

ABSTRACT
The l-arabinose-inducible araBAD promoter (PBAD) enables tightly controlled and tunable expression of genes of interest in a broad range of bacterial species. It has been used successfully to study bacterial sRNA regulation, where PBAD drives expression of target mRNA translational fusions. Here we report that in Escherichia coli, Spot 42 sRNA regulates PBAD promoter activity by affecting arabinose uptake. We demonstrate that Spot 42 sRNA represses araF, a gene encoding the AraF subunit of the high-affinity low-capacity arabinose transporter AraFGH, through direct base-pairing interactions. We further show that endogenous Spot 42 sRNA is sufficient to repress araF expression under various growth conditions. Finally, we demonstrate this posttranscriptional repression has a biological consequence, decreasing the induction of PBAD at low levels of arabinose. This problem can be circumvented using strategies reported previously for avoiding all-or-none induction behavior, such as through constitutive expression of the low-affinity high-capacity arabinose transporter AraE or induction with a higher concentration of inducers. This work adds araF to the set of Spot 42-regulated genes, in agreement with previous studies suggesting that Spot 42, itself negatively regulated by the cyclic AMP (cAMP) receptor protein-cAMP complex, reinforces the catabolite repression network.
IMPORTANCE The bacterial arabinose-inducible system is widely used for titratable control of gene expression. We demonstrate here that a posttranscriptional mechanism mediated by Spot 42 sRNA contributes to the functionality of the PBAD system at subsaturating inducer concentrations by affecting inducer uptake. Our finding extends the inputs into the known transcriptional control for the PBAD system and has implications for improving its usage for tunable gene expression.
KEYWORDS: regulatory small RNA, posttranscriptional regulation, arabinose transporter, arabinose-inducible promoter
INTRODUCTION
The Escherichia coli arabinose-inducible araBAD promoter (PBAD) system has been widely used for controlled gene expression in a broad range of bacterial hosts ever since its first application (1) 2 decades ago, owing to the fine control of expression, wide range of induction, tight repression in the absence of an inducer, and broad host range. In this system, the master dual transcriptional regulator AraC tightly controls the arabinose transporter genes (araE and araFGH) and arabinose catabolic genes (araBAD) in an arabinose-inducible manner. In the absence of arabinose, dimeric apo-AraC serves as a repressor that loops DNA and blocks transcription from PBAD. In the presence of arabinose, arabinose-bound AraC works as a transcriptional activator at the PBAD and transporter gene promoters (PE and PFGH) (reviewed in reference 2). The increased catabolism of arabinose by the AraB, AraA, and AraD enzymes downregulates intracellular arabinose levels, leading to attenuated induction of PBAD, PE, and PFGH. On the other hand, the expression of transporters increases intracellular arabinose concentrations, further activating the transcription of the arabinose catabolic and transporter promoters. These regulatory circuits contribute to PBAD activity by forming negative- (for the catabolic enzymes) or positive- (for the transporters) feedback loops. Additionally, the cyclic AMP receptor protein (CRP) is critical for the activation of the ara genes.
The arabinose transporters are essential for arabinose uptake, and thus are critical for PBAD promoter activity. AraE is a low-affinity (140 to 320 μM) high-capacity arabinose/proton symporter, and AraFGH is a high-affinity (∼10 μM) low-capacity ATP binding cassette (ABC) transporter, where AraF is the periplasmic arabinose-binding protein (3). The araE and araFGH genes are transcriptionally regulated by AraC and cyclic AMP (cAMP)-CRP transcription factors (2). At suboptimal arabinose concentrations, the positive feedback of these transporters leads to all-or-none induction of the PBAD promoter (4). Decoupling of araE promoter activity from the intracellular arabinose levels using a constitutive or isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible Plac promoter overcomes the all-or-none induction pattern of the PBAD system, significantly improving the application of the PBAD system (5–7). However, no posttranscriptional control has previously been described for the PBAD system.
Bacterial regulatory small RNAs (sRNA) are posttranscriptional regulators that control gene expression by affecting mRNA stability and/or translation initiation through limited complementary base pairing with cognate mRNA targets (reviewed in references 8 and 9). These base-pairing interactions are catalyzed by the sRNA chaperone Hfq, which binds both sRNA and mRNA targets (reviewed in references 10 and 11). The expression of Spot 42 sRNA, encoded by spf, is negatively regulated by the cAMP-CRP complex, and Spot 42 sRNA is highly expressed in early exponential phase or in glucose-containing medium (12). Recent studies suggest Spot 42 is a global RNA regulator for central carbon metabolism in E. coli (13) and Aliivibrio salmonicida (14). Through base pairing with mRNAs encoding sugar transporters and catabolic enzymes for secondary carbon sources, Spot 42 reduces the leaky expression of genes for the utilization of alternative carbon sources in the presence of a preferred carbon source (glucose) and contributes to expression dynamics in changing nutrient conditions by reinforcing the role of the transcriptional activator cAMP-CRP complex for many target genes (13). Spot 42 uses three conserved unstructured regions to base-pair with cognate mRNA targets for regulation (15).
Our lab developed a genetic approach in E. coli for screening sRNA regulators of specific mRNA targets. The approach utilizes PBAD-driven lacZ fused to an mRNA target of interest in combination with a library of plasmids that express given sRNAs (16, 17). Using this approach, we successfully identified numerous sRNA regulators for a number of mRNAs (16, 18–20). By varying the level of arabinose, the inducer for PBAD, this approach enables tuning of fusions harboring different ribosome binding-site sequences to an intermediate level of lacZ reporter expression (∼500 Miller units) for the screening of both positive and negative sRNA regulators.
This work began as part of a project to examine the effect of sRNAs on genes identified as likely and interesting targets of sRNAs based on their enrichment by Hfq immunoprecipitation. One such gene was recA. The RecA protein is essential for repairing double-strand breaks (DSBs) by homologous recombination and inducing the SOS response to DNA damage in cells (reviewed in reference 21). In experiments to identify sRNA regulators of recA mRNA using our sRNA library approach, we initially identified Spot 42 as a strong negative regulator. However, we found that Spot 42 sRNA affects PBAD-recA-lacZ expression indirectly by affecting arabinose uptake. Spot 42 sRNA achieves this by directly targeting araF mRNA, which encodes the periplasmic arabinose-binding protein of the high-affinity arabinose transporter. In this article, we discuss possible solutions to overcome this obstacle for using the PBAD system.
RESULTS
Spot 42 sRNA affects PBAD-driven genes under a low l-arabinose induction condition.
We and others successfully identified sRNA regulators for a variety of mRNA targets using an sRNA library approach combined with a chromosomally encoded, inducible translational reporter system in E. coli (16, 18–20, 22). In previous work (23), we found that recA mRNA coimmunoprecipitated with the sRNA chaperone Hfq, with a 20-fold enrichment compared to that of the total RNA sample, suggesting that Hfq-dependent sRNAs likely contribute to recA expression. Using the sRNA library approach to identify sRNA regulators for recA mRNA, we monitored recA expression using a PBAD-recA-lacZ chromosomal translational fusion and found that overexpressing only Spot 42 strongly repressed the fusion (Fig. 1A). The overexpression of Spot 42 sRNA reduced recA-lacZ expression 10-fold compared with that of the vector control (Fig. 1B). In these experiments, 0.0002% (13 μM) l-arabinose was used for induction (Fig. 1A and B). Mutational analysis of Spot 42 identified single-stranded regions I and III′, described previously (13, 15), as required for recA-lacZ repression, but there were no additive effects when both were mutated (Fig. 1B).
FIG 1.

Spot 42 sRNA regulates PBAD-recA-lacZ expression by indirectly affecting araBAD promoter activity. (A) Overexpression of individual sRNAs from an E. coli sRNA plasmid library in the PBAD-recA-lacZ reporter strain JC1005 identified Spot 42 as an sRNA repressor (>2-fold repression compared to the vector control as cutoff, shown by dotted lines). The reporter strain with sRNA plasmids was induced with 0.0002% l-arabinose and 50 μM IPTG at 37°C with shaking for 6 h before being assayed for β-galactosidase activity (Vmax/OD600). Expression of recA-lacZ was normalized to cells carrying the vector control, which gave a level of ∼40 specific units. (B) β-Galactosidase activity assay of recA-lacZ expression in JC1005 carrying Spot 42 mutants that disrupted putative base-pairing interactions with recA leader sequences. Bacterial cells were grown at 37°C in LB with ampicillin (100 μg/ml), 0.0002% l-arabinose, and 50 μM IPTG for 6 h before being assayed for β-galactosidase activity. (C) Northern blot (upper panel) to measure endogenous recA transcripts in JC1041 (lacIq Δspf) after a brief induction of Spot 42 expression (lanes 5 to 8) compared to the vector control (lanes 1 to 4). ssrA mRNA served as a loading control. Western blot (lower panel) to measure endogenous RecA protein in the WT (NM525,) the Δspf strain (JC1041), the Δspf strain carrying the vector, and the Δspf strain carrying the Spot 42 plasmid treated with IPTG and grown to an OD600 of 0.9. EF-Tu was used as a protein-loading control. (D) β-Galactosidase activity assays to measure recA-lacZ expression in JC1005 (PBAD-recA-lacZ) and JC1058 (CP12b-recA-lacZ) with overproduction of Spot 42. Bacterial cells were induced with 50 μM IPTG for 6 h at 37°C in the presence (for JC1005) or absence (for JC1058) of 0.0002% l-arabinose before being assayed for β-galactosidase activity. Results of assays are expressed in Miller units. Biological triplicates were measured, and means ± SEMs are presented.
However, ectopic expression of Spot 42 did not substantially affect the levels of endogenous recA mRNA or protein (Fig. 1C), suggesting that the regulation observed might be an artifact from the PBAD-recA-lacZ reporter. To test this notion, we constructed a second recA-lacZ reporter driven by the synthetic constitutive promoter CP12b (24) and compared it with PBAD-recA-lacZ. Consistent with the results from the endogenous recA mRNA and protein, this CP12b-driven recA-lacZ reporter was completely inert to Spot 42 overexpression as compared with the vector control, whereas expression was reduced 14.6-fold with the PBAD-recA-lacZ reporter (Fig. 1D), reinforcing the idea that Spot 42 sRNA repressed recA-lacZ expression indirectly by affecting expression from the araBAD promoter.
Spot 42 sRNA represses araF expression through direct base-pairing interactions.
Spot 42 sRNA controls multiple genes involved in secondary carbon utilization through direct base-pairing interactions (13). To determine how Spot 42 sRNA affects the PBAD-driven genes, we checked potential base-pairing interactions between Spot 42 sRNA and mRNAs in the PBAD system, including araE, -F, -G, -H, -C, -B, -A, and -D, using the CopraRNA tool (25). CopraRNA predicted a conserved base-pairing interaction between Spot 42 region I (nucleotides [nt] 1 to 8) and the araF 5′ untranslated region (UTR) (nt −30 to −37) (Fig. 2A). To verify the potential base-pairing interactions, we constructed an araF-lacZ translational fusion reporter under the control of the CP12b synthetic constitutive promoter by fusing the araF leader and initial translated region (nt −110 to 30 relative to the ATG) to the lacZ gene and then overproduced wild-type (WT) Spot 42 and six Spot 42 mutants (I, II, II′, III, III′, and I + III′) (13, 15) in the reporter strain. Overproduction of Spot 42 reduced araF-lacZ expression 2-fold, and only the Spot 42 region I mutant significantly alleviated the repression (Fig. 2B). We further confirmed that Spot 42 represses araF through a direct base-pairing interaction by testing repression in the chromosomal compensatory mutant araF*-lacZ (Fig. 2C). Ectopic expression of WT Spot 42 slightly decreased araF*-lacZ expression, and Spot 42 mutant I, which reestablished base pairing with araF*-lacZ, completely restored the repression. Collectively, these results demonstrate that Spot 42 represses araF expression by forming a short antisense interaction between its single-stranded 5′ end and the araF leader.
FIG 2.

Spot 42 sRNA represses araF expression through direct base-pairing interactions. (A) The CopraRNA-predicted RNA duplex formed between nt −37 to −30 relative to ATG of the araF 5′ UTR and nt 1 to 8 of Spot 42 sRNA, including wobble G-U base pairs (colon), is shown. Point mutations and compensatory mutations for Spot 42-I and araF* are in bold. (B) β-Galactosidase activity assay of araF-lacZ expression in JC1100 (CP12b-araF-lacZ) with the vector, the WT, and six Spot 42 mutants. Assay conditions are described as for Fig. 1B except that no arabinose was added. (C) sRNA and compensatory mRNA mutagenesis analyses demonstrate direct araF repression by Spot 42 via base-pairing interactions. Bacterial reporter strains JC1100 (araF-lacZ) and JC1102 (araF*-lacZ) transformed with the WT, a Spot 42 mutant, or the vector were induced with 50 μM IPTG at 37°C for 6 h before being assayed for β-galactosidase activities. (D) β-Galactosidase activity assays to measure PBAD-recA-lacZ expression in WT araF (JC1191) and specificity-mutant araF* (JC1192) strains overproducing Spot 42. Bacterial cells were induced with 100 μM IPTG for 6 h at 37°C in the presence of 0.0002% l-arabinose before being assayed for β-galactosidase activity. Biological triplicates were assayed, and data are plotted as means ± SEMs. Results of assays are expressed in Miller units.
Inhibition of araF by Spot 42 should reduce arabinose import and thus block induction of the promoter of the PBAD-recA-lacZ reporter at low arabinose concentrations. To confirm this, the araF* mutation that abolishes Spot 42 repression in the araF*-lacZ fusion (Fig. 2C) was introduced into the native araF gene in the PBAD-recA-lacZ reporter strain. In this strain (araF*), Spot 42 no longer repressed PBAD-recA-lacZ (Fig. 2D), demonstrating that repression of PBAD by Spot 42 fully depends on the pairing with araF. The induction level of PBAD-recA-lacZ in this strain increased (1.5-fold compared with that in WT araF), presumably reflecting the failure of endogenous Spot 42 to repress arabinose import (see below, Fig. 3B). In addition, we tested cells without the araFGH operon in the PBAD-recA-lacZ background to determine how overproducing Spot 42 affects recA-lacZ expression (see Fig. S1 in the supplemental material). Consistent with the notion that AraFGH is the major transporter at low concentrations of arabinose, the deletion of araFGH drastically decreased (14-fold) recA-lacZ induction when 0.0002% arabinose was used (compare open bars in Fig. S1A). Overexpressing Spot 42 in the ΔaraFGH strain reduced recA-lacZ expression 2.9-fold, much less than the 33.6-fold reduction in the araFGH+ host, consistent with AraFGH as the major target for Spot 42-mediated inhibition of PBAD activity at low arabinose concentrations. The remaining repression in the ΔaraFGH strain also suggests that, in the absence of AraFGH, Spot 42 likely has other targets in this regulon. Interestingly, region III′ of Spot 42, important for PBAD-recA-lacZ repression (Fig. 1B), was completely dispensable for araF-lacZ repression (Fig. 2B), further indicating that Spot 42 might affect PBAD activity through base pairing with other unknown targets involved in arabinose transport or utilization. At higher concentrations of arabinose, the effect of deleting araFGH was much less dramatic (see Fig. S1B), which is consistent with participation from other transporters in arabinose entry.
FIG 3.

Endogenous levels of Spot 42 sRNA are sufficient to repress araF expression. β-Galactosidase activity assays of araF-lacZ expression in WT (JC1100) and Δspf (JC1109) strains and araF*-lacZ expression in WT (JC1102) and Δspf (JC1110) strains in early exponential (A) and stationary (B) phases. Bacterial cells were grown at 37°C in LB or LB plus 0.2% glucose (LB+Glu) and assayed at an OD600 of 0.3 (exponential phase) or 3.0 (stationary phase) for β-galactosidase activity. Results of assays are expressed in Miller units. Fold changes in β-galactosidase activity in Δspf cells compared with that in the WT are shown above the bars. (C) Northern blot to measure Spot 42 sRNA levels during growth of JC1100 cells in the presence or absence of 0.2% glucose. ssrA mRNA served as a loading control.
Endogenous Spot 42 sRNA is sufficient to affect araF expression during early exponential cell growth or growth in glucose medium.
In E. coli, the cellular levels of Spot 42 sRNA are high in the early exponential phase and low during the remaining phases of cell growth (12). To test if endogenous levels of Spot 42 are sufficient to affect araF expression, arabinose uptake, and thus araBAD promoter activity, we compared araF-lacZ expression in WT and Spot 42 deletion (Δspf) strains grown in LB medium at exponential and stationary phases (blue bars in Fig. 3A and B). In early exponential phase, deletion of Spot 42 increased araF-lacZ expression by 2.2-fold compared with that of the WT (Fig. 3A, compare bars 1 and 2), whereas only a slight increase (1.3-fold) was observed in stationary phase (Fig. 3B, bars 1 and 2), consistent with high levels of Spot 42 in early exponential phase and low in stationary phase (Fig. 3C). Importantly, in the araF*-lacZ reporter strains, where WT Spot 42 cannot form complementary base pairing with araF*, reporter expression in the WT Spot 42 background was 2.3-fold higher than that of araF-lacZ in exponential phase (Fig. 3A, bar 5 versus bar 1), and deletion of Spot 42 did not further increase araF*-lacZ expression (Fig. 3A, compare bar 6 with bar 5). A similar pattern was seen in stationary phase, and the modest increase seen for the araF-lacZ fusion (Fig. 3B, compare bar 2 with bar 1) was eliminated in the araF*-lacZ fusion (Fig. 3B, compare bar 6 with bar 5). In summary, deleting Spot 42 (Δspf) and removing the pairing site (araF*) in both growth stages similarly increased the expression of the fusion. Taken together, these results support a base-pairing interaction between Spot 42 and araF that is required for repression and is seen most dramatically in exponential phase, where levels of Spot 42 are high.
The transcription of spf, encoding Spot 42, is negatively regulated by the cAMP-CRP complex and is highly induced in glucose medium (12) (Fig. 3C). We also tested how the endogenous Spot 42 affects araF expression in the same strains discussed above, when cells were grown in LB with 0.2% glucose (green bars in Fig. 3A and B). There was no significant effect of glucose on either Spot 42 levels (Fig. 3C) or Spot 42-dependent repression of araF-lacZ in early exponential phase (Fig. 3A). In the WT strain, araF-lacZ was expressed at the same level as in LB and was increased 2.4-fold with deletion of Spot 42 (Δspf) in LB with glucose (Fig. 3A, compare bar 4 with bar 3), similar to the 2.2-fold increase in expression in LB (Fig. 3A, compare bar 2 with bar 1). In contrast, in stationary phase, Spot 42, which was barely detectable without glucose in the WT strain, was highly abundant in cells grown with glucose (Fig. 3C). Consistent with the higher level of Spot 42, the araF-lacZ fusion was expressed at a lower level than in LB (Fig. 3B, compare bar 3 with bar 1). Therefore, the deletion of spf had a larger effect, increasing the expression by 1.9-fold (Fig. 3B, compare bar 4 with bar 3). The effects of glucose on the araF-lacZ fusion were entirely abolished in the spf null mutant (Fig. 3B, compare bar 4 with bar 2). Notably, araF*-lacZ expression was not affected by the presence of glucose or Spot 42, and expression levels were similar to those of araF-lacZ cells deleted for spf (Fig. 3A and B, bars 7 and 8 compared with bars 2 and 4). Altogether, these results demonstrate that when levels of endogenous Spot 42 are high, either in early exponential phase or when cells are grown with glucose, Spot 42 is sufficient to repress araF expression in a sequence-specific manner.
Since either multicopy or endogenous Spot 42 was sufficient to repress araF expression, we further tested the effect of Spot 42 on E. coli cell adaptation and growth in low arabinose medium. We first compared the growth of E. coli transformed with vector or Spot 42 plasmids in rich (LB) medium and in minimal medium with low (0.0002%) or high (0.2%) arabinose. The growth of cells was unaffected by Spot 42 overexpression in LB (see Fig. S2A in the supplemental material), was slightly slower in minimal 0.2% arabinose medium (Fig. S2B), and was dramatically slower in minimal 0.0002% arabinose medium (Fig. S2C) compared with the growth of the vector controls and cells in which Spot 42 was not induced. This growth defect is fully consistent with the role of Spot 42 in controlling AraFGH-mediated arabinose transport at low levels of arabinose. In a second experiment, we tested the adaptations of WT and Δspf cells when switched from minimal glucose to minimal arabinose medium (see Fig. S3). The Spot 42 deletion strain and the WT strain grew in identical manners after shifts from glucose minimal medium to either LB or minimal glucose medium. The Δspf strain had a slight but insignificant growth advantage in 0.2% (high) arabinose medium. At the 0.0002% (low) arabinose concentration, there was a modest but statistically significant growth advantage of the cells devoid of Spot 42 (see Fig. S3A and B). This growth advantage of Δspf cells is consistent with Spot 42 repressing the AraFGH transporter, causing a somewhat slower shift from glucose to arabinose.
Bypass of Spot 42 sRNA effect on the PBAD system by high arabinose concentrations or by constitutive expression of the AraE arabinose transporter.
Our lab has successfully used the PBAD system to identify and test sRNA regulators (16, 18–20, 26), and we found that Spot 42 was not always repressive in these other cases, suggesting that something about the conditions used for PBAD-recA-lacZ caused the problem. We noticed that in several cases, Spot 42 strongly affected PBAD-driven genes at low concentrations of l-arabinose but direct base-pairing interactions were hard to verify (19; unpublished data). The translation of recA was apparently robust, and thus, low levels of arabinose (0.0002%) were used for induction (Fig. 1). AraFGH is a high-affinity low-capacity ABC family arabinose transporter and is very efficient at transporting low concentrations of arabinose. Once extracellular arabinose surpasses a threshold concentration, the low-affinity high-capacity transporter AraE takes over (3) (see Fig. S1B). We measured the expression of PBAD-recA-lacZ under various concentrations of l-arabinose in the presence of multicopy Spot 42 (Fig. 4A). The 24-fold repression of recA-lacZ with Spot 42 overexpression in LB containing 0.0002% (13 μM) l-arabinose was largely eliminated (1.2-fold repression) with 0.002% (130 μM) l-arabinose and completely ablated with 0.02% (1.3 mM) l-arabinose (Fig. 4A). This arabinose dose-dependent control of recA-lacZ expression by Spot 42 is consistent with our finding that it is an indirect effect, caused by Spot 42 repression of AraF, inhibiting arabinose uptake (and thus induction of the PBAD-recA-lacZ fusion) at low levels of arabinose. This striking result also demonstrates that higher concentrations of arabinose might eliminate the effect on the expression of PBAD-driven genes from Spot 42. Such higher concentrations were used in many of our previous studies (16, 18, 19, 26), explaining why this problem was not previously appreciated.
FIG 4.
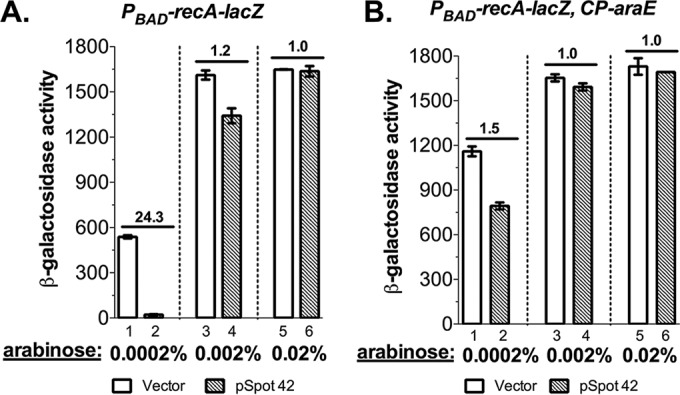
High arabinose induction concentrations or constitutive expression of the AraE transporter is sufficient to prevent Spot 42 repression of the araBAD promoter. (A) β-Galactosidase activity assays of PBAD-recA-lacZ expression in JC1005 harboring a plasmid encoding Spot 42 in the presence of 100 μM IPTG and increasing doses of arabinose (0.0002%, 0.002%, and 0.02%). (B) β-Galactosidase activity assays of recA-lacZ expression in a strain constitutively expressing the low-affinity high-capacity arabinose transporter AraE (JC1095) with the Spot 42 plasmid in the presence of 100 μM IPTG and increasing doses of arabinose (0.0002%, 0.002%, and 0.02%). Biological triplicates were assayed, and data are plotted as means ± SEMs. Results of assays are expressed in Miller units.
The arabinose-inducible promoter has an all-or-none expression pattern (1, 4), and constitutive expression of the low-affinity high-capacity AraE transporter overcomes this problem (6, 7). We tested if constitutive expression of the AraE symporter is sufficient to bypass the requirement for the AraFGH transporter, thus overcoming the inhibition by Spot 42 at low levels of arabinose. We introduced the araE gene controlled by the constitutive promoter CP18 (6) into the PBAD-recA-lacZ reporter strain and measured the effect of Spot 42 on recA-lacZ repression under various concentrations of arabinose (Fig. 4B). Constitutive expression of araE largely eliminated the inhibition on PBAD-recA-lacZ expression by Spot 42 even at the very low concentration (0.0002%) of l-arabinose (compare 1.5-fold repression in Fig. 4B [bar 2 versus bar 1] with 24-fold repression in Fig. 4A [bar 2 versus bar 1]). When induced with higher concentrations of arabinose (0.002% and 0.02%), recA-lacZ expression was completely independent of Spot 42 expression (Fig. 4B). Induction of the PBAD promoter was also better at the very low arabinose concentration in the araE constitutive strain, which is reflected by the higher level of PBAD-recA-lacZ expression (compare bar 1 in Fig. 4B with bar 1 in Fig. 4A). In both strains, 0.002% (130 μM) arabinose was sufficient for full induction (Fig. 4A and B, bars 3). Altogether, these data illustrate that we can manage the side effect of Spot 42 sRNA on PBAD-driven genes either by using higher concentrations of arabinose for induction or by working in a strain that constitutively expresses the AraE symporter.
DISCUSSION
Here we described the unexpected finding that Spot 42 translational repression of araFGH, encoding the high-affinity arabinose transporter, perturbs induction of the PBAD system in E. coli at subsaturating concentrations of the inducer. Induction of the PBAD system with either saturating concentrations of inducers or constitutive expression of the low-affinity high-capacity transporter AraE is sufficient to overcome the effect of Spot 42.
The araF gene encodes the periplasmic arabinose-binding protein of the high-affinity low-capacity arabinose transporter AraFGH and is the first gene in the operon encoding all components of this system. Our results are in good agreement with the consensus role of Spot 42 sRNA in controlling the utilization of alternative carbon sources. In the presence of glucose, the preferred carbon source, Moller et al. (27) demonstrated that highly induced Spot 42 downregulates GalK, the galactokinase in the galETKM operon essential for galactose catabolism, through base pairing with the galK mRNA Shine-Dalgarno sequence, thereby blocking galK translation. More recently, Beisel and Storz expanded the role of Spot 42 in carbon catabolite repression by identifying Spot 42-repressed genes from multiple operons involved in the uptake and utilization of nonfavored carbon sources, including xylose, fucose, galactose, sorbitol, and lactate (13). As the cAMP-CRP complex activates many of these sugar utilization genes and also represses spf (Spot 42 encoding gene), these regulatory pathways form a feedforward loop where Spot 42 reduces the leaky expression of target genes in glucose medium and contributes to expression dynamics of target genes under changing nutrient conditions. By identifying the high-affinity arabinose transporter as a new target of Spot 42, our finding further expands the repertoire of Spot 42-controlled carbon utilization genes, reinforcing it as a global RNA regulator for central carbon metabolism. It seems quite possible that the use of other catabolite-repressed promoters with coregulated permeases is also subject to Spot 42 regulation in E. coli and related bacteria. For instance, it was shown that the xylose ABC transporter xylF is repressed by Spot 42 (13). Because many of these genes are not well expressed in the absence of the inducing sugar, they may not be detected in most experiments, as was the case for the araFGH operon in previous studies (13, 23).
The PBAD system is a popular tool for inducible gene expression (28), and understanding the regulatory details will help improve the use of this system. It was previously recognized that one of the issues for the PBAD system was the all-or-none induction pattern at low concentrations of inducers (4). This problem can be solved by decoupling the transport of the inducer from the induction of the transporter genes (araE and araFGH). This has been achieved via constitutive expression of the transporter genes (6, 7) and by expressing a promiscuous LacY mutant (LacYA177C) transporter with relaxed specificity in an araE, araFGH, and araBAD-null strain (29). We demonstrated here that Spot 42 sRNA downregulates the high-affinity transporter AraFGH, thus affecting PBAD activity at low concentrations of inducers. To produce recombinant proteins with the PBAD expression system, high concentrations of an inducer will likely be used, and thus the effect of endogenous Spot 42 on AraF will be subtle (Fig. 3). However, for experiments in which levels of Spot 42 are high (early exponential phase or growth in glucose medium), endogenous levels of Spot 42 will reinforce the all-or-none behavior of the ara regulon by repressing basal levels of araFGH expression and thus may confound the interpretation of effects expected from the PBAD-controlled protein. Many other sugar utilization pathways are regulated by Spot 42 (13), and some of them (e.g., d-xylose and l-rhamnose) also show all-or-none responses (30). Thus, it is very likely that Spot 42 also contributes to complex single-cell behaviors in these pathways. Our results also suggest that Spot 42-mediated inhibition of PBAD at low inducer concentrations may involve other components of the arabinose utilization system. This is suggested by the observation that the III′ mutant of Spot 42 interfered with repression (Fig. 1B), although it did not affect araF repression (Fig. 2B) and moderately repressed PBAD-recA-lacZ in cells lacking araFGH (see Fig. S1A). However, these other effects were not seen when the AraFGH system was resistant to Spot 42 (Fig. 2D), suggesting that Spot 42 likely affects the expression of lower-affinity transporters. Nevertheless, given the effectiveness of constitutive araE expression for homogeneous induction of the PBAD genes (6, 7) as well as bypassing Spot 42 effects, incorporating constitutive expression of arabinose transporters is likely to be the best general solution.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are derivatives of the E. coli K-12 strain MG1655. Bacterial strains and plasmids are listed in Table S1 in the supplemental material. Primers, probes, and synthetic gene fragments are listed in Table S2. Spot 42 deletion strains and araE constitutive expression strains were constructed using phage P1vir transductions as previously described by Miller (31). The chromosomal inducible translational fusion PBAD-recA-lacZ reporter strain (JC1005) was generated by λ Red recombineering in strain PM1805 with PCR products of the recA 5′ UTR and the first 10 codons (nt −50 to 30 relative to the ATG) amplified using oligonucleotides JC33/JC28 and selected on sucrose minimal agar plates (32). The constitutive CP12b-recA-lacZ reporter strain (JC1058) was generated by replacing the araBAD promoter in JC1005 with a kan-CP12b gene fragment amplified from JC1051 using primers JC63/JC71 and selected on LB agar plates with kanamycin (Kan). The CP12b-araF-lacZ (JC1100) and CP12b-araF*-lacZ (JC1102) reporter strains were generated using synthetic DNA gene fragments, gblocks (IDT, Coralville, IA) named uPBAD-zeo-CP12b-araF-lacZ and uPBAD-zeo-CP12b-araF*-lacZ (DNA sequences are listed in Table S2), which contain the araF 5′ UTR and the first 10 codons (nt −110 to 30 relative to the ATG). These were introduced into PM1805 for recombineering (32, 33) and selected on LB agar plates with zeocin (Zeo). These gblocks contain a sequence of 40 bp homologous to the region upstream of PBAD (uPBAD) in PM1805, enabling replacement of PBAD with the zeo-CP12b cassette. The ΔaraFGH derivative of JC1005 (JC1180) was generated via λ Red recombineering in JC1005 with a PCR product amplified from JCS1001, using oligonucleotides JC149/JC150, and selected on LB agar plates with Kan. Derivatives of JC1005 harboring the chromosomal WT araF (JC1191) or specificity mutant araF* (JC1192) were constructed by homologous recombination of PCR products amplified from JC1100 or JC1102, respectively, using oligonucleotides JC164/JC165 into JC1186. JC1186 is a derivative of JC1005 in which a kan-sacB cassette, amplified from NM543 using oligonucleotides JC167/JC163, was inserted in the native araF leader. This enabled the counterselection of final recombinants containing araF or araF* on minimal sucrose agar. All chromosomal constructs in the reporter strains were verified by PCR and Sanger sequencing.
sRNA genes in the library were cloned in the pBRplac vector plasmid under the control of an isopropyl-β-d-1-thiogalactopyranoside (IPTG)-inducible promoter (16). Various Spot 42 sRNA mutants were either obtained from G. Storz (NIH) or generated by QuikChange mutagenesis as described by Beisel et al. (13, 15). Specifically, the pSpot 42 I + III′ mutant was constructed by QuikChange site-directed mutagenesis (Agilent Technologies) using pSpot 42 I as the DNA template and Spot 42-III′.fwd/Spot 42-III′.rev as primers (15). The construct was verified by Sanger sequencing.
All bacterial strains were grown in Lennox broth medium or M63 salt (KD Medical, Columbia, MD). The M63 medium was supplemented with 0.001% vitamin B1, 0.0001% biotin, 0.1% Casamino Acids, and either glucose or arabinose as the carbon source, as indicated. When needed, glucose was supplemented at a final concentration of 0.2%. A 0.0002% (13 μM) concentration of arabinose was used for PBAD induction except where otherwise indicated, and 50 μM or 100 μM IPTG was used for sRNA induction. The following concentrations of antibiotics were used: ampicillin, 100 μg/ml; chloramphenicol, 10 μg/ml; kanamycin, 25 μg/ml; and zeocin, 25 μg/ml.
sRNA library screen and β-galactosidase activity assay.
The sRNA overexpression library screen was carried out as described by Mandin and Gottesman (16). Briefly, 29 E. coli sRNAs were individually transformed into the PBAD-recA-lacZ reporter strain JC1005 using the transformation and storage solution (TSS) method (34). Transformants were inoculated into a 96-well microtiter plate containing 100 μl LB plus 100 μg/ml ampicillin, 0.0002% arabinose, and 50 μM IPTG to induce expression of the sRNAs and reporter gene and were grown at 37°C for 6 h with shaking at 225 rpm. The optical densities of the cells at 600 nm (OD600) were measured at the endpoint, and bacterial cells were permeabilized by adding 50 μl permeabilization buffer (100 mM Tris-HCl [H 7.8], 32 mM Na2HPO4, 8 mM dithiothreitol [DTT], 8 mM EDTA, 4% Triton X-100, 200 μg/ml polymyxin B). The β-galactosidase activities were measured as a function (Vmax) of OD420 versus time after the addition of 50 μl of an o-nitrophenyl-β-d-galactopyranoside (ONPG; 4 mg/ml) solution by using the SpectraMax 250 microtiter plate reader (Molecular Devices) as described by Majdalani et al. (35) and Zhou et al. (36). The final β-galactosidase activities for this assay were determined as specific units calculated as the Vmax normalized to OD600. These units, used only in Fig. 1A, are about 25 times smaller than Miller units.
lacZ expression in all other experiments, in various reporter strains, was determined by β-galactosidase activity assay as described by Miller (31). Unless otherwise noted, bacterial cultures were assayed at an OD600 of ∼3.0. Biological triplicates were assayed, and data were plotted as means ± standard errors (SEMs).
RNA extraction and Northern blot analysis.
To measure the accumulation of endogenous Spot 42 sRNA during cell growth, overnight cultures were diluted 500-fold into fresh LB medium in the presence or absence of 0.2% glucose and incubated at 37°C with shaking at 225 rpm. A 700-μl sample was collected from each culture when the OD600 reached ∼0.2, and then additional samples were taken every 15 min until cells reached an OD600 of ∼3.0. To determine the effect of ectopic expression of Spot 42 on endogenous recA mRNA levels, cells carrying vector plasmid control (pBRplac) or pSpot 42, in which Spot 42 is expressed from the lac promoter, were induced with IPTG (100 μM) at an OD600 of ∼0.4, and a 700-μl sample was collected at time zero and at 1, 5, and 10 min postinduction. Cellular total RNA was extracted from each sample using the hot phenol method as described by Masse et al. (37) and probed with the biotinylated recA probe (see Table S2).
Northern blot analyses of Spot 42 sRNA were conducted as previously described (38). Briefly, 3 μg of total RNA from each sample were resolved on a Bio-Rad Criterion 10% Tris-borate-EDTA (TBE)–urea polyacrylamide gel in 1× TBE buffer at 100 V for 1 h. The resolved RNA samples were then transferred to a Zeta-probe GT membrane (Bio-Rad) by wet electroblotting at 200 mA for 2 h in 0.5× TBE and cross-linked to the membrane by UV irradiation. The resulting membrane was hybridized with the biotinylated Spot 42 and ssrA probes (see Table S2) in ULTRAhyb solution (Ambion) at 42°C overnight and further incubated with a streptavidin-conjugated alkaline phosphatase. The blot was then developed using the BrightStar BioDetect kit (Ambion) according to the manufacturer's instructions. Northern blot to detect recA mRNA was performed as previously described (38). Northern blots were imaged by capturing the chemifluorescence using the image analyzer LAS-3000 (Fujifilm Life Science).
Western blotting.
Western blotting to determine the endogenous RecA protein levels was performed using standard procedures. Bacterial cells were diluted 500-fold from overnight cultures into fresh LB plus 0.2% glucose medium. For overexpressing sRNAs from plasmids, ampicillin and IPTG (100 μM) were added to the medium.
Bacterial cultures were collected at an OD600 of ∼0.9, and a volume equivalent to 1 ml of cells at an OD600 of 1 was pelleted, washed once with phosphate-buffered saline (PBS) (KD Medical), and lysed by adding 100 μl of 1× SDS sample buffer (New England BioLabs) and boiling for 10 min. A 10-μl sample of the supernatant was resolved on a 12% bis-Tris polyacrylamide NuPAGE gel (Invitrogen) in 1× morpholinepropanesulfonic acid (MOPS) buffer (Invitrogen). Protein samples were then transferred onto a 0.2-μm nitrocellulose membrane using an iBlot gel transfer device (Invitrogen). The membrane was blocked for 30 min in PBS with 0.1% Tween 20 (PBST) and 5% nonfat dry milk and probed with rabbit polyclonal anti-RecA antibodies (Abcam; 1:1,000) in PBST with 5% nonfat dry milk at room temperature for 1 h. After three PBST washes, the membrane was probed with alkaline phosphatase-conjugated anti-rabbit IgG antibodies (Cell Signaling Technology; 1:2,500) for another hour. The membrane was washed three additional times with PBST and then developed using the Novex AP chemiluminescent substrate (Thermo Fisher Scientific). Western images were acquired by capturing the chemifluorescence using the image analyzer LAS-3000 (Fujifilm Life Science).
Supplementary Material
ACKNOWLEDGMENTS
We thank Gottesman lab members K. Kavita and F. De Mets, as well as G. Storz and T. Updegrove, for their comments on the manuscript. We also thank other lab members for helpful discussion of the project.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00691-16.
REFERENCES
- 1.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34:779–796. doi: 10.1111/j.1574-6976.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- 3.Hogg RW. 1977. l-Arabinose transport and the l-arabinose binding protein of Escherichia coli. J Supramol Struct 6:411–417. doi: 10.1002/jss.400060314. [DOI] [PubMed] [Google Scholar]
- 4.Siegele DA, Hu JC. 1997. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci U S A 94:8168–8172. doi: 10.1073/pnas.94.15.8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khlebnikov A, Risa O, Skaug T, Carrier TA, Keasling JD. 2000. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J Bacteriol 182:7029–7034. doi: 10.1128/JB.182.24.7029-7034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. 2001. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology 147:3241–3247. doi: 10.1099/00221287-147-12-3241. [DOI] [PubMed] [Google Scholar]
- 7.Afroz T, Biliouris K, Boykin KE, Kaznessis Y, Beisel CL. 2015. Trade-offs in engineering sugar utilization pathways for titratable control. ACS Synth Biol 4:141–149. doi: 10.1021/sb400162z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottesman S, Storz G. 2011. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3:pii=a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Updegrove TB, Zhang A, Storz G. 2016. Hfq: the flexible RNA matchmaker. Curr Opin Microbiol 30:133–138. doi: 10.1016/j.mib.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sahagan BG, Dahlberg JE. 1979. A small, unstable RNA molecule of Escherichia coli: spot 42 RNA. II. Accumulation and distribution. J Mol Biol 131:593–605. [DOI] [PubMed] [Google Scholar]
- 13.Beisel CL, Storz G. 2011. The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol Cell 41:286–297. doi: 10.1016/j.molcel.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen GA, Ahmad R, Hjerde E, Fenton CG, Willassen NP, Haugen P. 2012. Expression profiling reveals Spot 42 small RNA as a key regulator in the central metabolism of Aliivibrio salmonicida. BMC Genomics 13:37. doi: 10.1186/1471-2164-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beisel CL, Updegrove TB, Janson BJ, Storz G. 2012. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J 31:1961–1974. doi: 10.1038/emboj.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandin P, Gottesman S. 2010. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J 29:3094–3107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandin P. 2012. Genetic screens to identify bacterial sRNA regulators. Methods Mol Biol 905:41–60. doi: 10.1007/978-1-61779-949-5_4. [DOI] [PubMed] [Google Scholar]
- 18.De Lay N, Gottesman S. 2012. A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol Microbiol 86:524–538. doi: 10.1111/j.1365-2958.2012.08209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee HJ, Gottesman S. 2016. sRNA roles in regulating transcriptional regulators: Lrp and SoxS regulation by sRNAs. Nucleic Acids Res 44:6907–6923. doi: 10.1093/nar/gkw358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker A, Gottesman S. 2016. Small RNA regulation of TolC, the outer membrane component of bacterial multidrug transporters. J Bacteriol 198:1101–1113. doi: 10.1128/JB.00971-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox MM. 2003. The bacterial RecA protein as a motor protein. Annu Rev Microbiol 57:551–577. doi: 10.1146/annurev.micro.57.030502.090953. [DOI] [PubMed] [Google Scholar]
- 22.Bak G, Lee J, Suk S, Kim D, Young Lee J, Kim KS, Choi BS, Lee Y. 2015. Identification of novel sRNAs involved in biofilm formation, motility, and fimbriae formation in Escherichia coli. Sci Rep 5:15287. doi: 10.1038/srep15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. 2013. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol 425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen PR, Hammer K. 1998. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol 64:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. 2013. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 110:E3487–E3496. doi: 10.1073/pnas.1303248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon K, Gottesman S. 2011. Competition among Hfq-binding small RNAs in Escherichia coli. Mol Microbiol 82:1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Møller T, Franch T, Udesen C, Gerdes K, Valentin-Hansen P. 2002. Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon. Genes Dev 16:1696–1706. doi: 10.1101/gad.231702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan-Kiss RM, Wadler C, Cronan JE Jr. 2002. Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc Natl Acad Sci U S A 99:7373–7377. doi: 10.1073/pnas.122227599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Afroz T, Biliouris K, Kaznessis Y, Beisel CL. 2014. Bacterial sugar utilization gives rise to distinct single-cell behaviours. Mol Microbiol 93:1093–1103. doi: 10.1111/mmi.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 32.Mandin P, Gottesman S. 2009. A genetic approach for finding small RNAs regulators of genes of interest identifies RybC as regulating the DpiA/DpiB two-component system. Mol Microbiol 72:551–565. doi: 10.1111/j.1365-2958.2009.06665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Court DL, Swaminathan S, Yu D, Wilson H, Baker T, Bubunenko M, Sawitzke J, Sharan SK. 2003. Mini-lambda: a tractable system for chromosome and BAC engineering. Gene 315:63–69. doi: 10.1016/S0378-1119(03)00728-5. [DOI] [PubMed] [Google Scholar]
- 34.Chung CT, Niemela SL, Miller RH. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A 86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. 1998. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc Natl Acad Sci U S A 95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Gottesman S. 1998. Regulation of proteolysis of the stationary-phase sigma factor RpoS. J Bacteriol 180:1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massé E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Lay N, Gottesman S. 2009. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 191:461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.