

ABSTRACT
Caseinolytic peptidases (ClpPs) regulate diverse aspects of cellular physiology in bacteria. Some species have multiple ClpPs, including the opportunistic pathogen Pseudomonas aeruginosa, in which there is an archetypical isoform, ClpP1, and a second isoform, ClpP2, about which little is known. Here, we use phenotypic assays to investigate the biological roles of ClpP1 and ClpP2 and biochemical assays to characterize purified ClpP1, ClpP2, ClpX, and ClpA. Interestingly, ClpP1 and ClpP2 have distinct intracellular roles for motility, pigment production, iron scavenging, and biofilm formation. Of particular interest, ClpP2, but not ClpP1, is required for microcolony organization, where multicellular organized structures first form on the pathway to biofilm production. We found that purified ClpP1 with ClpX or ClpA was enzymatically active, yet to our surprise, ClpP2 was inactive and not fully assembled in vitro; attempts to assist ClpP2 assembly and activation by mixing with the other Clp components failed to turn on ClpP2, as did solution conditions that have helped activate other ClpPs in vitro. We postulate that the active form of ClpP2 has yet to be discovered, and we present several potential models to explain its activation as well as the unique role ClpP2 plays in the development of the clinically important biofilms in P. aeruginosa.
IMPORTANCE Pseudomonas aeruginosa is responsible for severe infections of immunocompromised patients. Our work demonstrates that two different isoforms of the Clp peptidase, ClpP1 and ClpP2, control distinct aspects of cellular physiology for this organism. In particular, we identify ClpP2 as being necessary for microcolony organization. Pure active forms of ClpP1 and either ClpX or ClpA were characterized as assembled and active, and ClpP2 was incompletely assembled and inactive. By establishing both the unique biological roles of ClpP1 and ClpP2 and their initial biochemical assemblies, we have set the stage for important future work on the structure, function, and biological targets of Clp proteolytic enzymes in this important opportunistic pathogen.
KEYWORDS: Clp peptidase, ClpP1, ClpP2, Pseudomonas aeruginosa, biofilm, microcolonies
INTRODUCTION
Regulated protein degradation is a mainstay of life. In bacteria, multiple intracellular proteases share responsibility for protein quality control through the degradation of damaged or misfolded proteins and disposal of truncated translation products, in part in conjunction with the SsrA tagging system (1–3). Proteases also degrade specific regulatory substrates, like sigma factors, other transcription factors, metabolic enzymes, and structural proteins (4). This wide substrate profile in turn provides control over important adaptive and developmental processes, such as sporulation, the heat shock response, cell cycle progression, biofilm formation, antibiotic resistance, and motility; notably, all of these processes potentially contribute to bacterial virulence (5–7). Bacterial proteases therefore have key importance for physiological regulation and constitute promising targets for antimicrobial development (8).
Clp proteolytic enzymes are conserved in bacteria and in the organelles of eukaryotes and work together to identify, unfold, and degrade particular intracellular substrates. Substrates often have short N- or C-terminal degron tag sequences that are recognized by either small adaptor proteins or directly by ATPases associated with diverse cellular activities (AAA+) protein unfoldases, including ClpA, ClpC, and ClpX (3, 9). These unfoldases form hexameric rings that utilize energy from ATP hydrolysis to unfold substrates, thread them through a central pore, and deliver them to an associated peptidase, ClpP, for hydrolysis (10). ClpP assembles into a barrel made of two stacked heptamer rings, with the active-site serine, histidine, and aspartate residues facing the inner chamber. Entry pores at the top and bottom of the barrel are constricted to exclude large substrates (>10 amino acids) until association with an unfoldase effectively opens and enlarges the pores (11, 12). At the unfoldase-peptidase binding interface, conserved IGF/L loop motifs on the ATPase dock in hydrophobic surface pockets on ClpP, and the unfoldase pore 2 loops interact with the flexible N-terminal gating loops of ClpP to open them and prepare for substrate delivery (13) (Fig. 1).
FIG 1.

Alignment of ClpP isoforms from E. coli and P. aeruginosa. A single subunit of E. coli ClpP (PDB ID 1YG6) (50) is shown adjacent to a cartoon of a protease complex composed of AAA+ unfoldase ClpX (green) and ClpP (blue). Protein sequences are shown for E. coli ClpP (EcClpP), P. aeruginosa ClpP1 (PaClpP1), and P. aeruginosa ClpP2 (PaClpP2). Residues are colored blue to indicate conservation, except for catalytic residues (red, *) and hydrophobic docking pocket residues (black, ϕ). Additionally, N-terminal loop region residues are colored to indicate conservation (gray) or nonconservation (yellow). E. coli ClpP and P. aeruginosa ClpP1 have 70% sequence identity, while P. aeruginosa ClpP1 and ClpP2 have 40% sequence identity. The ClpP subunit was rendered with PyMOL (51), and sequences were aligned with MUSCLE (52), using default settings, and presented with JalView (53).
Some bacteria, like Escherichia coli, have a single type of ClpP subunit, but other species have multiple isoforms, and the assembly and functional characteristics of these isoforms can vary. For example, Mycobacterium tuberculosis has two ClpP paralogs, ClpP1 and ClpP2, that form a single functional enzyme complex, an active tetradecamer assembled from one heptameric ring of each isoform (14). In contrast, the cyanobacterium Synechococcus elongatus has two ClpP assemblies made of four isoforms: ClpP3/R is associated with ClpX, and ClpP1/P2 is associated with ClpC. (The “R” isoform is so named due to its lack of a functional active site and the assumption that it therefore has a regulatory role.) Thus, this cyanobacterium has two completely different Clp unfoldase-peptidase combinations: ClpXP1/P2 and ClpCP3/R. These two classes of ClpP assemblies interestingly have distinct expression patterns and biological functions, most likely at least in part because they work with different unfoldase partners, which in turn have different substrate specificities. Notably, the ClpP3/R barrel is composed of two identical rings with mixed subunit composition (15–17).
Another species of bacterium with multiple ClpP isoforms is Pseudomonas aeruginosa, an opportunistic pathogen that is a major cause of morbidity and mortality in nosocomial infections and chronic cystic fibrosis lung infections. It has two ClpP isoforms, ClpP1 and ClpP2 (Fig. 1). The gene encoding ClpP1 (PA1801) shares an operon with that encoding the ClpX unfoldase (PA1802), whereas the gene encoding ClpP2 (PA3326) is in a different chromosomal region. ClpP1 is similar to the well-characterized E. coli ClpP and has conserved residues that form the active peptidase site, the hydrophobic unfoldase docking pockets, and the N-terminal gating motifs (Fig. 1); ClpP1 also has a well-established function in cellular motility (18, 19). In contrast, less is known about ClpP2. It has conserved residues for the catalytic triad and hydrophobic docking sites but contains a unique sequence for the region that usually encodes the N-terminal loop module that docks with the ATPase pore 2 loops (Fig. 1). Genetic experiments by Qiu and colleagues showed that ClpX, ClpP1, and ClpP2 were all required for the degradation of a truncated anti-sigma factor called MucA25, which is found in a specific mucoid P. aeruginosa strain, PAO581 (20). These results indicate that ClpP2 might cooperate with ClpX and ClpP1 to promote degradation of at least some substrates. However, the biological role of ClpP2 in nonmucoid strains and its specific relationships with ClpX and ClpP1 have remained unclear, and there is currently no evidence for cooperation between ClpA and ClpP2.
Here, we use microbiological and biochemical assays to show that ClpP1 and ClpP2 are differentially expressed and control distinct aspects of physiology in P. aeruginosa. Whereas purified ClpP1, ClpX, and ClpA were active and assembled, purified ClpP2 was incompletely assembled and inactive, even though the ClpP2 gene is required by cells for appropriate microcolony organization. We discuss different models for how ClpP2 might be activated and influence the choices for substrate degradation in vivo.
RESULTS
ClpP1 and ClpP2 have distinct patterns of expression.
ClpP1 and ClpP2 have distinct genomic locations, and we therefore hypothesized that they might also have different expression patterns, perhaps being differently expressed during different stages of growth and development. Prior observations from genome-wide microarray studies suggest that ClpP1 is constitutively expressed, whereas ClpP2 expression is dramatically higher in stationary-phase cells and biofilms than in exponential-phase cells (21). To obtain information about intracellular protein levels, we monitored ClpP1 and ClpP2 expression by harvesting cells growing in liquid culture (37°C). SDS-PAGE and Western blotting with antibodies specific for the two isoforms revealed that ClpP1 levels appeared essentially unchanged throughout exponential phase and stationary phase (through 50 h of growth) (Fig. 2). In contrast, ClpP2 became convincingly detectable only as cells entered stationary phase (optical density at 600 nm [OD600] greater than ∼1.6). Our results thus demonstrate that ClpP1 and ClpP2 have different patterns of expression, a finding consistent with the suggestion that they may have distinct intracellular roles.
FIG 2.
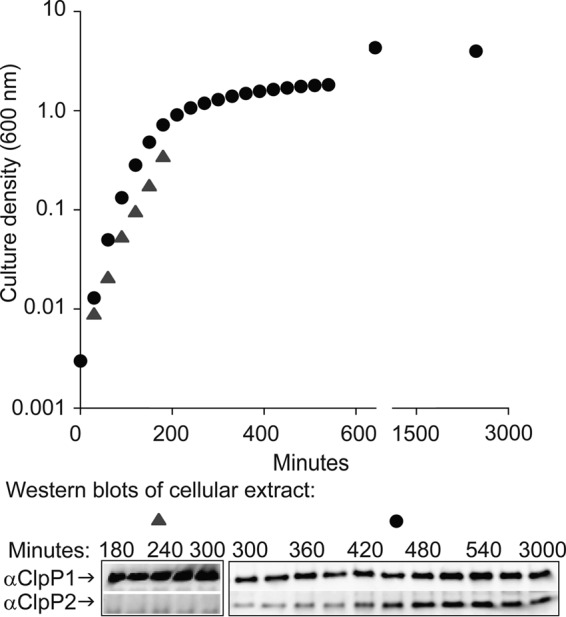
Expression of ClpP1 and ClpP2. Detection of ClpP1 and ClpP2 protein in aliquots of cells growing in LB liquid culture at 37°C. Aliquots were adjusted for density and subjected to SDS-PAGE and Western blot (bottom) using primary antibody specific for ClpP1 or ClpP2.
ClpP1, but not ClpP2, strongly influences cellular motility and pyoverdine and pyocyanin production.
To investigate the functions of ClpP1 and ClpP2 in P. aeruginosa, we constructed deletion strains for the open reading frames (ORFs) of ClpP1 (ΔP1), ClpP2 (ΔP2), or both genes (ΔP1P2). These otherwise isogenic deletion strains were used for phenotypic assays to search for biological processes that were dependent on each of the Clp peptidases.
Bacteria use their surface appendages for motility and environmental interactions. Flagella are used for power swimming, chemotaxis, and interaction with surfaces, whereas pili are used for twitching motility on solid surfaces and during initial surface attachment. To test for swimming and twitching motility, we inoculated each variant strain onto specialized medium plates and, after a defined incubation time, measured the size of the motility zone. For both motility assays, there was no significant difference in performance between the wild-type (WT) and ΔP2 mutant strains (Fig. 3). In contrast, ΔP1 and ΔP1P2 mutant strains were significantly defective in twitching and swimming compared to WT (P < 0.0001), although not as defective as nonmotile strains included as negative controls. These results reveal that ClpP1, but not ClpP2, is either directly or indirectly important for flagellum-dependent swimming motility and pilus-dependent twitching motility.
FIG 3.
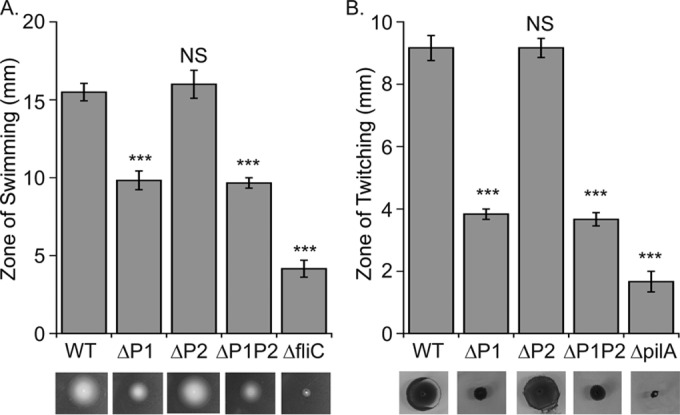
The role of ClpP1 and ClpP2 in swimming and twitching. (A) Swimming is controlled by ClpP1 and, to a lesser extent, ClpP2. Plates were grown ∼16 h at 30°C and motility measured directly with a NIST traceable ruler. Values are normalized averages (n = 6) ± 1 standard error of the mean (SEM). Data were compared to control values (WT) by analysis of variance (ANOVA) with Dunnett's post hoc test (***, P < 0.0001; NS, not significant). Images are representative examples for display purposes only and acquired as described in Materials and Methods. (B) Twitching motility is controlled by ClpP1 but not ClpP2. Plates were incubated ∼48 h at room temperature and dyed with crystal violet. Twitching was quantitated, reported, and displayed as described for swimming. Nonmotile strains lacking either flagella (ΔfliC mutant) or pili (ΔpilA mutant) were used as negative controls.
Next, we investigated the ability of these ClpP-deficient strains to produce pyoverdine and pyocyanin. Pyoverdine is a siderophore used to scavenge iron from the environment, and pyocyanin is a toxic blue-green pigment that inhibits the respiration of eukaryotic host cells during bacterial infection (22, 23). Both molecules are secreted by P. aeruginosa as virulence factors during mammalian infections and function in cellular signaling pathways (24). Interestingly, we found that the accumulation of both molecules in the growth medium was significantly higher (P < 0.0001) for the ΔP1 and ΔP1P2 mutant strains than for the WT and ΔP2 mutant strains (Fig. 4). Also, the ΔP2 mutant strain had slightly elevated levels of pyoverdine compared to the WT (P < 0.05). We confirmed the role of ClpP1 in pyoverdine abundance through a complementation experiment in which overproduction of pyoverdine by the ΔP1 mutant strains was complemented by the expression of ClpP1 from a plasmid (see Fig. S3A in the supplemental material). We conclude that ClpP1 has an important influence on pyoverdine and pyocyanin abundance, whereas ClpP2 has little or no role except perhaps for a slight effect on pyoverdine levels.
FIG 4.
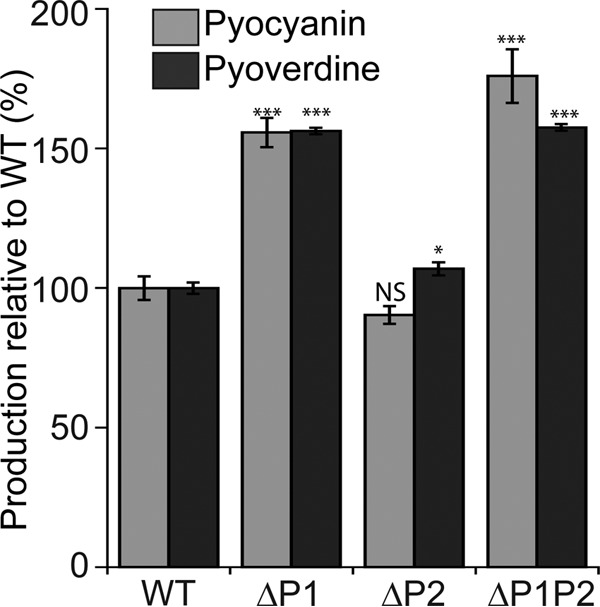
ClpP1, but not ClpP2, controls pyocyanin and pyoverdine production. Pyocyanin was measured using absorbance at 530 nm and pyoverdine measured using excitation at 405 nm and emission at 460 nm. Values are normalized averages (n = 6) ± SEM. Data were compared to control values (WT) by ANOVA with Dunnett's post hoc test (***, P < 0.0001; *, P < 0.05; NS, not significant). Comparison of ΔP1 and ΔP1P2 mutants for both assays, P > 0.05.
Surface adherence and microcolony formation.
Biofilm formation is strongly associated with chronic infections by Pseudomonas aeruginosa (25). Distinct stages of biofilm formation include (i) initial attachment to a surface, (ii) firm attachment and growth of cells into microcolonies, (iii) structured biofilm growth, and (iv) dispersal to reseed new biofilm colonies (26) (see also Fig. 8A and Discussion). To compare the ability of our ClpP-deficient strains to complete the early stages in biofilm formation, we initially used a simple assay in which cultures were incubated in 96-well plates for either 6 h or overnight. Cells that adhered to the plate surface were stained with crystal violet dye, and their relative abundances were measured by absorbance. We found that after 6 h of incubation, both the ΔP1 and ΔP1P2 mutant strains had significantly reduced levels of attachment compared to the WT, whereas the ΔP2 mutant strain was not significantly attachment impaired (Fig. 5A). Interestingly, attachment by the ΔP1P2 mutant strain was significantly worse at 6 h compared to the ΔP1 mutant strain. Furthermore, after overnight incubation, the ΔP1 and ΔP2 mutant strains both displayed modestly reduced levels of surface attachment (∼80% of WT), whereas the ΔP1P2 mutant strain had a much stronger defect (∼40% of WT) (Fig. 5A).
FIG 8.
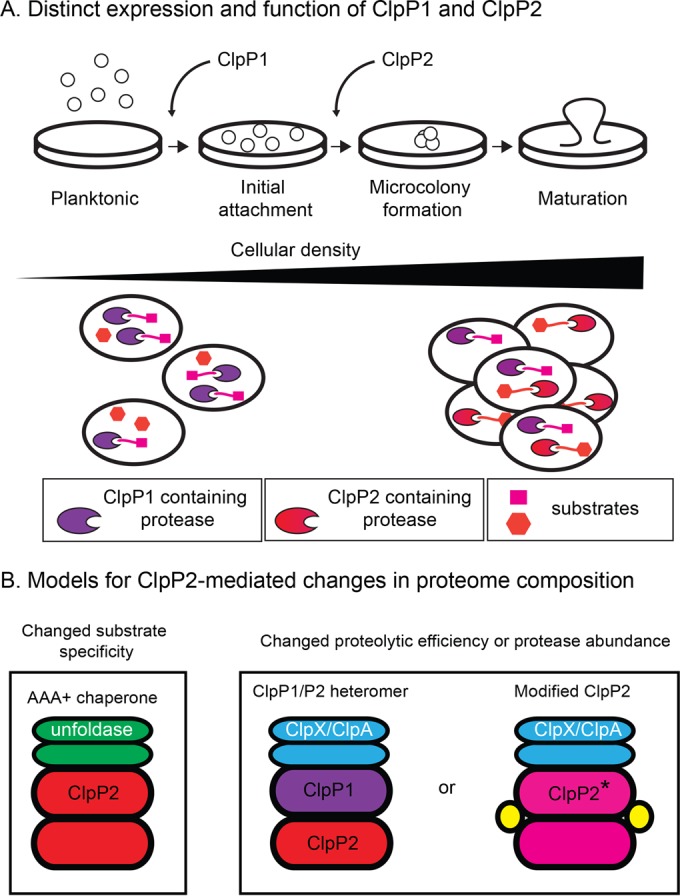
Models for ClpP2 assembly and function. (A) ClpP1 and ClpP2 have distinct expression patterns and biological roles, especially during the transition from planktonic growth and establishment of mature biofilm. The long triangle represents both increased cell density and increased expression of ClpP2 (although ClpP2 expression is not likely to continually rise throughout growth). Notice, however, the presence of ClpP2-containing proteases in cells only in the right portion of the figure, which represents cultures at high cell density and under biofilm formation conditions. (B) ClpP2 could change cellular proteomic composition by coupling with an AAA+ unfoldase with different substrate specificity compared to ClpX and ClpA, or function in either a ClpP1/P2 heteromer or a modified form that has either increased or decreased proteolytic efficiency or that changes the abundance of protease inside cells. Modified ClpP2 is denoted with a star, and potential assembly assistance proteins are shown in yellow.
FIG 5.
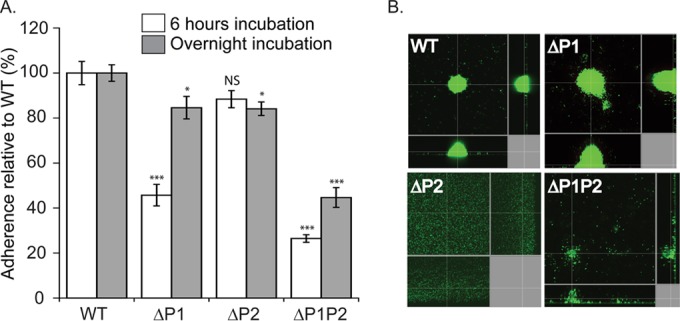
The role of ClpP1 and ClpP2 in surface adherence. (A) ClpP1 controls the level of adherence after 6 h of incubation, while ClpP2 does not, and both ClpP1 and ClpP2 control levels of adherence after overnight incubation. Cell adherence was measured with crystal violet dye at 37°C in 96-well plates. Values were normalized averages (n = 24) ± SEM. Data were compared to the WT values by ANOVA with Dunnett's post hoc test (***, P < 0.0001; *, P < 0.05; NS, not significant). Comparison of ΔP1 and ΔP1P2 mutants for 6 h of incubation, P = 0.0004. (B) ClpP2 controls microcolony organization, whereas ClpP1 does not. Microcolonies in flow cells were visualized with the LIVE/DEAD BacLight bacterial viability kit and imaged with confocal laser scanning microscopy at ×60 magnification after 72 h of growth in flow cells at 37°C. Replicate images are shown in Fig. S2 in the supplemental material, and results from flow cell experiments with complementation by plasmid expression of ClpP2 are shown in Fig. S3.
These results demonstrate that both ClpP1 and ClpP2 contribute to surface attachment, and the absence of both genes has a greater impact than the absence of just one or the other. However, crystal violet-based assays do not provide insights into potential differences in biofilm structure or organization. We therefore used a more sophisticated biofilm assembly assay and investigated microcolony formation, a hallmark step in the generation of biofilm, where cells that adhere to a surface begin to organize themselves into structured cell aggregates. Flow cells were inoculated with cultures and then subjected to a continuous buffer flow for 72 h. The resultant adherent cells were then visualized by fluorescent staining and confocal light microscopy. We observed that the parental WT and ΔP1 mutant strains assembled into well-structured microcolonies (Fig. 5B and S2). In contrast, the ΔP2 mutant strain adhered to flow cell walls but formed an essentially even and unorganized layer rather than microcolonies, and the ΔP1P2 mutant strain displayed a clear deficiency in organized microcolony formation as well. We repeated the assay using ΔP2 or ΔP1P2 mutant strains transformed with a plasmid expressing either ClpP2 or ClpP1, respectively (Fig. S3B). Plasmid-borne expression of ClpP2 restored microcolony organization in the ΔP2 mutant strain. In contrast, in the ΔP1P2 mutant cells, ClpP1 expression restored cell adherence but not microcolony organization. Taken together, our different assays for key steps in biofilm formation demonstrate that both ClpP1 and ClpP2 participate in distinct aspects of biofilm assembly: ClpP1 affects the rate/success of surface adherence (e.g., when assayed after 6 h), which is consistent with its strong effects on pilus-dependent twitching motility, whereas ClpP2 is required for microcolony formation, and the absence of both ClpP1 and ClpP2 substantially impairs the bacterium's ability to form organized microcolonies and thus almost certainly also fully developed biofilms.
Enzymatic activities of purified ClpP1, ClpP2, ClpX, and ClpA.
ClpP1 has 80% sequence identity with ClpP from E. coli, whereas ClpP2 is only 40% identical and carries clearly nonconserved sequences in both its N- and C-terminal regions (Fig. 1). Genetic experiments by Qiu and colleagues suggested that ClpX, ClpP1, and ClpP2 might work together to degrade intracellular substrates, but it was unclear if ClpP2 successfully interacts with ClpX and ClpP1 (20). To gain more specific information about ClpP2 and its relationship with ClpP1, ClpX, and ClpA, we overexpressed and purified each enzyme to >95% purity for biochemical and biophysical experiments.
P. aeruginosa ClpX has 76% sequence identity with Escherichia coli ClpX, including the conservation of multiple functional motifs required for enzyme activity (Fig. S4). ClpX hexamer formation is stabilized by ATP; therefore, assembly can be followed indirectly by monitoring changes in the rate of ATP hydrolysis (27). As expected, our purified P. aeruginosa ClpX had higher rates of hydrolysis with increasing ATP concentrations (Fig. 6A). Graphic analysis of the data indicated a half-maximal ATP concentration of around 70 μM and a Hill coefficient of 1.5, indicating some cooperativity in assembly or loading of ATP. From these results, we concluded that our purified ClpX assembled and hydrolyzed ATP as expected. We next assayed whether the ATP hydrolysis rate of ClpX was repressed upon association with ClpP, as it is for the E. coli enzymes (28). The ATP hydrolysis rate of ClpX decreased slightly (∼15%) in the presence of ClpP1 but remained unchanged in the presence of ClpP2 (Fig. 6A, inset, light gray bars). A mixture of ClpP1 and ClpP2 gave results similar to those with ClpP1 alone. We repeated the assay using purified E. coli ClpX with ClpP1 and ClpP2 from P. aeruginosa (Fig. 6A, inset, dark gray bars). The results were similar, although the decrease in ATP hydrolysis rates was more dramatic (an ∼60% reduction, which is similar to that observed with E. coli ClpXP). Our results thus suggest that ClpP1 functionally interacts with ClpX, whereas ClpP2 shows no sign of interaction.
FIG 6.

Enzymatic activity of purified ClpX, ClpP1, and ClpP2. (A) The rate of ATP hydrolysis by P. aeruginosa ClpX increases as a function of ATP concentration. Data were fit to the Hill version of the Michaelis-Menten equation (line). Inset, ATP hydrolysis of ClpX from P. aeruginosa and E. coli is repressed by ClpP1 but not by ClpP2. Mixtures of ClpP1 and ClpP2 sat overnight at room temperature before use. Values are normalized averages (n = 3) ± SEM. Data were compared to control values (WT) by ANOVA with Dunnett's post hoc test (***, P < 0.0001; **, P < 0.01; *, P < 0.05; N.S., not significant). P. aeruginosa ClpX hydrolyzed ATP at a rate of 274.9 ± 1.2 min−1 · ClpX6−1, and E. coli ClpX hydrolyzed ATP at a rate of 104.1 ± 2.2 min−1 · ClpX6−1. (B) ClpP1 is enzymatically active, whereas ClpP2 is not. Degradation of small fluorogenic dipeptide substrate is shown with light gray bars, and degradation of degron-tagged GFP in conjunction with ClpX is shown with dark gray bars (n.d., not detected). The reaction rates measured for E. coli enzymes are shown for comparison. (C) Increasing sodium citrate concentration increases the peptidase activity of ClpP1 but has no effect on ClpP2. (D) ActivX TAMRA-FP serine hydrolase probe labels ClpP1 but not ClpP2. For all assays, values with error bars are averages (n = 3) ± SEM.
P. aeruginosa ClpA has 60% sequence identity with E. coli ClpA and has conserved functional motifs (Fig. S5). To investigate if ClpA, rather than ClpX, might interact with ClpP2, we expressed and purified the enzyme for biochemical assays. We observed that purified P. aeruginosa ClpA exhibited increased rates of ATP hydrolysis with increasing ATP concentration (Fig. S6A) and found that ClpA with ClpP1 was autodegraded in an ATP-dependent manner (Fig. S6B), as is also observed with E. coli ClpAP (29). However, none of these biochemical experiments provided evidence for ClpA association with ClpP2.
We next tested the ability of ClpP1 and ClpP2 to hydrolyze peptide substrates independent of AAA+ unfoldase. Clp peptidases have “closed” pore conformations and exclude large substrates until activated by a partner AAA+ unfoldase, but they are able to cleave small peptide substrates of ∼10 amino acids or fewer by themselves. A common ClpP fluorogenic substrate, suc-LY-AMC, was used to monitor peptidase activity based on its change in fluorescence upon peptide-bond cleavage. We found that ClpP1 digested this substrate (although more slowly than E. coli ClpP), whereas ClpP2 had no detectable activity (Fig. 6B, light gray bars). To look for evidence that ClpP1 and ClpP2 might interact during protease assembly, we also tested a mixture of ClpP1 and ClpP2 that had been incubated overnight to allow for possible subunit mixing. The mixture showed no additional gain or loss of activity compared to ClpP1 by itself (Fig. 6B). We next tested if ClpP1 or ClpP2 activity changed in the presence of sodium citrate, a salt that promotes multimeric complex formation and that is known to increase ClpP peptidase activity in some cases (Fig. 6C) (30). We found that ClpP1 activity increased in the presence of sodium citrate, but there was no effect on ClpP2, which remained inactive (Fig. 6C).
In some species, ClpP is only active in a proteolytic complex; an example is the human mitochondrial ClpP, which is inactive and heptameric until it assembles into an active 14-mer peptidase when in complex with the ClpX ATPase (31). We therefore hypothesized that ClpP2 might gain activity in the presence of ClpX. To test this hypothesis, we carried out degradation assays with ClpX and an SsrA degron-tagged variant of green fluorescent protein (GFP). Under these conditions, we observed degradation by ClpX plus ClpP1 but not by ClpX plus ClpP2 (Fig. 6B, dark gray bars). Again, a mixture of ClpP1 and ClpP2 gave a degradation rate similar to that observed for ClpP1 in the absence of ClpP2. To test more directly for active-site assembly, we exposed ClpP1 and ClpP2 to a 6-carboxytetramethylrhodamine (TAMRA)-based fluorescent serine hydrolase probe. ClpP1 was substantially labeled by this probe, whereas ClpP2 was only slightly labeled (Fig. 6D). These data strongly suggest that purified ClpP2 lacks properly assembled catalytic sites.
Together, our results demonstrate that purified P. aeruginosa ClpX, ClpA, and ClpP1 are functional enzymes that behave as expected compared to characterized homologs from other bacteria. ClpP2, however, presents a conundrum, as the purified form used for experiments is a soluble well-behaved purified protein but shows no detectable activity either by itself or in conjunction with ClpX, ClpA, or ClpP1.
Oligomeric states of ClpP1 and ClpP2.
Tetradecamer formation is required for ClpP catalytic activity, and we therefore suspected that ClpP2 might be incompletely assembled. To test this hypothesis, we first used analytical size exclusion chromatography. ClpP1 and ClpP2 eluted as single peaks with elution volumes at 1.21 and 1.28 ml, respectively (Fig. 7A). Based on comparison with size standards, we concluded that these data are most consistent with a ClpP1 tetradecamer (predicted molecular mass, ∼323 kDa) and a ClpP2 heptamer (predicted molecular mass, ∼163 kDa). To further confirm that ClpP2 was heptameric, we used sedimentation velocity analytical ultracentrifugation (SV-AUC) and observed a predominant single sedimentation species that was also consistent with the heptamer (Fig. 7B). Similar results were observed with size exclusion chromatography with multiangle light scattering (SEC-MALS) (Fig. S7). Thus, we conclude that at least one reason purified ClpP2 lacks catalytic activity in vitro is because it forms heptamers rather than assembled active tetradecamers. Based on activity assays, this assembly defect is not rescued by coincubation with the AAA+ unfoldase partners that are known to pair with ClpP, as neither ClpX nor ClpA activated ClpP2.
FIG 7.
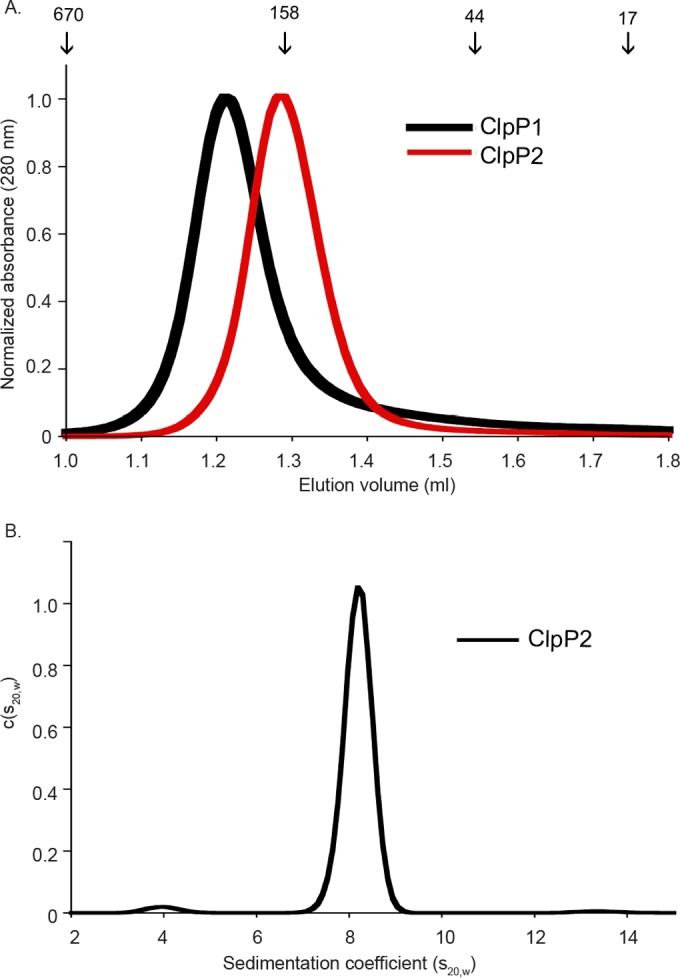
Oligomeric state of ClpP1 and ClpP2. (A) Analytical gel filtration of ClpP1 and ClpP2 at 10 μM load concentration showed tetradecamer (expected molecular mass, ∼323 kDa) and heptamer (expected molecular mass, ∼163 kDa), respectively. Size standard elution volumes are marked with arrows and values given in kilodaltons. (B) The distribution of ClpP2 by SV-AUC [c(s20,w)] is consistent with heptamer. Centrifugation was performed with 1 mg/ml protein in 0.1 M Tris (pH 8.5) and 0.15 M NaCl at 20°C. SEC-MALS data shown in Fig. S7 were also consistent with ClpP2 being a stable heptamer.
DISCUSSION
There are several examples of bacteria with multiple ClpP isoforms that are assembled into a single, biologically active, heteromeric, and barrel-shaped enzyme. For example, in Mycobacterium tuberculosis and Listeria monocytogenes, the active ClpP assembly is made of one ring of ClpP1 and one ring of ClpP2, and, in contrast, homomeric forms of either ClpP1 and ClpP2 alone are unassembled, inactive, or only marginally active (14, 32). This structural coupling of different subunit isoforms into a single active peptidase suggests that the biological roles of the two isoforms should be very similar or identical. However, here, we present an exception to this expectation, as the two P. aeruginosa ClpP isoforms, ClpP1 and ClpP2, clearly have different expression profiles and distinct biological roles (Fig. 2 to 5). Furthermore, ClpP1 assembles on its own into an active ClpP1 14-mer that forms a functional protease with the ClpX unfoldase (Fig. 6B). Thus, numerous lines of investigation strongly suggest that P. aeruginosa uses its two ClpP isoforms not simply to make a single heteromeric peptidase but rather to have a “menu” of different ClpP enzymes to use during different growth phases and/or under different environmental conditions (Fig. 2 and 8).
ClpP1 is constitutively expressed and influences pyocyanin and pyoverdine production, twitching, swimming, and initial surface attachment (this work and reference 19). In P. aeruginosa, pyoverdine and pyocyanin production and twitching motility are all regulated by the major stress response and stationary-phase sigma factor RpoS or σS (33). In several bacteria, σS levels are tightly regulated in part by proteolysis, and both ClpXP and its adaptor protein, RssB, are essential for σS degradation in E. coli. Interestingly, P. aeruginosa lacks an RssB homolog with the same function, indicating that the “control circuits” for σS regulation are distinct. In contrast to ClpP1, ClpP2 is expressed in stationary-phase cells and in biofilms (21) and is required for microcolony organization. ClpP2 expression is regulated by quorum sensing (34, 35), a cell density-dependent signaling mechanism that is also required for proper biofilm organization (36, 37).
The distinct roles of ClpP1 and ClpP2 imply that ClpP2 somehow modulates the composition of the proteome in stationary-phase cells and biofilm. Although the active form of ClpP2 has yet to be identified, we assume because of its sequence characteristics (Fig. 1) and previous work (20) that ClpP2 alters the proteome by working within a functional protease and promoting changes in protein degradation (Fig. 8). There are multiple models for how this alteration of proteolysis might be achieved (Fig. 8B). One possibility is that ClpP2 associates with a yet-to-be-characterized AAA+ unfoldase that has distinct substrate specificity compared to ClpA and ClpX (Fig. 8B, left). The very different N-terminal loop sequence of ClpP2 (a region that makes key contacts between ClpP and the pore loops of ClpX in the E. coli enzyme [Fig. 1]) compared to that of the ClpP1/ClpP family supports the idea that ClpP2 is unlikely to directly bind ClpX/ClpA in a manner similar to that used in E. coli, as these assemblies require coordination between the N-terminal loops in ClpP and the pore 2 loops of the Clp ATPase/unfoldase. Interestingly, there are several candidate genes that could encode an alternative AAA+ unfoldase partner, for example, PA0459 (19). A second class of models to explain how ClpP2 may alter the proteome is that ClpP2 either increases or decreases the overall efficiency of proteolysis, or it changes the abundance of active proteases available inside cells. For example, ClpP2 could form an active heteromer with ClpP1 (Fig. 8B, middle). In such a complex, ClpP2 could act to accelerate the rate of proteolysis, like ClpP1 from L. monocytogenes, and this rate change could cause degradation of substrates that were stable (or only very slowly degraded) by ClpP1 14-mer-containing proteases. Notably, if ClpP2 instead decreases the efficiency of proteolysis of substrates that were degraded efficiently by ClpP1-containing enzymes, some protein substrates might be markedly stabilized in the presence of ClpP2. Alternatively, ClpP2 could change the abundance of available proteases, for example, if a ClpP1/ClpP2 complex has either increased or reduced binding affinity for ClpA and ClpX, compared to the ClpP1 14-mer alone, the intracellular concentration of active Clp proteases could rise or fall when ClpP2 is expressed.
One problem, however, with any model evoking a ClpP1/P2 heteromer is that it is not straightforward to explain how the presence of ClpP2, regardless of the presence or absence of ClpP1, is required for microcolony organization (Fig. 5B). Therefore, a final attractive possibility (Fig. 8A, right) is that ClpP2 is modified somehow in vivo to “switch on” its activity. ClpP2 could have a yet-to-be-characterized prosequence or require another type of modification, such as phosphorylation or alkylation, to assemble and associate with ClpA or ClpX. Or, ClpP2 could require a protein or small-molecule cofactor for assembly, just as ClpCP from Bacillus subtilis needs MecA for efficient assembly and ClpP/R from Arabidopsis thaliana chloroplasts needs ClpT1 and ClpT2 (38, 39). Like a ClpP1/P2 heteromer, modified active ClpP2 might increase or decrease the efficiency of proteolysis or change the abundance of available proteases. These models are certainly not mutually exclusive, and it may be most attractive to consider some combination of these classes of scenarios to correctly explain the phenotypic consequences that are dependent on the presence of ClpP2.
Our work thus describes the robust importance for Clp proteolysis for the physiology and virulence of P. aeruginosa and opens new avenues of investigation into the roles of protein turnover and its inhibition in this medically important opportunistic pathogen.
MATERIALS AND METHODS
Strains and plasmids.
The plasmids, strains, and primers used are listed in Table 1 and in Table S1 in the supplemental material. PCR was carried out on P. aeruginosa PAO1 genomic DNA provided by the American Type Culture Collection (ATCC). Plasmids were selected using 100 μg/ml ampicillin, 50 μg/ml kanamycin, 10 μg/ml gentamicin (E. coli), or 30 μg/ml gentamicin (P. aeruginosa), as appropriate. Deletion strains of P. aeruginosa PAO1 were made by homologous recombination using vector pMQ30, yeast strain Saccharomyces cerevisiae InvSc1, and E. coli mating strain ST18. Briefly, pMQ30 deletion constructs were obtained by cotransforming cut pMQ30 plasmid and PCR-amplified regions flanking P. aeruginosa genes of interest into S. cerevisiae InvSc1. Reconstituted deletion plasmids were isolated, purified, concentrated, and transformed into E. coli strain ST18 for mating with P. aeruginosa PAO1. Genetic knockouts were confirmed by colony PCR and phenotypic assays. Deletion strains were healthy and divided at a rate similar to that of WT in liquid culture, although the ΔP1 and ΔP1P2 mutant strains formed smaller colonies on plates. For overexpression plasmids, genes of interest were PCR amplified from genomic DNA flanked by specific restriction sites for ligation into pET23b, pET28b (Novagen), or pMQ71 (18). For purification purposes, ClpP1 and ClpP2 were expressed with C-terminal polyhistidine (H6) and Strep II (IBA) affinity tags, respectively. ClpX was expressed with an N-terminal thrombin-cleavable polyhistidine tag and ClpA with an N-terminal polyhistidine–small-ubiquitin-like modifier (SUMO) combination tag that is cleaved off during purification.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference and/or source |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Invitrogen |
| JK10 | clpP::cat Δlon slyD::kan λDE3 | 45 |
| ST18 | S17 λpir ΔhemA | 46 |
| ER2556(pLysS) | F− λ− fhuA2 lon ompT lacZ::T7 gene I gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::mini-Tn10) | 47 |
| Pseudomonas aeruginosa | ||
| PAO1 | University of Washington Collection, 48 | |
| ΔP1 mutant | ΔClpP1 (ΔPA1801) | This study |
| ΔP2 mutant | ΔClpP2 (ΔPA3326) | This study |
| ΔP1P2 mutant | ΔClpP1 ΔClpP2 (ΔPA1801 ΔPA3326) | This study |
| ΔfliC mutant | PW2970 transposon mutant | 48 |
| ΔpilA mutant | PW8622 transposon mutant | 48 |
| Saccharomyces cerevisiae InvSc1 | MATa his3D1 leu2 trp1-289 ura3-52 MAT his3D1 leu2 trp1-289 ura3-52 | Life Technologies |
| Plasmids | ||
| pET23b | Apr, expression plasmid | Novagen |
| pET28b | Kmr, expression plasmid | Novagen |
| pMQ30 | Gmr, tool for gene deletion | 18 |
| ClpP1-pMQ30 | Gene deletion tool | This study |
| ClpP2-pMQ30 | Gene deletion tool | This study |
| pMQ71 | Apr Gmr, shuttle expression plasmid | 18 |
| ClpP1-pMQ71 | ClpP1 complementation plasmid | This study |
| ClpP2-pMQ71 | ClpP2 complementation plasmid | This study |
| pET23b.smt3 | Polyhistidine-tagged Smt3 construct | 49 |
| pBH111-pET23b | ClpP1 with C-terminal polyhistidine tag | This study |
| pBH213-pET23b | ClpP2 with C-terminal Strep II tag | This study |
| pBH304-pET28b | ClpX with N-terminal thrombin-cleavable polyhistidine tag | This study |
| pBH402-pET23b | ClpA with N-terminal Ulp1-cleavable polyhistidine tag and Smt3 SUMO tag | This study |
R, rearrangement; Apr, ampicillin resistance; Kmr, kanamycin resistance; Gmr, gentamicin resistance.
Antibody production, growth curve, and Western blotting.
The following peptides were synthesized for the purpose of polyclonal antibody production via conjugation (Covance Research Products): GGLVPMVVEQSARGERKKC and KTDDKDREGGDSHGAIKKC. The resulting antibodies were specific for their targets on ClpP1 and ClpP2, respectively, and did not cross-react (Fig. S1). WT P. aeruginosa PAO1 was grown in LB at 37°C. To determine ClpP1 and ClpP2 levels during different growth phases, growth curves were performed, cell density-adjusted aliquots were taken at select time points and pelleted by centrifugation, the supernatant was removed, and the cell pellet was resuspended in SDS loading dye. Samples were heated at 95°C for 10 min and cooled. Samples (10 μl) were loaded onto 8% Tris-Tricine polyacrylamide gels. Gels were transferred onto polyvinylidene difluoride (PVDF) membranes using a semidry apparatus (Thermo), probed with anti-ClpP1 or anti-ClpP2 antibody at a 1:6,000 or 1:3,000 dilution, respectively; incubation with these primary antibodies continued for 3 h at room temperature, followed by incubation with anti-rabbit IgG–alkaline phosphatase (AP) conjugate (Bio-Rad) at 1:5,000 dilution for 1 h at room temperature, and blots were then developed with alkaline phosphatase ECF substrate (GE Healthcare).
Motility and pyocyanin and pyoverdine assays.
Twitching plates containing 5 ml of T broth (10 g/liter tryptone, 5 g/liter NaCl) plus 1.5% agar were inoculated by stab with a sterile pipette tip and grown for 48 h at room temperature. Agar was removed and the plate was stained with 1% crystal violet dye for 5 min, washed with water, and allowed to air dry. The surface of swimming plates containing T broth with 0.3% agarose were inoculated from a single colony transferred with a sterile toothpick, wrapped in plastic wrap, and incubated face up at 30°C for 14 h. Both motility assay measurements were taken directly from plates using a National Institute of Standards and Technology (NIST)-certified ruler, and representative images were taken for display purposes only using an Alpha Innotech imager. To measure relative pyoverdine production levels, cultures were grown at 37°C for 2 days in 5 g/liter Casamino Acids, 1.54 g/liter potassium phosphate, and 0.25 g/liter magnesium sulfate. Cells were removed by centrifugation, the supernatant was diluted 1:200 in 10 mM Tris-HCl (pH 7.5), the samples were excited at 405 nm, and the emission was measured from 420 to 490 nm; values were measured at the fluorescence peak for each curve, which was always a value between 460 and 470 nm. Pyocyanin was assayed as previously described (40).
Crystal violet surface adherence assays.
Microtiter surface attachment assays were performed as previously described (41). After either 6 h or overnight incubation at 37°C in LB medium without agitation, nonattached cells were discarded and the wells washed with water. Adhered cells were stained with 0.1% crystal violet for 20 min. Again, the wells were washed with water, the crystal violet bound to the attached cells was dissolved with 70% ethanol, and the extent of adherence was assayed by measuring the absorbance of this ethanol wash at 600 nm.
Flow cell growth assays.
Microcolony or early biofilms were analyzed by growing cells for 72 h at 37°C in flow chambers with channel dimensions of 1 by 4 by 40 mm. Silicone tubing (inside diameter [i.d.], 0.062 mm; outer diameter [o.d.], 0.125 mm; walls, 0.032 mm; VWR) was autoclaved, and the system was assembled and sterilized by pumping through a 0.5% hypochlorite solution at 6 rpm for 30 min using a Watson Marlow 205S multichannel peristaltic pump. The system was then rinsed at 6 rpm with sterile water and medium for 30 min each. Flow chambers were inoculated by injecting 400 μl of an overnight culture diluted to an OD600 of 0.05. After inoculation, chambers were left without flow for 2 h, after which medium was pumped through the system at a constant rate of 0.75 rpm (3.6 ml/h). Cells were stained using the LIVE/DEAD BacLight bacterial viability kit (Molecular Probes, Eugene, OR) prior to microscopy. A 1:5 ratio of SYTO-9 (green fluorescence, live cells) to propidium iodide (PI) (red fluorescence, dead cells) was used. Microscopy was done using a confocal laser scanning microscope (FluoView FV1000; Olympus) at ×60 magnification, and three-dimensional reconstructions were generated using the Imaris software package (Bitplane AG).
Protein overexpression and purification.
ClpP1 and ClpP2 were expressed in E. coli strain JK10 grown at 37°C in Luria-Bertani broth supplemented with antibiotic. At an OD600 of ∼0.6, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to 1 mM and cells harvested after 3 h. Cell pellets were resuspended in buffer S (50 mM sodium phosphate [pH 8.0], 5 mM imidazole, 1 M NaCl, 10% glycerol), lysed by French press, and centrifuged for 20 min at 30,000 relative centrifugal force (RCF). For polyhistidine-tagged proteins, lysates were applied to nickel-nitrilotriacetic acid (Ni-NTA) resin equilibrated in buffer S, washed extensively with buffer S plus 20 mM imidazole, and protein was eluted with buffer S plus 500 mM imidazole. For Strep-II tag variant proteins, lysate was applied to Strep-Tactin resin (IBA) equilibrated in buffer W (100 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA), washed 5× with buffer W, and eluted with buffer W plus 1 mg/ml desthiobiotin. Eluates were concentrated to ∼1 ml volume and further purified using a HiPrep Sephacryl S300 HR gel filtration column equilibrated in either buffer W without EDTA for ClpP2 or buffer Q200 (50 mM Tris-Cl [pH 8.0], 1 mM dithiothreitol [DTT], 10 mM MgCl2, 10% glycerol, 0.2 M KCl) for ClpP1. Fractions containing protein of >95% purity as judged by SDS-PAGE were combined, concentrated, flash-frozen in aliquots, and stored at −80°C. P. aeruginosa ClpX was produced and purified as previously described for Caulobacter crescentus ClpX, with the exception that E. coli ER2556(pLysS) was used for overexpression (42). P. aeruginosa ClpA was overexpressed and purified with Ni-NTA, as described for ClpP1. Protein was then incubated 2 h at 30°C with His-tagged Ulp1 to cleave between the SUMO domain and ClpA, eluted from Ni-NTA resin, dialyzed into buffer Q50 (50 mM Tris [pH 8], 1 mM DTT, 10 mM MgCl2, 10% glycerol, 50 mM KCl), and purified on a Mono Q column (GE Healthcare). Selected fractions were then further purified by size exclusion in buffer Q200 as for ClpP1. GFP with a C-terminal SsrA degron tag was expressed and purified as previously described (43).
Biochemical assays.
ATPase, protein, and peptide degradation assays were carried out in 25 mM HEPES-KOH (pH 7.6), 5 mM KCl, 5 mM MgCl2, 0.032% NP-40, and 10% glycerol at 30°C. Peptidase assays used 10% dimethyl sulfoxide (DMSO), 2.0 mM N-succinyl-Leu-Tyr-7-amido-4-methylcoumarin (Suc-LY-AMC) (Bachem), and 0.4 μM ClpP tetradecamer (ClpP14), and degradation was monitored by fluorescence with excitation at 345 nm and emission at 440 nm. Degradation of GFP with a C-terminal SsrA degron tag was monitored as previously described using 0.2 μM ClpP14 and 0.1 μM ClpX6 and an ATP regeneration system (44). ATP hydrolysis rates of ClpX were monitored with 0.1 μM ClpX hexamer (ClpX6) and 0.2 μM ClpP14, as previously described (27). For active-site labeling, 20 μM ActivX TAMRA-FP serine hydrolase probe (Thermo) was mixed with 1 μM ClpP14 for 15 min at room temperature, and the protein was subjected to SDS-PAGE and visualized with a Typhoon imager, according to the manufacturer's instructions.
Ultracentrifugation and analytical gel filtration.
Analytical gel filtration was performed by loading 25 μl of protein in buffer W without EDTA onto a Superdex 200 PC 3.2/30 column (GE Healthcare) running at 0.05 ml/min. Molecular weight markers were loaded at 10 mg/ml (catalog no. 151-1901; Bio-Rad). SV-AUC was performed with a Beckman Optima XL-1 analytical ultracentrifuge (Biophysical Instrumentation Facility, Massachusetts Institute of Technology, Cambridge, MA). Sample was collected after gel filtration purification in buffer W without EDTA, loaded into a dual-sector charcoal-filled Epon centerpiece, and run at 30,000 rpm in an An-50 Ti rotor at 20°C. Data were analyzed using a solvent density of 1.00728, viscosity of 0.01048, and a ClpP2 partial specific volume of 0.7375, as calculated with SEDNTER. SEDFIT was used to calculate the continuous distribution of sedimentation coefficients, with a confidence ratio of 0.95.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Hunter for strains, E. Spear for advice on Pseudomonas aeruginosa and knockout construction, and K. Schmitz and members of the laboratory for helpful comments on the manuscript.
Analytical ultracentrifugation experiments were performed in the Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (grant NSF-0070319). Work by R.E.W.H. was supported by the Canadian Institute for Health Research. Work in the laboratory of T.A.B. was supported by U.S. National Institutes of Health (NIH) grant GM 049224, in part by NIH predoctoral training grant T32GM007287, and by the Howard Hughes Medical Institute. R.E.W.H. holds a Canadian Research Chair. T.A.B. is an employee of the Howard Hughes Medical Institute.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00568-16.
REFERENCES
- 1.Himeno H, Nameki N, Kurita D, Muto A, Abo T. 2015. Ribosome rescue systems in bacteria. Biochimie 114:102–112. doi: 10.1016/j.biochi.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Gur E, Sauer RT. 2008. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev 22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gur E, Ottofueling R, Dougan DA. 2013. Machines of destruction–AAA+ proteases and the adaptors that control them. Subcell Biochem 66:3–33. doi: 10.1007/978-94-007-5940-4_1. [DOI] [PubMed] [Google Scholar]
- 4.Gur E, Biran D, Ron EZ. 2011. Regulated proteolysis in Gram-negative bacteria—how and when? Nat Rev Microbiol 9:839–848. doi: 10.1038/nrmicro2669. [DOI] [PubMed] [Google Scholar]
- 5.Jenal U. 2009. The role of proteolysis in the Caulobacter crescentus cell cycle and development. Res Microbiol 160:687–695. doi: 10.1016/j.resmic.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Molière N, Turgay K. 2013. General and regulatory proteolysis in Bacillus subtilis. Subcell Biochem 66:73–103. doi: 10.1007/978-94-007-5940-4_4. [DOI] [PubMed] [Google Scholar]
- 7.Konovalova A, Sogaard-Andersen L, Kroos L. 2014. Regulated proteolysis in bacterial development. FEMS Microbiol Rev 38:493–522. doi: 10.1111/1574-6976.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brötz-Oesterhelt H, Sass P. 2014. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int J Med Microbiol 304:23–30. doi: 10.1016/j.ijmm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 10.Baker TA, Sauer RT. 2012. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexopoulos JA, Guarne A, Ortega J. 2012. ClpP: a structurally dynamic protease regulated by AAA+ proteins. J Struct Biol 179:202–210. doi: 10.1016/j.jsb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Lee ME, Baker TA, Sauer RT. 2010. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol 399:707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin A, Baker TA, Sauer RT. 2007. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell 27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz KR, Carney DW, Sello JK, Sauer RT. 2014. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci U S A 111:E4587–E4595. doi: 10.1073/pnas.1417120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tryggvesson A, Stahlberg FM, Mogk A, Zeth K, Clarke AK. 2012. Interaction specificity between the chaperone and proteolytic components of the cyanobacterial Clp protease. Biochem J 446:311–320. doi: 10.1042/BJ20120649. [DOI] [PubMed] [Google Scholar]
- 16.Stanne TM, Pojidaeva E, Andersson FI, Clarke AK. 2007. Distinctive types of ATP-dependent Clp proteases in cyanobacteria. J Biol Chem 282:14394–14402. doi: 10.1074/jbc.M700275200. [DOI] [PubMed] [Google Scholar]
- 17.Mikhailov VA, Stahlberg F, Clarke AK, Robinson CV. 2015. Dual stoichiometry and subunit organization in the ClpP1/P2 protease from the cyanobacterium Synechococcus elongatus. J Struct Biol 192:519–527. doi: 10.1016/j.jsb.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernández L, Breidenstein EB, Song D, Hancock RE. 2012. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:1128–1132. doi: 10.1128/AAC.05336-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu D, Eisinger VM, Head NE, Pier GB, Yu HD. 2008. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 154:2119–2130. doi: 10.1099/mic.0.2008/017368-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waite RD, Paccanaro A, Papakonstantinopoulou A, Hurst JM, Saqi M, Littler E, Curtis MA. 2006. Clustering of Pseudomonas aeruginosa transcriptomes from planktonic cultures, developing and mature biofilms reveals distinct expression profiles. BMC Genomics 7:162. doi: 10.1186/1471-2164-7-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schalk IJ, Guillon L. 2013. Pyoverdine biosynthesis and secretion in Pseudomonas aeruginosa: implications for metal homeostasis. Environ Microbiol 15:1661–1673. doi: 10.1111/1462-2920.12013. [DOI] [PubMed] [Google Scholar]
- 23.Jayaseelan S, Ramaswamy D, Dharmaraj S. 2014. Pyocyanin: production, applications, challenges and new insights. World J Microbiol Biotechnol 30:1159–1168. doi: 10.1007/s11274-013-1552-5. [DOI] [PubMed] [Google Scholar]
- 24.Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. 2012. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev 76:46–65. doi: 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tolker-Nielsen T. 2014. Pseudomonas aeruginosa biofilm infections: from molecular biofilm biology to new treatment possibilities. APMIS 122(Suppl 138):1–51. doi: 10.1111/apm.12335. [DOI] [PubMed] [Google Scholar]
- 26.Kostakioti M, Hadjifrangiskou M, Hultgren SJ. 2013. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med 3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burton RE, Baker TA, Sauer RT. 2003. Energy-dependent degradation: linkage between ClpX-catalyzed nucleotide hydrolysis and protein-substrate processing. Protein Sci 12:893–902. doi: 10.1110/ps.0237603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi SA, Hersch GL, Baker TA, Sauer RT. 2004. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol 11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- 29.Maglica Z, Striebel F, Weber-Ban E. 2008. An intrinsic degradation tag on the ClpA C-terminus regulates the balance of ClpAP complexes with different substrate specificity. J Mol Biol 384:503–511. doi: 10.1016/j.jmb.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 30.Gersch M, Stahl M, Poreba M, Dahmen M, Dziedzic A, Drag M, Sieber SA. 2016. Barrel-shaped ClpP proteases display attenuated cleavage specificities. ACS Chem Biol 11:389–399. doi: 10.1021/acschembio.5b00757. [DOI] [PubMed] [Google Scholar]
- 31.Kang SG, Dimitrova MN, Ortega J, Ginsburg A, Maurizi MR. 2005. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J Biol Chem 280:35424–35432. doi: 10.1074/jbc.M507240200. [DOI] [PubMed] [Google Scholar]
- 32.Dahmen M, Vielberg MT, Groll M, Sieber SA. 2015. Structure and mechanism of the caseinolytic protease ClpP1/2 heterocomplex from Listeria monocytogenes. Angew Chem Int Ed Engl 54:3598–3602. doi: 10.1002/anie.201409325. [DOI] [PubMed] [Google Scholar]
- 33.Suh SJ, Silo-Suh L, Woods DE, Hassett DJ, West SE, Ohman DE. 1999. Effect of rpoS mutation on the stress response and expression of virulence factors in Pseudomonas aeruginosa. J Bacteriol 181:3890–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arevalo-Ferro C, Hentzer M, Reil G, Gorg A, Kjelleberg S, Givskov M, Riedel K, Eberl L. 2003. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ Microbiol 5:1350–1369. doi: 10.1046/j.1462-2920.2003.00532.x. [DOI] [PubMed] [Google Scholar]
- 35.Chugani S, Kim BS, Phattarasukol S, Brittnacher MJ, Choi SH, Harwood CS, Greenberg EP. 2012. Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc Natl Acad Sci U S A 109:E2823–2831. doi: 10.1073/pnas.1214128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 37.Davey ME, Caiazza NC, O'Toole GA. 2003. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J Bacteriol 185:1027–1036. doi: 10.1128/JB.185.3.1027-1036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Kimber MS, Nishimura K, Friso G, Schultz L, Ponnala L, van Wijk KJ. 2015. Structures, functions, and interactions of ClpT1 and ClpT2 in the Clp protease system of Arabidopsis chloroplasts. Plant Cell 27:1477–1496. doi: 10.1105/tpc.15.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K. 2003. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci U S A 100:2306–2311. doi: 10.1073/pnas.0535717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlsson M, Shukla S, Petersson AC, Segelmark M, Hellmark T. 2011. Pseudomonas aeruginosa in cystic fibrosis: pyocyanin negative strains are associated with BPI-ANCA and progressive lung disease. J Cyst Fibros 10:265–271. doi: 10.1016/j.jcf.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 41.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 42.Chien P, Perchuk BS, Laub MT, Sauer RT, Baker TA. 2007. Direct and adaptor-mediated substrate recognition by an essential AAA+ protease. Proc Natl Acad Sci U S A 104:6590–6595. doi: 10.1073/pnas.0701776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dougan DA, Reid BG, Horwich AL, Bukau B. 2002. ClpS, a substrate modulator of the ClpAP machine. Mol Cell 9:673–683. doi: 10.1016/S1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- 44.Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. 2014. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol 21:871–875. doi: 10.1038/nsmb.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenniston JA, Burton RE, Siddiqui SM, Baker TA, Sauer RT. 2004. Effects of local protein stability and the geometric position of the substrate degradation tag on the efficiency of ClpXP denaturation and degradation. J Struct Biol 146:130–140. doi: 10.1016/j.jsb.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 46.Thoma S, Schobert M. 2009. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol Lett 294:127–132. doi: 10.1111/j.1574-6968.2009.01556.x. [DOI] [PubMed] [Google Scholar]
- 47.Neher SB, Villen J, Oakes EC, Bakalarski CE, Sauer RT, Gygi SP, Baker TA. 2006. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol Cell 22:193–204. doi: 10.1016/j.molcel.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang KH, Sauer RT, Baker TA. 2007. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev 21:403–408. doi: 10.1101/gad.1511907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bewley MC, Graziano V, Griffin K, Flanagan JM. 2006. The asymmetry in the mature amino-terminus of ClpP facilitates a local symmetry match in ClpAP and ClpXP complexes. J Struct Biol 153:113–128. doi: 10.1016/j.jsb.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrödinger, LLC. The PyMOL molecular graphics system, version 1.2r3pre. Schrödinger, LLC. [Google Scholar]
- 52.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.