concentration-time profile in brainECF using the final model captured the external acetaminophen concentration in brainECF well (SMAPE < 61%). Prediction of the remoxipride concentration-time profile in brainECF, CSFLV and CSFCM using the final model also captured the external remoxipride concentrations in brainECF, CSFLV and CSFCM concentrations well (SMAPE < 67, 77, 56%, respectively).
Fig. 5.

Model prediction versus external acetaminophen and morphine data in rat. Individual concentration-time profile of the external data (circles) and prediction from the brain PK model (red lines: median, shaded area is 95% prediction interval). (a) Acetaminophen data were obtained after 200 mg administration, (b) remoxipride data were obtained from the dose group of 0.7, 5.2 and 14 mg/kg. The x-axis represents the time in minutes and the y-axis represents the dose-normalized acetaminophen and remoxipride concentration. The panels are stratified by brain compartments and compounds.
The model was successfully scaled to predict concentration-time profiles of acetaminophen and morphine in human brain compartments. Table IV summarizes the parameter values that were used for the prediction of human plasma, CSFEVD, CSFSAS and brainECF. In Figure 6, the human predictions versus human observations are depicted. The acetaminophen human CSFSAS concentration in the patients with nerve-root compression pain and CSFEVD concentration in the patients with TBI were predicted relatively well (SMAPE < 90 and 66% respectively), even though there is a slightly faster elimination in CSFSAS. Morphine brainECF concentrations in the physiologically “normal” brain tissue of TBI patients were predicted very well (SMAPE < 35%). However, morphine brainECF concentrations were underpredicted when the brainECF concentrations were taken from “injured” brain tissue of TBI patients (SMAPE < 56%).
Fig. 6.

Human brainECF and CSF concentration-time profiles (circles) and prediction from the translational model (red lines: median, shaded area is 95% prediction interval). (a) Acetaminophen data was obtained from plasma, CSFSAS and CSFEVD, (b) morphine data was obtained from plasma and brainECF in “normal” brain and “injured” brain. The x-axis represents the time in minutes and the y-axis represents the acetaminophen and morphine concentration in ng/ml. The panels are stratified by brain compartments and brain conditions.
Discussion
The developed multi-compartmental brain PK model could describe the data of the nine compounds in the rat adequately in the absence and presence of active transport blockers (Fig. 4). After scaling of the model, human brain concentration-time profiles of acetaminophen and morphine could be adequately predicted in several physiological compartments under normal physiological conditions.
The model structure we have derived differs from the ones published earlier by: (i) a combined drug dispersion parameter was estimated to capture the CSF and brainECF flow and turbulence flow of the drug molecules; and (ii) drug transfer across the BCSFB was excluded (23–25). The final model has four different CSF compartments. This model is developed to predict human brain concentration profiles using rat data. In our analysis, rat data was sampled from CSFLV and CSFCM. Since in rats it is anatomically easier to access the CSFCM compartment to obtain drug concentration by microdialysis and by the cisternal puncture methods, there are more data available from CSFCM (51). Through keeping the CSFCM compartment in the model structure, it will be easier to apply the model to additional compounds’ data obtained in animals. Furthermore, substantial differences between CNS compartments may exist, such as a concentration difference between CSFLV and CSFCM for methotrexate and quinidine in rat (24,25). Thus, to predict the drug target site concentration, the location of the CSF sampling site should be taken into account. For human, in clinical studies most CSF samples are taken from other CSF compartments, such as CSFSAS and CSFLV where samples are taken by EVD. Hence, we think that our model structure is a minimal, necessary model structure for translation.
We found that the brain intracellular fluid compartment (brainICF) is required for the description of drug distribution of quinidine and paliperidone, and likely associated with the lipophilic basic nature of quinidine (pKa 13.9, log P 3.4) and paliperidone (pKa 13.7, log P 1.8). For other compounds with a less distinct lipophilic-basic nature, such as for acetaminophen and phenytoin, we have shown that brainICF was not required for the description of concentration-time profiles in the brain. However, for a generally applicable brain PK model, inclusion of this compartment would still be required since prediction of intracellular drug concentrations would be of relevance for CNS drug development as well as prediction of extracellular drug concentrations. Our model and the microdialysis methodology used only allow quantification of extracellular concentrations. However, in combination with PBPK modeling based principles to predict intracellular partitioning, our model will be of significant relevance as it provides the required predictions for unbound extracellular drug concentration kinetics.
A drug exchange parameter across the BCSFB (QLV_PL) was identified for methotrexate only, even though it could not be identified for the other 8 compounds. This suggests that an additional efflux transporter might be present at the BCSFB for which methotrexate is a substrate. It is known that methotrexate is indeed a substrate of various transporters, such as RFC1, MRP, BCRP, OATP and OAT transporters (25), which are not involved in the drug transfer of the other 8 compounds. This result indicates that drug transport at BCSFB still needs to be investigated using data on compounds which are substrates for those transporters. The current model delineates the process that can be used to arrive to the best-performing model for such drugs. We took care to design the modeling process such that the total number of models that need to be fitted is minimal.
We identified a drug dispersion rate parameter that captures drug dispersion from brainECF to CSF. The median estimated drug dispersion flow was 0.0237 mL/min. The magnitude of the drug dispersion rate was approximately ten times faster than the reported physiological CSF flow rate alone (52), and about 100 times faster than the reported physiological brain ECF bulk flow rate (53,54). Since similar values across drugs were identified, the parameter may be considered a system-specific parameter that could be fixed in further analyses (see Table III), to allow for estimation of other processes of interest.
P-gp transport for quinidine, risperidone, paliperidone, morphine and phenytoin was confirmed as efflux transporter at the BBB which were in line with literature (55,56). P-gp transporter effects were not identified at the BCSFB for these 5 P-gp substrates, i.e. CSF concentrations for these compounds were well-described solely by the BBB mediated P-gp transport. The role and contribution of P-gp transporters at the BCSFB is still inconsistent, and both efflux and influx processes have been reported (57–59). Our results however suggest that the function of P-gp may be ignored, since its potential magnitude likely is negligible compared to transport at the BBB, and drug dispersion processes prevail. Nonetheless, overall, we envision that the combination of our dynamical modeling approach with the incorporation of in-vitro assays to characterize active transport across the BBB or BCSFB, may be a fruitful direction to further characterize and disentangle the precise contribution to the brain drug disposition of different drug transport.
The developed model adequately predicted the external acetaminophen and remoxipride rat data, confirming the reliability of the model. Both of these drugs were also used for model development, but the experiments were different and applied somewhat different designs. Since we aimed to generate mean predictions, the variation in numbers of animals is expected to result in limited bias in the modeling. Furthermore, sampling time points were very informatively distributed and any inter-experimental differences in these time points are therefore also considered to be of limited impact on model development. The external validation results indicated that the model is robust with respect to variations in experimental designs and conditions (i.e. the number of rats, sampling times, infusion times, and flow rates of microdialysis).
We consider the developed model structure suited for translational predictions of human brain (target site) concentrations such as required during drug development. The predictive performance in human data ranged between SMAPE of 35–90%. Even though errors <90% may appear large, such < two-fold error is not considered unacceptable when compared to for instance QSAR studies, which are used to predict unbound brain partition coefficients of drugs in drug development (60,61). Secondly, the prediction error is likely inflated because of the use of human data obtained from patients with traumatic brain injury or with nerve-root compression pain. Therefore, larger variability in their physiological condition is expected.
Body weight in combination with allometric scaling was used to scale the parameters to humans, and this resulted in adequate predictions of human brain concentrations for physiologically “normal” brains. Different translational methods for estimation of CNS PK parameters have been reported in the literature. For instance, system-based scaling was applied using volume of brain tissue or brain endothelial surface area (25,62), but allometric scaling using body weight (our approach) was supported by work from others in the literature (63–66). Based on our current approach, reasonable predictions were obtained. Therefore, we suggest that the allometric scaling approach may indeed be appropriate although it would be worthwhile to investigate alternative approaches.
Our model was developed based on healthy rats and then translated to human data that was partly based on patients with severe brain injuries. Indeed, observed human morphine concentrations in brainECF obtained from the “injured” side of the brain of the TBI patients was higher than the prediction from the translational model (Fig. 6). It is known that the BBB permeability is increased after TBI, which may be the reason for the under-prediction of our translational model for those data (67,68). Therefore, for predictions in patients with pathological conditions that alter the integrity of BBB or BCSFB barriers, or brain fluid flows, our model should be further extended with additional physiological details.
Conclusion
A multi-compartmental brain PK model structure was developed across a wide range of drugs with different physicochemical properties. The model structure was shown to be of relevance for the scaling of brain concentrations in humans. As such, the developed model structure can be used to inform the prediction of relevant target site concentrations in humans and aid in the translational development of CNS targeted drugs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Model prediction (solid lines) versus observation (lines and circles) of the nine compounds in rat for each dose and without and with co-administration of active transport blockers. The x-axis represents the time in minutes and the y-axis represents the concentration of the nine compounds in ng/ml. The panel is stratified by brain compartments and by active transport blockers (colors). (EPS 532 kb)
Weighted residuals versus time of the nine compounds in rat. The x-axis represents the time in minutes and the y-axis represents the weighted residuals of the nine compounds. The panel is stratified by brain compartments and by active transport blockers (colors). (EPS 263 kb)
(DOCX 33 kb)
(DOCX 38 kb)
(DOCX 34 kb)
(DOCX 40 kb)
ACKNOWLEDGMENTS AND DISCLOSURES
This research article was prepared within the framework of project no. D2-501 of the former Dutch Top Institute Pharma, currently Lygature (Leiden, the Netherlands; www.lygature.org). The authors have no conflicts of interest that are directly relevant to the contents of this research article.
ABBREVIATIONS
- BBB
Blood–brain barrier
- BCSFB
Blood–cerebrospinal fluid barrier
- brainECF
Brain extracellular fluid compartment
- brainICF
Brain intracellular fluid compartment
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- CSFCM
Compartment of cerebrospinal fluid in cisterna magna
- CSFEVD
Compartment of cerebrospinal fluid obtained by external-ventricular drainage
- CSFLV
Compartment of cerebrospinal fluid in lateral ventricle
- CSFSAS
Compartment of cerebrospinal fluid in subarachnoid space
- CSFTFV
Compartment of cerebrospinal fluid in third and fourth ventricle
- ECF
Extracellular fluid
- EVD
External-ventricular drainage
- ICF
Intracellular fluid
- IPRED
Individual prediction
- PF
Perfusion fluid
- PK
Pharmacokinetic
- PRED
Population predictions
- SABBB
Surface area of blood–brain barrier
- SABCSFSB
Surface area of blood–cerebrospinal barrier
- SMAPE
Symmetric mean absolute percentage error
- SPE
Solid phase extraction
- TBI
Traumatic brain injury
References
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates. Nat Rev Drug Discov. 2004;3:711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Hurko O, Ryan JL. Translational research in central nervous system drug discovery. J Am Soc Exp Neurother. 2005;2(4):671–82. doi: 10.1602/neurorx.2.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov Nat Publ Group. 2014;13(6):419–31. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 4.Danhof M, de Jongh J, De Lange ECM, Della Pasqua O, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol. 2007;47(1):357–400. doi: 10.1146/annurev.pharmtox.47.120505.105154. [DOI] [PubMed] [Google Scholar]
- 5.De Lange ECM. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS. Fluids Barriers CNS. 2013;10(1):1–16. doi: 10.1186/2045-8118-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westerhout J, Danhof M, Lange ECMDE. Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J Pharm Sci. 2011;100(9):3577–93. doi: 10.1002/jps.22604. [DOI] [PubMed] [Google Scholar]
- 7.Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25(8):1737–50. doi: 10.1007/s11095-007-9502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbott NJ. Prediction of blood-brain barrier permeation in drug discovery from in vivo, in vitro and in silico models. Drug Discov Today Technol. 2004;1(4):407–16. doi: 10.1016/j.ddtec.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 9.de Lange ECM, Hammarlund-Udenaes M. Translational aspects of blood-brain barrier transport and central nervous system effects of drugs: from discovery to patients. Clin Pharmacol Ther. 2015;97(4):380–94. doi: 10.1002/cpt.76. [DOI] [PubMed] [Google Scholar]
- 10.Loryan I, Fridén M, Hammarlund-Udenaes M. The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS. 2013;10(6):1–9. doi: 10.1186/2045-8118-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loryan I, Sinha V, Mackie C, Van Peer A, Drinkenburg W, Vermeulen A, et al. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm Res. 2014;32(8):2203–19. doi: 10.1007/s11095-014-1319-1. [DOI] [PubMed] [Google Scholar]
- 12.Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7(1):84–96. doi: 10.1016/S1474-4422(07)70326-5. [DOI] [PubMed] [Google Scholar]
- 13.Hammarlund-Udenaes M. The use of microdialysis in CNS drug delivery studies: pharmacokinetic perspectives and results with analgesics and antiepileptics. Adv Drug Deliv Rev. 2000;45(2-3):283–94. doi: 10.1016/S0169-409X(00)00109-5. [DOI] [PubMed] [Google Scholar]
- 14.Van Hasselt JGC, Van Der Graaf PH. Towards integrative systems pharmacology models in oncology drug development. Drug Discov Today Technol. 2015;15:1–8. doi: 10.1016/j.ddtec.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Collins JM, Dedrick RL. Distributed model for drug delivery to CSF and brain tissue. Am J Physiol. 1983;245(3):R303–10. doi: 10.1152/ajpregu.1983.245.3.R303. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, et al. Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther. 2005;313(3):1254–62. doi: 10.1124/jpet.104.079319. [DOI] [PubMed] [Google Scholar]
- 17.Kielbasa W, Kalvass JC, Stratford R. Microdialysis evaluation of atomoxetine brain penetration and central nervous system pharmacokinetics in rats. Drug Metab Dispos. 2009;37(1):137–42. doi: 10.1124/dmd.108.023119. [DOI] [PubMed] [Google Scholar]
- 18.Kielbasa W, Stratford RE. Exploratory translational modeling approach in drug development to predict human brain pharmacokinetics and pharmacologically relevant clinical doses. Drug Metab Dispos. 2012;40(5):877–83. doi: 10.1124/dmd.111.043554. [DOI] [PubMed] [Google Scholar]
- 19.Badhan RKS, Chenel M, Penny JI. Development of a physiologically-based pharmacokinetic model of the rat central nervous system. Pharmaceutics. 2014;6(1):97–136. doi: 10.3390/pharmaceutics6010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball K, Bouzom F, Scherrmann J-M, Walther B, Declèves X. Comparing translational population-PBPK modelling of brain microdialysis with bottom-up prediction of brain-to-plasma distribution in rat and human. Biopharm Drug Dispos. 2014;25(8):485–99. doi: 10.1002/bdd.1908. [DOI] [PubMed] [Google Scholar]
- 21.Deo AK, Theil F-P, Nicolas J-M. Confounding parameters in preclinical assessment of blood-brain barrier permeation: an overview with emphasis on species differences and effect of disease states. Mol Pharm. 2013;10(5):1581–95. doi: 10.1021/mp300570z. [DOI] [PubMed] [Google Scholar]
- 22.Gaohua L, Neuhoff S, Johnson TN, Rostami-hodjegan A, Jamei M. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab Pharmacokinet. 2016;31(3):224–33. doi: 10.1016/j.dmpk.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Westerhout J, Ploeger B, Smeets J, Danhof M, de Lange ECM. Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J. 2012;14(3):543–53. doi: 10.1208/s12248-012-9366-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westerhout J, Smeets J, Meindert D, De Lange ECM. The impact of P-gp functionality on non-steady state relationships between CSF and brain extracellular fluid. J Pharmacokinet Pharmacodyn. 2013;40(3):327–42. doi: 10.1007/s10928-013-9314-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westerhout J, Van Den Berg D-J, Hartman R, Danhof M, De Lange ECM. Prediction of methotrexate CNS distribution in different species—influence of disease conditions. Eur J Pharm Sci. 2014;57:11–24. doi: 10.1016/j.ejps.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Stevens J, Ploeger BA, Van Der Graaf PH, Danhof M, De Lange ECM. Systemic and direct nose-to-brain transport pharmacokinetic model for remoxipride after intravenous and intranasal administration. Drug Metab Dispos. 2011;39(12):2275–82. doi: 10.1124/dmd.111.040782. [DOI] [PubMed] [Google Scholar]
- 27.Srikanth CH, Chaira T, Sampathi S, V B S, Bambal RB. Correlation of in vitro and in vivo plasma protein binding using ultracentrifugation and UPLC-tandem mass spectrometry. Analyst. 2013;138(20):6106–16. [DOI] [PubMed]
- 28.Widman M, Nilsson L, Bryske B, Lundström J. Disposition of remoxipride in different species. Species differences in metabolism. Arzneimittelforschung. 1993;43(3):287–97. [PubMed] [Google Scholar]
- 29.Ståhle L, Segersvärd S, Ungerstedt U. A comparison between three methods for estimation of extracellular concentrations of exogenous and endogenous compounds by microdialysis. J Pharmacol Methods. 1991;25(1):41–25. doi: 10.1016/0160-5402(91)90021-V. [DOI] [PubMed] [Google Scholar]
- 30.Dey M, Jaffe J, Stadnik A, Awad IA. External ventricular drainage for intraventricular hemorrhage. Curr Neurol Neurosci Rep. 2012;12(1):24–33. doi: 10.1007/s11910-011-0231-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannwarth B, Netter P, Lapicque F, Gillet P, Péré P, Boccard E, et al. Plasma and cerebrospinal fluid concentrations of paracetamol after a single intravenous dose of propacetamol. Br J Clin Pharmacol. 1992;34(1):79–81. doi: 10.1111/j.1365-2125.1992.tb04112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazzard B, Ford-Hutchinson A, Smith M, Williams R. The binding of paracetamol to plasma proteins of man and pig. J Pharm Pharmacol. 1973;25(12):964–7. doi: 10.1111/j.2042-7158.1973.tb09987.x. [DOI] [PubMed] [Google Scholar]
- 33.Bouw R, Ederoth P, Lundberg J, Ungerstedt U, Nordström C-H, Hammarlund-Udenaes M. Increased blood – brain barrier permeability of morphine in a patient with severe brain lesions as determined by microdialysis Case report. Acta Anaesthesiol Scand. 2001;45(3):390–2. doi: 10.1034/j.1399-6576.2001.045003390.x. [DOI] [PubMed] [Google Scholar]
- 34.Ederoth P, Tunblad K, Bouw R, Lundberg CJ, Ungerstedt U, Nordstrom CH, et al. Blood-brain barrier transport of morphine in patients with severe brain trauma. Br J Clin Pharmacol. 2003;57(4):427–35. doi: 10.1046/j.1365-2125.2003.02032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beal S, Sheiner L, Boeckmann A, Bauer R. NONMEM user’s guides. Ellicott City: Icon Development Solutions; 2010. [Google Scholar]
- 36.R Core Team. R: A Language and Environment for StatisticalComputing, R Foundation for Statistical Computing. 2013. Available from: http://www.R-project.org/.
- 37.Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetics parameters. I. Michaelis-Menten model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1980;8(6):553–71. doi: 10.1007/BF01060053. [DOI] [PubMed] [Google Scholar]
- 38.Hettiarachchi HDM, Hsu Y, Harris TJ, Penn R, Linninger AA. The effect of pulsatile flow on intrathecal drug delivery in the spinal canal. Ann Biomed Eng. 2011;39(10):2592–602. doi: 10.1007/s10439-011-0346-x. [DOI] [PubMed] [Google Scholar]
- 39.Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11(26):1–32. doi: 10.1186/2045-8118-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skipor J, Thiery JC. The choroid plexus – cerebrospinal fluid system: undervaluated pathway of neuroendocrine signaling into the brain. Acta Neurobiol Exp. 2008;68(3):414–28. doi: 10.55782/ane-2008-1708. [DOI] [PubMed] [Google Scholar]
- 41.Kumar G, Smith QR, Hokari M, Parepally J, Duncan MW. Brain uptake, pharmacokinetics, and tissue distribution in the rat of neurotoxic N-butylbenzenesulfonamide. Toxicol Sci. 2007;97(2):253–64. doi: 10.1093/toxsci/kfm057. [DOI] [PubMed] [Google Scholar]
- 42.Strazielle N, Ghersi-Egea JF. Choroid plexus in the central nervous system: biology and physiopathology. J Neuropathol Exp Neurol. 2000;59(7):561–74. doi: 10.1093/jnen/59.7.561. [DOI] [PubMed] [Google Scholar]
- 43.Makridakis S. Accuracv concerns measures: theoretical and practical concerns. Int J Forecast. 1993;9:527–9. doi: 10.1016/0169-2070(93)90079-3. [DOI] [Google Scholar]
- 44.Begley DJ, Bradbury MW, Kreuter J. The blood–brain barrier and drug delivery to the CNS. New York: Marcel Dekker, Inc.; 2000. [Google Scholar]
- 45.Condon P, Wyper D, Grant R, Patterson J, Hadley D, Teasdale G, et al. Use of magnetic resonance imaging to measure intracranial cerebrospinal fluid volume. Lancet. 1986;327(8494):1355–7. doi: 10.1016/S0140-6736(86)91666-1. [DOI] [PubMed] [Google Scholar]
- 46.Kohn MI, Tanna NK, Herman GT, Resnick SM, Mozley PD, Gur RE, et al. Analysis of brain and cerebrospinal fluid volumes with MR imaging. Part I. Methods, reliability, and validation. Radiology. 1991;178(1):115–22. doi: 10.1148/radiology.178.1.1984289. [DOI] [PubMed] [Google Scholar]
- 47.Dickey CC, Shenton ME, Hirayasu Y, Fischer I, Voglmaier MM, Niznikiewicz MA, et al. Large CSF volume not attributable to ventricular volume in schizotypal personality disorder. Am J Psychiatry. 2000;157(1):48–54. doi: 10.1176/ajp.157.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robertson EG. Developmental defects of the cisterna magna and dura mater. J Neurol Neurosurg Psychiatry. 1949;12(1):39–51. doi: 10.1136/jnnp.12.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adam R, Greenberg JO. The mega cisterna magna. J Neurosurg. 1978;48(2):190–2. doi: 10.3171/jns.1978.48.2.0190. [DOI] [PubMed] [Google Scholar]
- 50.Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS. BioMed Cen Ltd. 2011;8(7):1–7. doi: 10.1186/2045-8118-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahat MYA, Fakrudeen Ali Ahamed N, Chandrasekaran S, Rajagopal S, Narayanan S, Surendran N, et al. An improved method of transcutaneous cisterna magna puncture for cerebrospinal fluid sampling in rats. J Neurosci Methods. 2012;211(2):272–9. doi: 10.1016/j.jneumeth.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 52.Cserrl H. Potassium exchange between cerebrospinal fluid, plasma, and brain. Am J Physiol. 1965;209:1219–26. doi: 10.1152/ajplegacy.1965.209.6.1219. [DOI] [PubMed] [Google Scholar]
- 53.Cserr H, Cooper D, Suri P, Patlak C. Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am J Physiol. 1981;240(4):319–28. doi: 10.1152/ajprenal.1981.240.4.F319. [DOI] [PubMed] [Google Scholar]
- 54.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–52. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv Drug Deliv Rev. 1999;36(2-3):179–94. doi: 10.1016/S0169-409X(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 56.Tsuji A, Tamai I. Blood-brain barrier function of P-glycoprotein. Adv Drug Deliv Rev. 1997;25(2-3):287–98. doi: 10.1016/S0169-409X(97)00504-8. [DOI] [Google Scholar]
- 57.Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, et al. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood– cerebrospinal-fluid drug-permeability barrier. Med Sci. 1999;96(7):3900–5. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86(2):695–8. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kassem NA, Deane R, Segal MB, Chen R, Preston JE. Thyroxine (T4) transfer from CSF to choroid plexus and ventricular brain regions in rabbit: contributory role of P-glycoprotein and organic anion transporting polypeptides. Brain Res. 2007;1181:44–50. doi: 10.1016/j.brainres.2007.08.052. [DOI] [PubMed] [Google Scholar]
- 60.Chen H, Winiwarter S, Fridén M, Antonsson M, Engkvist O. In silico prediction of unbound brain-to-plasma concentration ratio using machine learning algorithms. J Mol Graph Model. 2011;29(8):985–95. doi: 10.1016/j.jmgm.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Loryan I, Sinha V, Mackie C, Van Peer A, Drinkenburg WH, Vermeulen A, et al. Molecular properties determining unbound intracellular and extracellular brain exposure of CNS drug candidates. Mol Pharm. 2015;12(2):520–32. doi: 10.1021/mp5005965. [DOI] [PubMed] [Google Scholar]
- 62.Johnson M, Kozielska M, Pilla Reddy V, Vermeulen A, Barton HA, Grimwood S, et al. Translational modeling in schizophrenia: predicting human dopamine D2 receptor occupancy. Pharm Res. 2016;33(4):1003–17. doi: 10.1007/s11095-015-1846-4. [DOI] [PubMed] [Google Scholar]
- 63.Li CH, Stratford RE, Velez de Mendizabal N, Cremers TI, Pollock BG, Mulsant BH, et al. Prediction of brain clozapine and norclozapine concentrations in humans from a scaled pharmacokinetic model for rat brain and plasma pharmacokinetics. J Transl Med. 2014;12(1):1–11. doi: 10.1186/1479-5876-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melhem M. Perspectives in pharmacology translation of central nervous system occupancy from animal models: application of pharmacokinetic / pharmacodynamic modeling. J Pharmacol Exp Ther. 2013;347:2–6. doi: 10.1124/jpet.112.199794. [DOI] [PubMed] [Google Scholar]
- 65.Stevens J, Ploeger BA, Hammarlund-Udenaes M, Osswald G, Van Der Graaf PH, Danhof M, et al. Mechanism-based PK-PD model for the prolactin biological system response following an acute dopamine inhibition challenge: quantitative extrapolation to humans. J Pharmacokinet Pharmacodyn. 2012;39(5):463–77. doi: 10.1007/s10928-012-9262-4. [DOI] [PubMed] [Google Scholar]
- 66.Lu Y, Barton HA, Leung L, Zhang L, Hajos-Korcsok E, Nolan CE, et al. Cerebrospinal fluid β-amyloid turnover in the mouse, dog, monkey and human evaluated by systematic quantitative analyses. Neurodegener Dis. 2013;12(1):36–50. doi: 10.1159/000341217. [DOI] [PubMed] [Google Scholar]
- 67.Beaumont A, Marmarou A, Hayasaki K, Barzo P, Fatouros P, Corwin F, et al. The permissive nature of Blood Brain Barrier (BBB) opening in edema formation following traumatic brain injury. Acta Neurochir Suppl. 2000;76:125–9. doi: 10.1007/978-3-7091-6346-7_26. [DOI] [PubMed] [Google Scholar]
- 68.Başkaya MK, Rao AM, Doğan A, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett. 1997;226(1):33–6. doi: 10.1016/S0304-3940(97)00239-5. [DOI] [PubMed] [Google Scholar]
- 69.de Lange EC, Danhof M, de Boer AG, Breimer DD. Critical factors of intracerebral microdialysis as a technique to determine the pharmacokinetics of drugs in rat brain. Brain Res. 1994;666(1):1–8. doi: 10.1016/0006-8993(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 70.Groenendaal D, Freijer J, de Mik D, Bouw MR, Danhof M, de Lange ECM. Population pharmacokinetic modelling of non-linear brain distribution of morphine: influence of active saturable influx and P-glycoprotein mediated efflux. Br J Pharmacol. 2007;151(5):701–12. doi: 10.1038/sj.bjp.0707257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bouw MR, Gårdmark M, Hammarlund-Udenaes M. Pharmacokinetic-pharmacodynamic modelling of morphine transport across the blood-brain barrier as a cause of the antinociceptive effect delay in rats—a microdialysis study. Pharm Res. 2000;17(10):1220–7. doi: 10.1023/A:1026414713509. [DOI] [PubMed] [Google Scholar]
- 72.Stevens J, Suidgeest E, van der Graaf PH, Danhof M, de Lange ECM. A new minimal-stress freely-moving rat model for preclinical studies on intranasal administration of CNS drugs. Pharm Res. 2009;26(8):1911–7. doi: 10.1007/s11095-009-9907-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thorne RG, Hrabe S, Nicholson C, Robert G. Diffusion of epidermal growth factor in rat brain extracellular space measured by integrative optical imaging. J Neurophysiol. 2004;92(6):3471–81. doi: 10.1152/jn.00352.2004. [DOI] [PubMed] [Google Scholar]
- 74.Levinger IM. The cerebral ventricles of the rat. J Anat. 1971;108(3):447–51. [PMC free article] [PubMed] [Google Scholar]
- 75.Bass NH, Lundborg P. Postnatal development of bulk flow in the cerebrospinal fluid system of the albino rat: clearance of carboxyl-(14 C)inulin after intrathecal infusion. Brain Res. 1973;52:323–32. doi: 10.1016/0006-8993(73)90668-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Model prediction (solid lines) versus observation (lines and circles) of the nine compounds in rat for each dose and without and with co-administration of active transport blockers. The x-axis represents the time in minutes and the y-axis represents the concentration of the nine compounds in ng/ml. The panel is stratified by brain compartments and by active transport blockers (colors). (EPS 532 kb)
Weighted residuals versus time of the nine compounds in rat. The x-axis represents the time in minutes and the y-axis represents the weighted residuals of the nine compounds. The panel is stratified by brain compartments and by active transport blockers (colors). (EPS 263 kb)
(DOCX 33 kb)
(DOCX 38 kb)
(DOCX 34 kb)
(DOCX 40 kb)