

Abstract
AIM
To develop a human in vitro model of non-alcoholic fatty liver disease (NAFLD), utilising primary hepatocytes cultured in a three-dimensional (3D) perfused platform.
METHODS
Fat and lean culture media were developed to directly investigate the effects of fat loading on primary hepatocytes cultured in a 3D perfused culture system. Oil Red O staining was used to measure fat loading in the hepatocytes and the consumption of free fatty acids (FFA) from culture medium was monitored. Hepatic functions, gene expression profiles and adipokine release were compared for cells cultured in fat and lean conditions. To determine if fat loading in the system could be modulated hepatocytes were treated with known anti-steatotic compounds.
RESULTS
Hepatocytes cultured in fat medium were found to accumulate three times more fat than lean cells and fat uptake was continuous over a 14-d culture. Fat loading of hepatocytes did not cause any hepatotoxicity and significantly increased albumin production. Numerous adipokines were expressed by fatty cells and genes associated with NAFLD and liver disease were upregulated including: Insulin-like growth factor-binding protein 1, fatty acid-binding protein 3 and CYP7A1. The metabolic activity of hepatocytes cultured in fatty conditions was found to be impaired and the activities of CYP3A4 and CYP2C9 were significantly reduced, similar to observations made in NAFLD patients. The utility of the model for drug screening was demonstrated by measuring the effects of known anti-steatotic compounds. Hepatocytes, cultured under fatty conditions and treated with metformin, had a reduced cellular fat content compared to untreated controls and consumed less FFA from cell culture medium.
CONCLUSION
The 3D in vitro NAFLD model recapitulates many features of clinical NAFLD and is an ideal tool for analysing the efficacy of anti-steatotic compounds.
Keywords: Non-alcoholic fatty liver disease, Liver disease, Three-dimensional cell culture, Organ-on-chip, Primary cell culture, Fatty liver, Hepatocytes
Core tip: We report the development of an in vitro, fully human, three-dimensional cell culture model of non-alcoholic fatty liver disease (NAFLD). The model recapitulates key features of clinical NAFLD, with primary human hepatocytes continuously loaded with fat, for up to 14 d of culture, without causing any hepatotoxicity. Fat loading caused transcriptomic, proteomic and metabolic changes to the hepatocytes, including reduced activity of CYP3A4 and CYP2C9 enzymes. Fat loading in the model could be modulated using known anti-steatotic drugs (e.g., metformin), demonstrating the utility of the model for screening the efficacy of novel anti-steatotic compounds.
INTRODUCTION
Due to the major increase in the prevalence of obesity, non-alcoholic fatty liver disease (NAFLD) is now considered to be among the most common liver diseases worldwide[1]. NAFLD is regarded as a broad spectrum of pathological conditions ranging from hepatic steatosis through to non-alcoholic steatohepatitis (NASH) and liver fibrosis. Simple steatosis is a relatively benign state of liver injury, defined by the presence of fat in the liver which accounts for more than 5% of the livers weight[2,3]. Fatty livers become vulnerable to further injury from oxidative stress, lipoapoptosis and inflammatory cytokines, which cause the development of NASH and cirrhosis[2]. Hepatic steatosis can often be a self-limiting state, but in up to 25% of cases the disease will progress to NASH and understanding why this transition takes place in certain individuals remains a key focus for research. The study of NAFLD also has important implications for drug metabolism and toxicity. Hepatocytes from steatotic livers often have altered metabolic capacity; with changes in expression of several cytochrome P450 (P450) enzymes and efflux transporter proteins[4,5]. Ultimately this can result in NAFLD patients having altered drug exposure and an elevated risk of adverse drug reactions[4,5].
Hepatic steatosis is characterised by the deposition of cytoplasmic triglycerides as macro- and/or micro-vesicular lipid vacuoles and this excessive accumulation of triglycerides arises from an imbalance in triglyceride acquisition and removal[3]. Investigating the molecular mechanisms that underlie steatosis and the hepatocellular consequences of triglyceride accumulation may be important in understanding the transition from steatosis to NASH and in the development of novel therapeutic interventions. Animal models (e.g., genetic models, diet-induced models) were first used to analyse the molecular basis of NAFLD and its development and many of these findings have been confirmed through clinical studies[6]. However, significant differences exist between humans and pre-clinical animal models in the way in which steatosis progresses, the inflammatory markers that are produced and the triglycerides that accumulate within hepatocytes[7]. Additionally, due to the complexity of these models it is difficult to analyse how the interplay between hepatic fat accumulation, environmental signals (oxidative stress and cytokines) and transcriptional regulation combine to cause the progression from steatosis to the more severe stages of NAFLD.
Some studies have also employed in vitro approaches to help elucidate the molecular mechanisms involved in disease progression, but compared to other liver diseases (e.g., viral hepatitis) only limited data has yet to be acquired using these approaches[8]. Various groups have analysed oleic acid-induced steatosis in hepatoma cell lines (e.g., HepG2 and HuH7) showing the accumulation of triglycerides reduces proliferation and transiently increases expression of inflammatory genes[9-12]. However, due to their immortalised nature, the capacity for drug metabolism of these cells is vastly reduced in comparison to primary cells[13]. They also lack expression of many key enzymes for lipid and fatty acid metabolism[14] and have altered intra- and extra-cellular signalling pathways[15].
Primary human hepatocyte cultures are a valuable model for analysing the mechanisms that underlie hepatic function as they most closely represent clinical conditions. The use of monolayer or sandwich in vitro cultures of hepatocytes has become common for metabolism and toxicology studies, but often these studies last for only a few days as hepatocytes lose metabolic capacity when cultured for longer[16]. Limited studies have looked at the development of steatosis with human hepatocytes in vitro. The combination of oleic and palmitic acid has been shown to promote intracellular fat accumulation in primary human hepatocytes[17] and IL-8 expression was observed to increase after treatment with palmitic acid[18]. These studies involved the culture of cells in the presence of excess free fatty acids (FFA) for short periods of time, generally 12-48 h, in simple monolayer systems. Subsequently, only transient cellular changes were analysed and more long lasting effects of triglyceride accumulation were not studied.
Numerous three-dimensional (3D) cell culture models have now been developed that are proposed to better maintain the differentiated state of primary cells and provide more complex tissue organisation that was not previously possible in conventional 2D systems[19,20]. Using organ-on-a-chip and microfluidic approaches, cell culture micro-environments can be created that recapitulate the mechanical forces, tissue-tissue interfaces, and spatiotemporal chemical gradients of living organs[21,22]. Numerous advanced liver models have been described, which improve the long-term functionality of hepatocytes compared with 2D monocultures or simple 3D spheroid cultures[20,21].
Here, we introduce a cryopreserved primary human hepatocyte model of NAFLD in which the cells are cultured in 3D microtissues on an engineered scaffold that recapitulates the liver capillary bed under perfusion (Figure 1)[23-25]. The microtissues are cultured in the LiverChip® platform which contains twelve isolated bioreactors, each of which has an integrated micro-pump for controlling flow of culture medium through the tissue, creating an oxygen gradient across the tissue, like that observed in the in vivo liver sinusoid (Figure 1)[24]. Liver microtissues were cultured in lean or fat conditions to analyse the effect of triglyceride accumulation and the development of a NAFLD phenotype. Metabolic, transcriptome and phenotypic changes were measured in the system and these were found to mimic many of the changes observed in the clinical condition.
Figure 1.
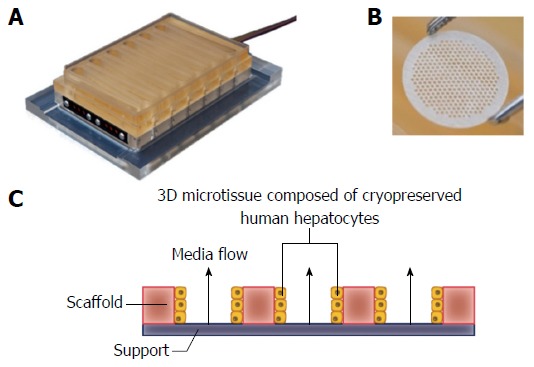
LiverChip® three-dimensional perfused cell culture system. A: The LiverChip® plate contains 12 independent bioreactors for the 3D culture of hepatocytes; B: Hepatocytes form 3D micro-tissue structures in an array of channels in a collagen I-coated scaffold that is contained in each LiverChip® well; C: Schematic representation of how media flows through the channels of each LiverChip scaffold due to the action of a pneumatically operated pumps in the base of the plate. The speed and direction of flow can be adjusted using an electronic controller. 3D: Three-dimensional.
MATERIALS AND METHODS
Hepatocyte culture
Cryopreserved human hepatocytes were purchased from Life Technologies (Paisley, United Kingdom). Cells were thawed according to the instructions provided by the supplier. Viability was assessed using the trypan blue exclusion test and was > 85% for all lots. Cells were seeded into LiverChip® platforms (CN Bio Innovations, United Kingdom) (Figure 1), which were housed in a humidified cell culture incubator at 37 °Cwith 5% CO2. The LiverChip® platforms consist of 12 individual bioreactors in which fluid is recirculated by a pneumatically driven micro-pump across a collagen-coated scaffold, which enables the formation of 3D microtissues in the channels of the scaffold (Figure 1). The platform is covered with a single loose lid, as per a standard microtiter plate enabling access to each of the 12 bioreactors for cell seeding, media change, and sampling.
Cells were seeded with flow in the downward direction through the scaffold for 8 h at a flow rate of 1.0 μL/s. Downward flow encourages cells to seed within the scaffold and form microtissues. Following cell attachment, the flow was changed to the upward direction and maintained at 1.0 μL/s for the remainder of the culture. Hepatocytes were seeded at a density of 6 × 105 viable cells in 1.6 mL of medium per well. The cells were maintained in Williams’ E medium (WEM) containing primary hepatocyte thawing and plating supplements (Life Technologies, United States) for the first 24 h of culture. Thereafter cells were cultured in lean or fat WEM, both of which contained physiologically relevant quantities of insulin (2 nmol/L) and glucose (5.5 mmol/L), as well as standard WEM supplements (0.5% Pen/Strep, 2 mmol GlutaMAX, 15 mmol/L HEPES, 6.25 ng/mL sodium selenite, 6.25 μg/mL transferrin, 1.25 mg/mL BSA, 5.35 μg/mL linoleic acid, 100 nmol/L dexamethasone). The fat media was further supplemented with 600 μmol/L FFA, containing a 2:1 mix of oleate acid and palmitate acid, conjugated to BSA. Complete media changes were performed on all wells every 48-72 h. Hepatocytes were also cultured in 2D in 96-well plates in lean and fat conditions. Post-thawing 5 × 104 viable cells were seeded into each well of a collagen-coated 96-well plate. Cells were cultured in 150 μL media and treated equivalently to 3D-perfused samples.
Oil Red O staining and quantitation
Scaffolds containing microtissues were removed from plates, washed with PBS and fixed in 4% PFA (in PBS) for 15 min. Scaffolds were washed twice with 70% isopropanol for 5 min and then stained for 1 h in Oil Red O stain, which was a 3:2 mix of Oil Red O solution (Sigma Aldrich, United Kingdom) and dH2O. Tissues were washed three times in dH2O and twice in 70% isopropanol to remove non-specifically bound stain. Colour bright field images of stained scaffolds were taken using an inverted light microscope (Leica, United Kingdom). Oil Red O stain was removed from tissues by incubating them with 100% isopropanol for 15 min. The level of Oil Red O in the microtissues was quantified by analysing absorbance at 515 nm. Total cellular protein was determined for each sample following staining. Each scaffold was washed once in PBS and lysed in 0.1 mol NaOH + 2% SDS. Total cellular protein was measured with a Pierce BCA protein assay kit (Thermo Fisher, United Kingdom). The relative fat content of each sample was determined by normalising the level of Oil Red O, expressed as absorbance at 515 nm to the quantity of total protein.
Free fatty acid consumption
The consumption of free fatty acid from cell culture medium was determined using the Free Fatty Acid Quantification kit (Abcam, United Kingdom), following the manufacturers protocol. Free fatty acid consumption was calculated by comparing freshly prepared lean and fat media to conditioned media removed from plates at each media change.
Hepatocyte phenotyping
Albumin secretion was measured in culture supernatant using a human albumin enzyme-linked immunosorbent assay (Assay Pro, United States). Urea was quantified with a colorimetric assay kit (BioAssay Systems, United States) and lactate dehydrogenase (LDH) activity was measured using the CytoTox 96® Non-radioactive cytotoxicity assay (Promega, United Kingdom). Aspartate transaminase (AST) and alanine aminotransferase (ALT) clinical chemistry assays were performed at the College of American Pathologists certified clinical laboratories in the University of Pittsburgh Medical Center (Pittsburgh, United States). Total glutathione was quantified using the GSH-Glo™ Glutathione Assay (Promega, United Kingdom). Microtissues were lysed from scaffolds in glutathione lysis buffer and each lysate was diluted 100-fold in fresh lysis buffer before luminescent quantification of total glutathione against a standard curve according to vendor instructions. The glutathione content was normalised to total cellular protein. Mitochondrial activity was assessed using Cell Proliferation Reagent WST-1 (Roche, United Kingdom) at a 20-fold dilution in cell culture medium.
CYP3A assay
CYP3A activity was measured with the P450-Glo CYP3A4 assay with Luciferin-IPA (Promega, United Kingdom). A complete medium change to medium containing a 1000-fold dilution of Luciferin-IPA was performed and the plate incubated for 1 h at 37 °C, under flow. Samples from each well were placed in a 96-well assay plate and mixed 1:1 with luciferin detection reagent. The plate was incubated at room temperature for 20 min and luminescence measured relative to a standard curve of beetle luciferin potassium salt (Promega, United Kingdom).
RNA isolation and gene expression analysis
Total RNA was extracted from scaffolds cultured for 7 d in fat or lean conditions using TRIzol® Reagent (Ambion, United States) and a chloroform phase separation. RNA was precipitated from aqueous phase samples using 100% isopropanol and RNA pellets were resuspended in dH2O. Reverse transcription and PCR were performed using RT2 First Strand Kit and RT2 Profiler PCR Arrays (Qiagen, United Kingdom) with 0.5 μg RNA analysed on each plate. Human Fatty Liver (PAHS-157Z) RT² Profiler™ PCR Arrays and Human Drug Metabolism (PAHS-002ZC-12) RT² Profiler™ PCR Arrays were used to analyse each sample. Samples were analysed using a Quantstudio 6 real time PCR system (Applied Biosystems, United Kingdom). Ct values from samples were compared and normalised to house-keeping gene expression. The fold-change in each transcript represented on the array plate was determined using RT2 Profiler™ PCR Array Data Analysis (v.3.5) Web Portal (Qiagen, United Kingdom).
Adipokine proteome arrays
Cells were cultured in either fat or lean conditions for 7 d and conditioned media samples were analysed using the Proteome Profiler™ Human Adipokine array kits (R&D Systems, United States), following the manufactures protocol. Arrays were imaged using the LAS-3000 imaging system (Fujifilm, Japan) and images were analysed for pixel density using the ImageQuant software (GE Life Sciences, United Kingdom).
Liquid chromatography tandem mass spectrometry analysis for drug and metabolite quantification
To determine the activities of CYP -1A2, -2C9, -2D6 and -3A4 respectively, the conversion of Tacrine (5 μmol) to 1-hydroxytacrine, Diclofenac (90 μmol) to 4-hydroxydiclodenac, Bufuralol (10 μmol) to hydroxybufuralol and Midazolam (5 μmol) to 1-hydroxymidazolam were analysed (compounds supplied by Sigma Aldrich, United Kingdom). A mixture of the four substrates was prepared at 1000-fold concentration in DMSO and added to microtissues during a full medium change. Cells were incubated for 2 h under standard culture conditions. Metabolites were quantified in cell culture medium by liquid chromatography tandem mass spectrometry. Samples were separated using a Waters Acquity UPLC system with an ACE Excel C18-AR column and analysed on a Waters TQ-S Micro mass spectrometer against quantitative standard curves.
Exposure of hepatocytes to anti-steatotic compounds
Microtissues were cultured for 7 d in lean or fat conditions, before dosing with anti-steatotic compounds. Pioglitazone hydrochloride and metformin hydrochloride (Sigma Aldrich, United Kingdom) were dissolved in DMSO, diluted in cell culture medium and added to microtissues during a full medium change. LiverChip® cultures were maintained in the same lean or fat conditions, in the presence of the compound, for a further 7 d.
Statistical analysis
All the experiments were performed with at least three replicates, with cells obtained from three different primary human hepatocyte donors, if not indicated otherwise. Values reported are mean ± SD, unless otherwise stated. Comparisons between groups were performed using Student’s t test. P values < 0.05 were considered statistically significant.
RESULTS
In the presence of excess free fatty acid, hepatocytes can accumulate fat deposits without inducing cytotoxicity
The 3D perfused in vitro model of NAFLD was created by seeding primary human hepatocytes into the LiverChip® platform and culturing the cells under fat and lean conditions. Both culture conditions contained physiologically relevant levels of glucose and insulin and fat media contained 600 μmol/L FFA. To assess the level of steatosis in microtissues, scaffolds were stained with Oil Red O, which demonstrated significant steatosis and triglyceride accumulation in samples cultured in fat media (Figure 2A). The absorbance of extracted Oil Red O from microtissues was spectrophotometrically determined and normalised to total cellular protein, to give a measure of relative fat content of each sample (Figure 2B). After 7 d of culture the fat media caused a significant increase in lipid accumulation and this further increased after 14 d (Figure 2B). Hepatocytes cultured in fat media also consumed more than four times the amount of FFA from the cell culture media, than equivalent cells in the lean condition (Figure 2C). Fatty acid consumption was determined by measuring changes in the concentration of FFA in the cell culture medium at each media change and comparing it to input media. The loss of FFA from the media was considered to be equivalent to the amount of FFA consumed by the cells.
Figure 2.
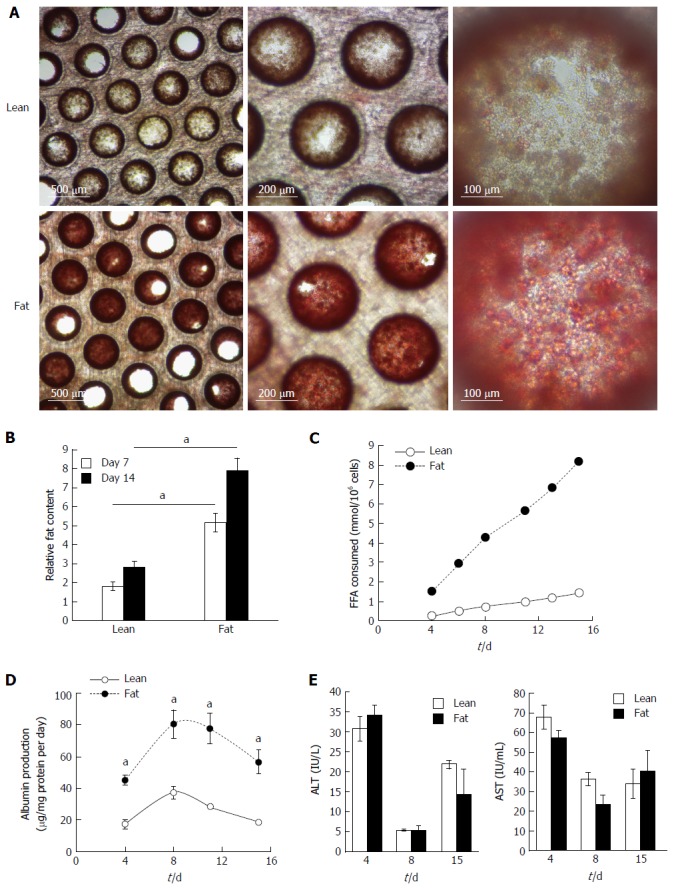
Hepatocytes accumulate intracellular fat over time. Hepatocytes were cultured for 14 d under fat and lean conditions. Fat loading was measured by Oil Red O staining of microtissues, which were observed by microscopy (A), and quantified by absorbance at 510 nm and normalised to total cellular protein to give a relative fat content (B). Fat consumed by cells over 14 d of culture was calculated by analysing culture medium at each media change for the presence of free fatty acids using enzyme-based colorimetric assay (C). Hepatocytes were compared for albumin (D) and ALT/AST production (E). Each time point is a mean of 6 independent cultures ± SD (aP < 0.01).
Cryopreserved human hepatocyte donors have varying levels of basal fat, but all donors studied were observed to accumulate fat when cultured under fat conditions in the model (Supplementary Figure 1). Cell from the same donor were also cultured in 2D static monolayer cultures alongside the 3D-perfused platforms and were observed to produce significantly less albumin, had reduced expression of key metabolic genes and consumed lower quantities of FFA (Supplementary Figure 2). On this basis, the 3D perfused platform was selected for the in vitro NAFLD model.
Overloading hepatic cells with FFA has previously been observed to cause cytotoxic effects[12,17], therefore we analysed the NAFLD model for cell death and liver-specific functions. Under fat conditions hepatocytes were fully viable and fat loading significantly increased their secretion of albumin (Figure 2D), whilst AST and alanine transaminase (ALT) levels were not affected (Figure 2E). LDH release, urea genesis and mitochondrial activity, as measured by WST-1 and cellular glutathione levels, were also not affected by fat loading (Supplementary Figure 3). Taken together this demonstrates hepatocytes cultured in the 3D perfused platform under fat conditions are fully viable, functional and continually become more steatotic.
Fat loading induces transcriptional and proteomic changes in hepatocytes
The development of NAFLD and hepatic steatosis are associated with a range of transcriptomic and proteomic changes, particularly in genes associated with inflammation, cholesterol/lipid metabolism and insulin signalling[3]. We therefore explored whether our 3D in vitro NAFLD model recapitulated the changes observed in vivo. We analysed the expression of key genes associated with fatty liver and a number of these were observed to be upregulated in cells cultured in fatty conditions, including CYP2E1, IGFβ1, PDK4 and CYP7A1 (Figure 3) (Supplementary Table 1). We also explored transcriptional changes in genes associated with hepatic metabolism and found upregulation of several P450 genes, as well as the gene for alcohol dehydrogenase C1 in fat loaded samples (Supplementary Figure 4, Supplementary Table 2). In addition to exploring changes at the transcriptional level we analysed the secretion of key adipokines from cells cultured in fat and lean conditions. Fat loaded hepatocytes secreted a range of adipokines, and in comparison to lean cultured cells, produced increased quantities of pro-inflammatory markers IL-8 and MIF; as well as adipokines associated with fibrosis and wound healing, including fibrinogen and TIMP-1 (Figure 4). The changes observed at the transcriptional and proteomic level in our 3D in vitro model mimic many of the changes commonly associated with NAFLD/steatosis[26,27].
Figure 3.
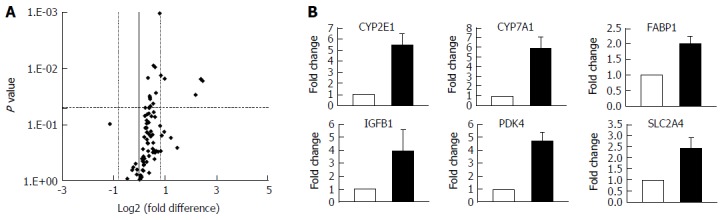
Hepatocytes cultured in fat media have altered gene expression profiles. Hepatocytes were cultured in fat or lean conditions for 7 d before total RNA was extracted and gene expression was compared using Fatty Liver RT2 Profiler PCR Arrays. A: Gene expression changes were defined by a fold change > 1.95 and P ≤ 0.05; B: Fold change in expression in fat vs lean condition of key genes, filled bars = fat, white bars = lean. Data are mean ± SD from nine independent cultures (three donors per condition and n = 3 per donor).
Figure 4.
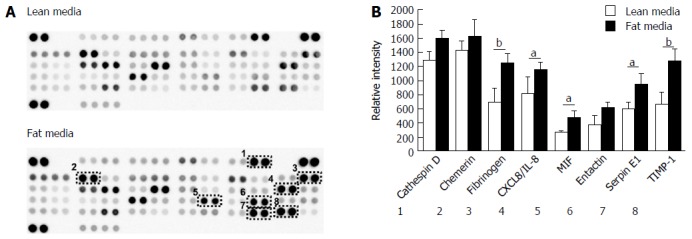
Hepatocytes cultured in fat media secrete increased concentration of adipokines. A: Hepatocytes were cultured in fat or lean conditions for 7 d and secreted protein production was compared by antibody-based adipokine array analysis; B: Adipokine levels were expressed as the relative densitometric intensity. Data are mean ± SEM from nine independent cultures (three donors per condition and n = 3 per donor); aP < 0.05, bP < 0.01.
Metabolic capacity of hepatocytes is affected by fat loading
As the transcription of numerous metabolic genes were altered in the NAFLD model, we investigated how fat loading affected the metabolic capacity of the hepatocytes. The 3D perfused platform has previously been shown to maintain the metabolic capacity of human hepatocytes for extended periods of culture[25]. Initially, we used the CYP3A-Glo assay and found for multiple hepatocyte donors fat loading for seven days caused a significant reduction in P450 activity (Figure 5A). The different donors had varying P450 activity, but when each donor was cultured in the fat media its CYP3A activity was reduced by ≥ 40% (Figure 5A). We explored the activity of CYP1A2, CYP2C9, CYP2D6 and CYP3A4, in more detail by measuring their ability to metabolise probe compounds. Fat loaded hepatocytes had significantly reduced CYP3A4 and CYP2C9 activity when compared to cells cultured in lean media (Figure 5B). CYP3A4 activity was reduced by almost half and CYP2C9 activity was reduced by 30%, however no differences were observed for CYP1A2 and CYP2D6 activity (Figure 5). These observations align with those of previous studies involving human hepatocytes and clinical samples from NAFLD patients, which for example have shown that CYP3A4 activity decreases with disease progression[28,29].
Figure 5.
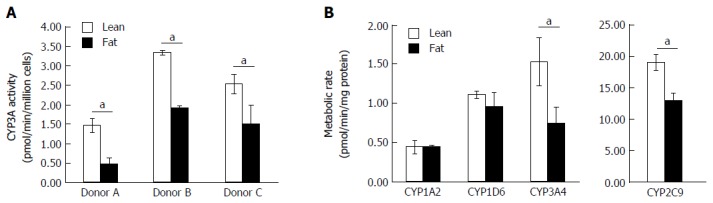
Fat alters the metabolic capacity of hepatocytes. Hepatocytes were cultured in fat or lean conditions for 7 d. A: CYP3A activity was measured in three donors by CYP3A-Glo assay (n = 3 per donor); B: P450 activity of cells was compared using an LC-MS/MS cocktail assay. Production of key metabolites from probe compounds was measured after 2 h exposure and normalised to total cellular protein content. Data is a mean (n = 3) ± SD, aP < 0.05.
Steatosis can be reduced using current NAFLD therapies
We explored whether the level of fat loading into hepatocytes could be modulated using known anti-steatotic compounds. Both pioglitazone and metformin have been shown to have anti-steatotic effects in vivo, ultimately leading to improved clinical outcomes[30,31]. Hepatocytes were cultured for one week in lean or fat conditions and then continued for a second week in the same conditions, but in the presence of pioglitazone, metformin or vehicle control (Figure 6).
Figure 6.
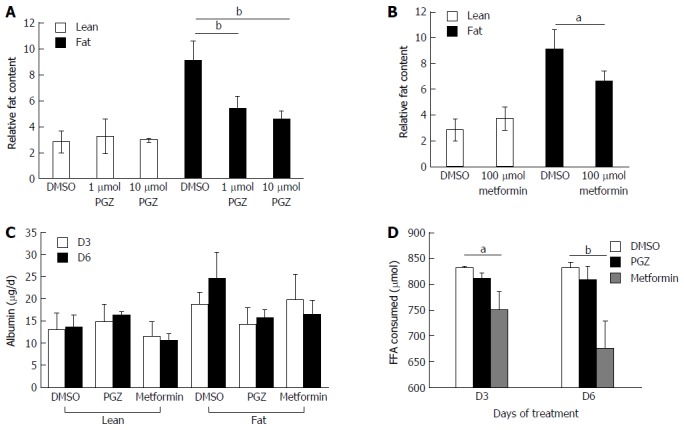
Pioglitazone and Metformin can reduce steatosis in three-dimensional non-alcoholic fatty liver disease model. Hepatocytes were cultured for 7 d in fat or lean conditions, before treatment with anti-steatotic compounds, Pioglitazone (PGZ) (A) and Metformin (B). Cells were treated for 7 d in the continued presence/absence of fat. Fat content in all wells was analysed by Oil Red O staining and normalised to total cellular protein content. Albumin production in treated cultures (data shown for 10 μmol PGZ treated samples) (C). Fat consumed by cells during treatment was calculated by analysing culture medium for the presence of free fatty acids using enzyme-based colorimetric assay (D). Data is a mean ± SD (n = 3), aP < 0.05, bP < 0.01.
Both pioglitazone (Figure 6A) and metformin (Figure 6B) significantly reduced fat loading of hepatocytes, whilst neither affected hepatic function, as determined by albumin production (Figure 6C). Physiologically relevant concentrations of both drugs, which were representative of therapeutic doses achievable in vivo, were used to test in the model. Pioglitazone had the most significant effect on hepatic fat accumulation, reducing the relative fat content by 50% when dosed at 10 μmol (Figure 6A), however we did not observe pioglitazone to affect inflammation or fibrosis markers in the model; the amount of IL-8 and fibrinogen expressed by dosed cells was the same as control cells (data not shown). Neither compound affected the cells cultured in lean media, which accumulate only small quantities of fat. The two compounds were demonstrated to have differing modes of action, as only metformin was observed to affect the consumption of FFA from the cell culture medium, with treated cells consuming up to 20% less FFA (Figure 6D). This data demonstrates the utility of the 3D in vitro NAFLD model for drug screening and its potential utility in discovering compounds that can prevent or reverse hepatic steatosis.
DISCUSSION
The utilisation of advanced or organ-on-a-chip type in vitro culture systems that more closely mimic the microenvironment of the liver are now established as a method for maintaining the functions of primary human hepatocytes in vitro and preventing rapid dedifferentiation[20,21]. We have utilised a 3D perfused platform to develop an all human in vitro model of NAFLD. The main hallmark of NAFLD is intrahepatic triglyceride accumulation (steatosis), which initially leads to a benign condition, but with the correct additional stimuli can progress to more advanced conditions of steatohepatitis and fibrosis.
We cultured primary human hepatocytes as 3D microtissues and exposed them to 600 μmol FFA, which caused significant triglyceride accumulation and lead to a range of phenotypic changes similar to those observed in NAFLD patients. Our model utilised a combination of palmitic and oleic acids, which are common dietary long-chain FFAs found to accumulate in excess in human livers with steatosis[32,33]. When cultured in vitro, hepatocytes are also known to accumulate fat when exposed to high levels of glucose[34]. Therefore the media conditions used in this model were selected to have lower, more physiologically relevant, glucose and insulin concentrations when compared to standard hepatocyte culture media[35]. This media composition allowed us to study the specific effect of triglyceride accumulation on hepatocytes.
In fat media conditions cells were observed to continually accumulate intracellular triglycerides without showing signs of cytotoxicity. However, it is well established that when lipids accumulate in non-adipose tissues they can enter non-oxidative, deleterious pathways, potentially leading to cell injury and death. Previous studies have shown that fat loading of hepatocytes cultured in vitro can cause cytotoxicity, particularly when high concentrations of saturated fatty acids are present[17]. The concentration of FFA and ratio of saturated to unsaturated FFAs (1:2) used in our model were selected as these had previously been previously shown to not cause hepatotoxicity, whilst inducing significant levels of intracellular triglyceride accumulation[9,11,17].
We observed a four-fold increase in hepatocyte triglyceride accumulation between fat and lean cultured cells, which is a similar increase to that previously reported for short term 2D cultures of hepatocytes cultured with FFAs[12,17] and for hepatocytes isolated from steatotic human livers compared to those obtained from healthy, non-steatotic livers[36]. Under fat conditions cells could maintain hepatic functions in the 3D culture system for up to two weeks. To our knowledge this is significantly longer than all previous in vitro steatosis studies, which have either analysed freshly isolated primary hepatocytes or cultured cells with FFA for less than 5 d. This allowed the analysis of longer term, progressive effects of exposing hepatocytes to excess FFA, rather than simply looking at the transient changes that may occur in the cells when they are first exposed. Interestingly, throughout the 14-d culture, albumin production was significantly higher from cells cultured with excess FFA. Similar observations have been made in vivo, where increased serum albumin concentrations have been associated with insulin resistance, diabetes and over nutrition[37,38]. However, such findings have not always been consistent, and some studies of NAFLD patient subsets suggest that serum albumin concentrations are not affected by the development of disease[39]. It may be that early stages of steatosis and intrahepatic fat accumulation lead to increased albumin production, which is balanced out in later stages of NAFLD when inflammation and fibrosis begin to more severely negatively impact hepatic function.
To more fully appreciate the other phenotypic changes that occur in hepatocytes during the accumulation of triglycerides we analysed both transcriptomic and proteomic changes in the model. Numerous transcriptional changes were observed after exposing the microtissues to FFA in the 3D-perfused platform, including a five-fold increase in CYP2E1 expression. Higher hepatic CYP2E1 expression and activity have been frequently observed in the context of NAFLD and obesity[40,41]. It has been suggested that this increase can result in reactive oxygen species overproduction which can promote NAFLD pathogenesis[40,42]. It should be noted that we only measured transcriptional changes in CYP2E1 and it is known that these do not always correlate well with enzyme activity[28] and future studies should ascertain how CYP2E1 enzyme activity is affected by fat loading in the model. CYP7A1, a key gene in cholesterol metabolism, was also upregulated in the model. This upregulation has previously been observed in NAFLD/NASH patients, who have increased free cholesterol levels and the dysregulation of cholesterol metabolism has been correlated with disease severity[43]. Further genes associated with insulin resistance and lipid metabolism, such as IGFβ1, PDK4 and FABP-1 were also upregulated in fat loaded hepatocytes. IGFβ1 has been identified as an early marker for metabolic liver disease[44], whilst PDK4 has been found to be directly linked to hepatic insulin resistance[45]. It is important to note that steatosis is the first stage in NAFLD pathogenesis and is not associated with gross hepatic functional changes. Therefore as expected we did not observe large numbers of transcriptional changes that have been reported for more advanced stages of NAFLD and NASH[46].
When we analysed the proteins secreted by the hepatocytes, the accumulation of fat also affected the adipokines released by the cells, influencing the secretion of proteins associated with fibrosis and liver injury (fibrinogen, TIMP-1) as well as inflammatory markers (IL-8 and MIF). It has been widely reported that NAFLD/NASH patients have elevated serum levels of pro-inflammatory cytokines, in particular IL-8[47] and previously palmitic acid has been shown to stimulate IL-8 expression in cultured human hepatocytes[18]. Increased MIF expression is associated with NAFLD development in vivo, with liver biopsies of NAFLD patients showing higher expression of MIF compared to healthy controls[48]. Animal studies have also shown an association between high fat diets, insulin resistance and the plasma level of MIF[49]. Fibrinogen expression is also associated with fatty liver disease as its expression increases with liver stress and damage[50].
Despite these changes other inflammatory cytokines associated with NAFLD, such as IL-1β and IL-6, were not observed to be secreted by fat loaded cells in the model. This is likely due to a lack of non-parenchymal cells in our model which are the primary sources of inflammatory cytokines during NAFLD development, in particular IL-6 is highly expressed by Kupffer cells[51]. The adipokines observed to change in the model including IL-8, fibrinogen, and MIF are known to be expressed by hepatocytes and are all associated with a NAFLD phenotype[18,47,49].
Hepatic steatosis is known to affect the metabolic capacity of hepatocytes, by affecting the expression and activity of P450 enzymes and cell membrane transporters[5,29]. In our NAFLD model the metabolic rates of both CYP3A4 and CYP2C9 were reduced, but the gene expression of both enzymes was not affected. Similar changes in P450 activity were previously observed by Donato et al[29] using 2D plated human hepatocytes, but these were only exposed to FFA for 14 h. Fisher et al[28] reported decreasing CYP3A4 protein expression and activity in hepatocytes isolated from NAFLD/NASH patients and this activity decreased with disease progression. Conversely the same study found that CYP2C9 activity is increased in more advanced stages of NAFLD/NASH, so the decrease in activity that we observed may be a specific feature of early stages of steatosis that reverts during disease progression. Being able to model the metabolic changes in NAFLD demonstrates one way in which our in vitro model could have important utility for analysing how novel compounds will behave in different patient populations. There remains a lack of studies relating to absorption, distribution, metabolism, and elimination (ADME) processes in human NAFLD. The study of metabolic changes associated with NAFLD will become an increasing issue as the proportion of the population with fatty livers continues to rise.
We further demonstrated the utility of our in vitro NAFLD model to be a tool for screening drugs as NAFLD therapeutics by analysing the activities of two known anti-steatotic compounds, pioglitazone and metformin. When dosed in the model both compounds reduced the level of triglyceride accumulation in the fat loaded cells, but appeared to function by different mechanisms. Pioglitazone has previously been reported to reduce hepatic steatosis in animal models of NAFLD and shown to have some benefit to NAFLD patients in clinical trials[52]. The direct effect of pioglitazone on hepatocytes, which can reduce triglyceride accumulation, is by its ability to alter lipid metabolism and mediate lipoprotein lipase expression through the activation of PPARγ[53-55]. Pioglitazone is also known to have indirect effects on hepatic steatosis by influencing the release of FFA from adipose tissue[56]. We observed its ability to reduce triglyceride accumulation, but it did not affect the consumption of FFA from the cell culture media. In contrast, metformin was able to reduce FFA consumption from the culture medium, leading to reduced intrahepatic triglyceride concentration. This finding was previously reported for metformin treatment of murine models of NAFLD[30] and metformin has been found to decrease the expression of lipid-droplet associated proteins such as ADRP[57] and inflammatory markers, including TNFα[58].
Our results demonstrate that the prolonged culture of human hepatocytes in vitro, under fat conditions mimics many of the effects observed in clinical NAFLD. The 3D perfused nature of the model allows for sustained culture of hepatocytes in vitro, allowing prolonged effects of FFA accumulation to be explored. This model is therefore well suited to exploring the molecular mechanisms that underlie steatosis and has the potential to be used as a tool for drug screening. Using the 3D-perfused platform multicellular liver models (e.g., hepatocytes and Kupffer cells) have already been developed[23] and could potentially be combined with our steatosis model to create a further more advanced model of NAFLD/NASH.
ACKNOWLEDGMENTS
The authors thank Dr Alan Wells and Dr Amanda Clark (University of Pittsburgh) for performing clinical chemistry assays for ALT and AST. Mass Spectrometry was performed by an independent contract research organisation Xenogesis Ltd (Nottingham, United Kingdom). Adipokine arrays were performed by contract research organisation Labstract Ltd (Cambridge, United Kingdom).
COMMENTS
Background
Non-alcoholic fatty liver disease (NAFLD) covers a spectrum of chronic liver diseases, ranging from hepatic steatosis or fatty liver, non-alcoholic steatohepatitis, liver fibrosis and liver cirrhosis. It is estimated that more than 1 billion people worldwide are now affected by some form of NAFLD, but currently no specific treatments options are available and the only curative therapy option available is liver transplantation. To understand the molecular basis of this disease in more detail better experimental models are required that fully recapitulate the phenotype of the human disease.
Research frontiers
Previous in vitro studies of NAFLD have generally involved in the use of hepatoma cell lines or the exposure of primary hepatocytes to triglycerides for short periods of time (< 48 h) and therefore do not capture the complexity of the disease.
Innovations and breakthroughs
The authors have generated a novel three-dimensional (3D) perfused, all human in vitro model of NAFLD, with primary human hepatocytes cultured under high fat conditions in 3D microtissues. The transcriptomic, metabolic and phenotypic changes caused by triglyceride accumulation are analysed in the system.
Applications
The model of NAFLD described is ideally suited to exploring the molecular changes that occur during the earliest stages of fatty liver disease and how this disease stage can progress to more advanced stages of disease. The model has the potential to be used to identify novel therapeutic targets in NAFLD and to be used as a tool for drug screening.
Terminology
NAFLD, is defined by the presence of hepatic steatosis in the absence of significant alcohol consumption, steatogenic medication or hereditary disorders. In the majority of patients, NAFLD is also associated with obesity, diabetes mellitus and high levels of cholesterol and triglycerides in the blood.
Peer-review
Authors showed develop a human in vitro model of NAFLD by using primary hepatocytes cultured in a 3D perfused platform. In this model, hepatocytes cultured in fat medium were found to accumulate fat more than lean cells although aspartate transaminase and alanine aminotransferase did not change. Furthermore, inflammatory and fibrotic genes associated with NAFLD were upregulated in hepatocyte cultured in fat medium. Finally, authors concluded that the 3D in vitro NAFLD model recapitulates many features of clinical NAFLD and is an ideal tool for analysing the efficacy of anti-steatotic compounds. This original paper is interesting, and manuscript is well written.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United Kingdom
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: At the time of this study all authors were employees of CN Bio Innovations Ltd.
Data sharing statement: No additional data are available.
Peer-review started: August 17, 2016
First decision: October 11, 2016
Article in press: November 16, 2016
P- Reviewer: Tokuyama S S- Editor: Yu J L- Editor: A E- Editor: Zhang FF
References
- 1.Moore JB. Non-alcoholic fatty liver disease: the hepatic consequence of obesity and the metabolic syndrome. Proc Nutr Soc. 2010;69:211–220. doi: 10.1017/S0029665110000030. [DOI] [PubMed] [Google Scholar]
- 2.Than NN, Newsome PN. A concise review of non-alcoholic fatty liver disease. Atherosclerosis. 2015;239:192–202. doi: 10.1016/j.atherosclerosis.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donato MT, Lahoz A, Jiménez N, Pérez G, Serralta A, Mir J, Castell JV, Gómez-Lechón MJ. Potential impact of steatosis on cytochrome P450 enzymes of human hepatocytes isolated from fatty liver grafts. Drug Metab Dispos. 2006;34:1556–1562. doi: 10.1124/dmd.106.009670. [DOI] [PubMed] [Google Scholar]
- 5.Lake AD, Novak P, Fisher CD, Jackson JP, Hardwick RN, Billheimer DD, Klimecki WT, Cherrington NJ. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:1954–1960. doi: 10.1124/dmd.111.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic Steatohepatitis: A Search for Factual Animal Models. Biomed Res Int. 2015;2015:574832. doi: 10.1155/2015/574832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willebrords J, Pereira IV, Maes M, Crespo Yanguas S, Colle I, Van Den Bossche B, Da Silva TC, de Oliveira CP, Andraus W, Alves VA, et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog Lipid Res. 2015;59:106–125. doi: 10.1016/j.plipres.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavez-Tapia NC, Rosso N, Tiribelli C. In vitro models for the study of non-alcoholic fatty liver disease. Curr Med Chem. 2011;18:1079–1084. doi: 10.2174/092986711794940842. [DOI] [PubMed] [Google Scholar]
- 9.Cui W, Chen SL, Hu KQ. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J Transl Res. 2010;2:95–104. [PMC free article] [PubMed] [Google Scholar]
- 10.Vidyashankar S, Sharath Kumar LM, Barooah V, Sandeep Varma R, Nandakumar KS, Patki PS. Liv.52 up-regulates cellular antioxidants and increase glucose uptake to circumvent oleic acid induced hepatic steatosis in HepG2 cells. Phytomedicine. 2012;19:1156–1165. doi: 10.1016/j.phymed.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Chavez-Tapia NC, Rosso N, Tiribelli C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012;12:20. doi: 10.1186/1471-230X-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janorkar AV, Harris LM, Murphey BS, Sowell BL. Use of three-dimensional spheroids of hepatocyte-derived reporter cells to study the effects of intracellular fat accumulation and subsequent cytokine exposure. Biotechnol Bioeng. 2011;108:1171–1180. doi: 10.1002/bit.23025. [DOI] [PubMed] [Google Scholar]
- 13.Donato MT, Lahoz A, Castell JV, Gómez-Lechón MJ. Cell lines: a tool for in vitro drug metabolism studies. Curr Drug Metab. 2008;9:1–11. doi: 10.2174/138920008783331086. [DOI] [PubMed] [Google Scholar]
- 14.Olsavsky KM, Page JL, Johnson MC, Zarbl H, Strom SC, Omiecinski CJ. Gene expression profiling and differentiation assessment in primary human hepatocyte cultures, established hepatoma cell lines, and human liver tissues. Toxicol Appl Pharmacol. 2007;222:42–56. doi: 10.1016/j.taap.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexopoulos LG, Saez-Rodriguez J, Cosgrove BD, Lauffenburger DA, Sorger PK. Networks inferred from biochemical data reveal profound differences in toll-like receptor and inflammatory signaling between normal and transformed hepatocytes. Mol Cell Proteomics. 2010;9:1849–1865. doi: 10.1074/mcp.M110.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dash A, Inman W, Hoffmaster K, Sevidal S, Kelly J, Obach RS, Griffith LG, Tannenbaum SR. Liver tissue engineering in the evaluation of drug safety. Expert Opin Drug Metab Toxicol. 2009;5:1159–1174. doi: 10.1517/17425250903160664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-Lechón MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O’Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, McClain CJ. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823–830. doi: 10.1002/hep.21752. [DOI] [PubMed] [Google Scholar]
- 19.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebrahimkhani MR, Neiman JA, Raredon MS, Hughes DJ, Griffith LG. Bioreactor technologies to support liver function in vitro. Adv Drug Deliv Rev. 2014;69-70:132–157. doi: 10.1016/j.addr.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin C, Ballinger KR, Khetani SR. The application of engineered liver tissues for novel drug discovery. Expert Opin Drug Discov. 2015;10:519–540. doi: 10.1517/17460441.2015.1032241. [DOI] [PubMed] [Google Scholar]
- 22.Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov. 2015;14:248–260. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarkar U, Rivera-Burgos D, Large EM, Hughes DJ, Ravindra KC, Dyer RL, Ebrahimkhani MR, Wishnok JS, Griffith LG, Tannenbaum SR. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Dispos. 2015;43:1091–1099. doi: 10.1124/dmd.115.063495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vivares A, Salle-Lefort S, Arabeyre-Fabre C, Ngo R, Penarier G, Bremond M, Moliner P, Gallas JF, Fabre G, Klieber S. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45:29–44. doi: 10.3109/00498254.2014.944612. [DOI] [PubMed] [Google Scholar]
- 26.Fitzpatrick E, Dhawan A. Noninvasive biomarkers in non-alcoholic fatty liver disease: current status and a glimpse of the future. World J Gastroenterol. 2014;20:10851–10863. doi: 10.3748/wjg.v20.i31.10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wruck W, Kashofer K, Rehman S, Daskalaki A, Berg D, Gralka E, Jozefczuk J, Drews K, Pandey V, Regenbrecht C, et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci Data. 2015;2:150068. doi: 10.1038/sdata.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, Cherrington NJ. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009;37:2087–2094. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donato MT, Jiménez N, Serralta A, Mir J, Castell JV, Gómez-Lechón MJ. Effects of steatosis on drug-metabolizing capability of primary human hepatocytes. Toxicol In Vitro. 2007;21:271–276. doi: 10.1016/j.tiv.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 30.Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. doi: 10.1038/79697. [DOI] [PubMed] [Google Scholar]
- 31.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 32.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 33.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y, Zhang G, Shen C, Uygun K, Yarmush ML, Meng Q. A novel 3D liver organoid system for elucidation of hepatic glucose metabolism. Biotechnol Bioeng. 2012;109:595–604. doi: 10.1002/bit.23349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shulman M, Nahmias Y. Long-term culture and coculture of primary rat and human hepatocytes. Methods Mol Biol. 2013;945:287–302. doi: 10.1007/978-1-62703-125-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amaro A, Fabbrini E, Kars M, Yue P, Schechtman K, Schonfeld G, Klein S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139:149–153. doi: 10.1053/j.gastro.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bae JC, Seo SH, Hur KY, Kim JH, Lee MS, Lee MK, Lee WY, Rhee EJ, Oh KW. Association between Serum Albumin, Insulin Resistance, and Incident Diabetes in Nondiabetic Subjects. Endocrinol Metab (Seoul) 2013;28:26–32. doi: 10.3803/EnM.2013.28.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishizaka N, Ishizaka Y, Nagai R, Toda E, Hashimoto H, Yamakado M. Association between serum albumin, carotid atherosclerosis, and metabolic syndrome in Japanese individuals. Atherosclerosis. 2007;193:373–379. doi: 10.1016/j.atherosclerosis.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 39.Fierbinteanu-Braticevici C, Baicus C, Tribus L, Papacocea R. Predictive factors for nonalcoholic steatohepatitis (NASH) in patients with nonalcoholic fatty liver disease (NAFLD) J Gastrointestin Liver Dis. 2011;20:153–159. [PubMed] [Google Scholar]
- 40.Aubert J, Begriche K, Knockaert L, Robin MA, Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin Res Hepatol Gastroenterol. 2011;35:630–637. doi: 10.1016/j.clinre.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 41.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665–674. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li HH, Doiron K, Patterson AD, Gonzalez FJ, Fornace AJ. Identification of serum insulin-like growth factor binding protein 1 as diagnostic biomarker for early-stage alcohol-induced liver disease. J Transl Med. 2013;11:266. doi: 10.1186/1479-5876-11-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao R, Xiong X, Harris RA, White MF, Dong XC. Genetic inactivation of pyruvate dehydrogenase kinases improves hepatic insulin resistance induced diabetes. PLoS One. 2013;8:e71997. doi: 10.1371/journal.pone.0071997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:645–655. doi: 10.1038/nrgastro.2013.182. [DOI] [PubMed] [Google Scholar]
- 47.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:727–735. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akyildiz M, Gunsar F, Nart D, Sahin O, Yilmaz F, Akay S, Ersoz G, Karasu Z, Ilter T, Batur Y, et al. Macrophage migration inhibitory factor expression and MIF gene -173 G/C polymorphism in nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2010;22:192–198. doi: 10.1097/MEG.0b013e328331a596. [DOI] [PubMed] [Google Scholar]
- 49.Morrison MC, Kleemann R. Role of Macrophage Migration Inhibitory Factor in Obesity, Insulin Resistance, Type 2 Diabetes, and Associated Hepatic Co-Morbidities: A Comprehensive Review of Human and Rodent Studies. Front Immunol. 2015;6:308. doi: 10.3389/fimmu.2015.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia. 2009;13:9–19. [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol. 2016;64:1403–1415. doi: 10.1016/j.jhep.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Hardy T, Anstee QM, Day CP. Nonalcoholic fatty liver disease: new treatments. Curr Opin Gastroenterol. 2015;31:175–183. doi: 10.1097/MOG.0000000000000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin S, Liu T, Kamanna VS, Kashyap ML. Pioglitazone stimulates apolipoprotein A-I production without affecting HDL removal in HepG2 cells: involvement of PPAR-alpha. Arterioscler Thromb Vasc Biol. 2007;27:2428–2434. doi: 10.1161/ATVBAHA.107.150193. [DOI] [PubMed] [Google Scholar]
- 54.Sakamoto J, Kimura H, Moriyama S, Odaka H, Momose Y, Sugiyama Y, Sawada H. Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun. 2000;278:704–711. doi: 10.1006/bbrc.2000.3868. [DOI] [PubMed] [Google Scholar]
- 55.Nagashima K, Lopez C, Donovan D, Ngai C, Fontanez N, Bensadoun A, Fruchart-Najib J, Holleran S, Cohn JS, Ramakrishnan R, et al. Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115:1323–1332. doi: 10.1172/JCI23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest. 2006;116:581–589. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu F, Wang C, Zhang L, Xu Y, Jang L, Gu Y, Cao X, Zhao X, Ye J, Li Q. Metformin prevents hepatic steatosis by regulating the expression of adipose differentiation-related protein. Int J Mol Med. 2014;33:51–58. doi: 10.3892/ijmm.2013.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Federico A, Zulli C, de Sio I, Del Prete A, Dallio M, Masarone M, Loguercio C. Focus on emerging drugs for the treatment of patients with non-alcoholic fatty liver disease. World J Gastroenterol. 2014;20:16841–16857. doi: 10.3748/wjg.v20.i45.16841. [DOI] [PMC free article] [PubMed] [Google Scholar]