

Abstract
Environmental mutagens cause DNA damage that disturbs replication and produces mutations, leading to cancer and other diseases. We discuss mechanisms of mutagenesis resulting from DNA damage, from the level of DNA replication by a single polymerase to the complex DNA replisome of some typical model organisms (including bacteriophage T7, T4, Sulfolobus solfataricus, E. coli, yeast and human). For a single DNA polymerase, DNA damage can affect replication in three major ways: reducing replication fidelity, causing frameshift mutations, and blocking replication. For the DNA replisome, protein interactions and the functions of accessory proteins can yield rather different results even with a single DNA polymerase. The mechanism of mutation during replication performed by the DNA replisome is a long-standing question. Using new methods and techniques, the replisomes of certain organisms and human cell extracts can now be investigated with regard to the bypass of DNA damage. In this review, we consider the molecular mechanism of mutagenesis resulting from DNA damage in replication at the levels of single DNA polymerases and complex DNA replisomes, including translesion DNA synthesis.
Keywords: environmentalmutagens, DNA damage, DNA replication, mutation, polymerase, replisome
1. Introduction
Large amounts of chemicals are produced in industry, agriculture, and transportation (automobile and truck exhausts). These environmental chemicals can be widespread in air, water, and soil. In 1915, Yamagiwa and Ishikawa [1] reported the formation of tumors in the ears of rabbits after treatment with tars, and in 1933 Cook et al. isolated benzo[a]pyrene as a component of coal tar [2]. Many chemicals have now been shown to cause mutations and cancer. Animals have produced tumors after exposure to many environmental mutagens [3]. Human exposure to specific chemical or physical carcinogens also produces characteristic mutational spectra, considered to be highly relevant not only to cancer but also to cardiovascular disease, teratology, and aging [4-6]. Therefore, studies on how chemicals can lead to mutations have received considerable attention.
Beginning in the 1940s, James and Elizabeth Miller showed that these environmental mutagens are converted to reactive products in the body [7]. These compounds react with DNA to form DNA adducts [8, 9]. In each human cell, > 50,000 DNA damage sites can occur every day [10]. DNA adducts lead to mutations during DNA replication that can further lead to cell death, aging, birth defects, and cancer. Several families of DNA polymerases are involved in DNA synthesis. At least seven families of DNA polymerases have been described, classified based on their sequences and structural similarities: A, B, C, D, X, Y, and reverse transcriptase. Each family has specific functions in DNA polymerization. In general, the A-family and C-family DNA polymerases in prokaryotes, the B-family DNA polymerases in eukaryotes, and the A-family mitochondrial Polγ in eukaryotes are much faster and accurate (and are termed replicative DNA polymerases) compared to other families of DNA polymerases. The Y-family polymerases carry out translesion DNA synthesis (TLS) and bypass DNA damage in an error-prone or error-free manner. Some TLS DNA polymerases preferentially insert the correct base and do not result in mutation, giving at least 3 outcomes from the process: no mutation, mutation, or cell death.
Generally, the replicative DNA polymerases that perform normal DNA synthesis are replaced by Y-family DNA polymerases when bypassing DNA damage and return back to the replication fork after bypass of the DNA damage [11]. Upon encountering DNA damage, polymerases may misincorporate, be blocked by these adducts, or produce frameshifts, each of which disturbs DNA replication and leads to mutations. The details of how DNA damage disturbs DNA replication will be summarized and analyzed in this review. DNA replication in vivo includes both leading- and lagging-strand DNA synthesis, generally not performed by only a single DNA polymerase but by the complex DNA replisome that contains DNA polymerase(s), helicase, primase, single-stranded DNA binding protein, and a number of other accessory proteins [12]. Thus, the DNA replisome plays a critical role in damage bypass, which may not be observed with a single DNA polymerase. The effects of DNA damage on replication by DNA replisomes have not been extensively investigated until recently. It has been accepted that polymerase exchange occurs when a DNA replisome encounters DNA damage. Besides polymerase exchange, other proteins in the replisome may also be involved in the bypass of DNA damage. In this review, we summarize and analyze current studies on the effects of DNA damage on replication by the DNA replisome of Escherichia coli and bacteriophages T4 and T7.
In addition to the bypass of DNA damage by DNA polymerases, other proteins in the DNA replisome are also involved in dealing with DNA damage. For instance, the E. coli DNA repair helicase UvrA2B protein complex allows limited ATP-dependent scanning of DNA to detect damaged bases in UV-induced lesions, and the UvrD helicase (helicase II) in E. coli nucleotide excision repair (NER) excises damage-containing oligonucleotides [13]. Semi-replicative simian virus 40 (large T antigen) DNA helicases are able to bypass a bulky adduct on the translocated strand [14], bringing a new understanding of the interaction between enzymes and DNA damage.
Overall, DNA damage has a wide range of effects on DNA replication and its related processes. In this review, we will address how DNA damage leads to mistakes in DNA replication from the viewpoint of an individual DNA polymerase (from bacteriophage T7, S. solfataricus, E. coli, yeast or human) and the DNA replisome (of E. coli, bacteriophage T7 or T4). The studies about single DNA polymerases were mainly contributed by Gugenerich’s lab. Because the human DNA replisome is far more difficult to study, the human DNA replication complex will be discussed only briefly at the end of this review. Mutations arising from DNA repair are not included in this review. DNA damage has a wider effect on living systems beyond DNA replication and the DNA polymerase and DNA replisome, scientific areas which should also be further explored. We have selected some typical DNA polymerases and types of DNA damage to describe mechanisms of mutation in the presence of DNA damage.
2. Environmental mutagens cause DNA damage
2.1. Environmental mutagens cause cancer
Environmental mutagens damage the genome by forming DNA adducts through different chemical reactions [15], including alkylation (which may involve cross-linking), oxidation, deamination, coordination, photo-addition, and hydrolysis (Table 1). Alkylating agents produce N2-alkyl-2′-deoxyguanosine (N2-alkylG), O6-alkylguanine (O6-alkylG), polycyclic aromatic hydrocarbon DNA adducts (PAH-DNA), and etheno (ε) DNA adducts. Bis-electrophilic agents form DNA-DNA and DNA-protein cross-links. Oxidizing agents, ionizing radiation, and UV irradiation can form 7,8-dihydro-8-oxo-guanine (8-oxoG). Arylamines and 1-nitropyrene (1-NP) produce several DNA adducts, e.g., 2-aminofluorene (AF-dG) and N-[deoxyguanosine-8-yl]-1-aminopyrene (APG) adducts following oxidation and esterification. Heavy metal ions, e.g. Cr(III), form DNA-DNA and DNA-protein cross-links by coordination. UV radiation leads to photoproducts (e.g., cyclobutane pyrimidine dimer, CPD). Spontaneous hydrolysis of glycosidic bonds results in the formation of abasic (apurinic/apyrimidinic, AP) sites. The various DNA damage products lead to mutation during DNA replication, altering oncogenes and tumor suppressor genes, leading to cancer.
Table 1.
Categories of DNA damage.
| DNA damage category | Structures and names | Agents | Ref. |
|---|---|---|---|
| N2-alkylG |
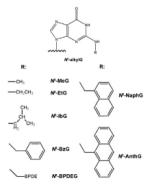
|
Alkylation agents: Formaldehyde, Ethanol, Polycyclic aromatic hydrocarbons, et al. |
[13,14,16,17] |
| N2,N2-dialkylG |
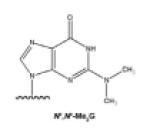
|
Alkylation agents: Formaldehyde |
[16] |
| N6-alkylA |
|
Polycyclic aromatic hydrocarbons: benzo[a]pyrene (B[a]P), et al. |
[20] |
| O6-alkylG |
|
Alkylation agents | [18,19] |
| Oxidized lesion |
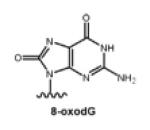
|
Oxidation agents, ionizing radiation, UV irradiation, et al. |
[25,26] |
| Etheno (ε) DNA adducts |
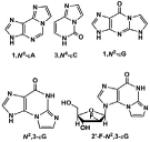
|
Bioactivated Vinyl monomers: mutagens vinyl chloride, vinyl carbamate, et al. |
[20,21] |
| DNA-DNA or DNA-protein cross-links |
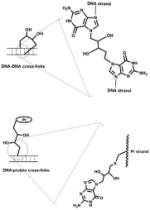
|
Bis- electrophilic agents: 1,3-butadiene, et al. |
[22–24] |
| Arylation adducts |
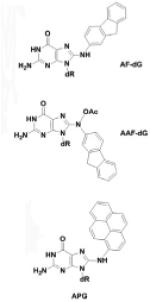
|
Arylamines and N-acetyl arylamines, 1-Nitropyrene, et al. |
[27,28] |
| DNA-DNA coordinated cross-links |
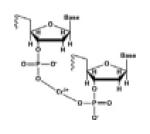
|
Heavy metal ions: Cr (III) ion |
[29] |
| Cyclobutane pyrimidine dimer (CPD) photoproduct |
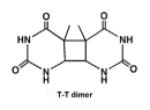
|
UV radiation, et al. |
[30,31] |
| Apurinic/ apyrimidinic (AP) sites |
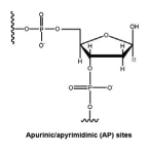
|
Spontaneous, chemically- induced, enzyme- catalyzed hydrolysis of the N-glycosyl bond |
[32] |
2.2. Environmental mutagens produce DNA damage via different chemical reactions
Alkylation
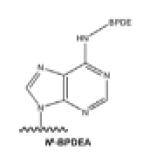
Alkylation agents result in the formation of N2-alkylG and other lesions. Formaldehyde, an environmental concern, reacts with the exocyclic amino group of deoxyguanosine (G), to produce N2-methylG [16]. Ethanol is enzymatically oxidized to acetaldehyde, which forms N2-ethylG, which has been observed in liver DNA and urine of alcoholic patients [17]. With both of these reactions [13, 14], 2-electron reduction of the intermediate imine is required to balance the stoichiometry. Alkylating agents also lead to the formation of O6-methylG (O6-MeG). O6-MeG is a common lesion formed after exposure to tobacco-specific nitrosamines. Chemotherapy, in which many alkylating agents are used, also produces this DNA adduct [18, 19]. Polycyclic aromatic hydrocarbons (PAHs) are toxic and mutagenic environmental pollutants produced from the combustion of carbon-containing materials, and alkylation of DNA yields PAH-DNA adducts. For instance, benzo[a]pyrene (B[a]P), a prototypical PAH, is initially oxidized to the epoxide (B[a]P-7,8-epoxide) which is then hydrolyzed to a dihydrodiol (B[a]P-7,8-dihydrodiol) and further converted to a diol-epoxide (B[a]P-7,8-dihydrodiol-9,10-epoxide, BPDE), which alkylates the exocyclic amino group at the N2 position of G or the N6 position of A. N2-BPDEG is the major BPDE-DNA adduct found in vivo [20]. Endogenous processes (e.g. lipid peroxidation) and exposure to bioactivated vinyl monomers, such as the epoxides formed from vinyl chloride and vinyl carbamate (an oxidation product of urethane), can lead to the formation of etheno (ε) DNA adducts, a series of exocyclic adducts including 1,N6-ethenoadenine (1,N6-εA), 3,N4-ethenocytosine (3,N4-εC), N2,3-ethenoguanine (N2,3-εG), and 1,N2ethenoguanine (1,N2-εG) [21].
Bis-electrophilic agents, such as 1,3-butadiene (BD) (present in automobile exhaust and cigarette smoke), can result in DNA-DNA and DNA-protein cross-links. BD can be oxidized to 1,2,3,4-diepoxybutane (DEB), a prominent bis-electrophilic carcinogenic metabolite (Fig. 1). Alkylation of adenine or guanine base by DEB produces 2-hydroxy-3,4-epoxybut-1-yl (HEB) DNA lesions that contain an inherently reactive oxirane group that can further alkylate neighboring nucleotide bases within the DNA duplex to form DNA-DNA cross-links [22, 23]. Alternatively, the 3,4-epoxy ring can also be subject to nucleophilic attack by amino acid groups in side chains of neighboring proteins, giving rise to DNA-protein cross-links [24]; the opposite order (DEB reacting with a protein such as O6-alkylG DNA-alkyl transferase or the tripeptide glutathione and then DNA) is also documented and probably more likely.
Fig. 1.

Formation of DNA-DNA or DNA-protein crosslinks involving an oxidation product of 1,3-butadiene (BD). (Pr: protein)
Oxidation
Oxidizing agents can produce 7,8-dihydro-8-oxo-2′-oxodeoxyguanosine (8-oxodG) lesions 8-oxodG is a ubiquitous lesion arising from the oxidation of the C8 atom of G to form a hydroxyl group by free radical intermediates of oxygen that are produced by chemical oxidation, ionizing radiation, or UV irradiation [25, 26]. The enol (a lactim) at the C8-N7 position of G is converted to the more stable 8-oxodG lactam form.
Amination
Arylamines and N-acetyl arylamines are well-studied mutagens used as models and found in numerous occupational settings, tobacco smoke, and chemical dyes, leading to the formation of adducts such as the models 2-aminofluorene (AF-dG) and N-acetyl-2-aminofluorene (AAF-dG) through amination of the C8 atom of guanine (via an initial N7 reaction, linking the amine group of the arylamine) [27] (Fig. 2A). 1-Nitropyrene (1-NP) is the most abundant nitro PAH, on a global basis. The active product of 1-NP (an ester of the reduction product hydroxylamine) forms a bulky N-[deoxyguanosine-8-yl]-1-aminopyrene (APG) adduct by reaction at the C8 atom of deoxyguanosine following an initial reaction at the N7 atom and rearrangement [28] (Fig. 2B).
Fig. 2.

Formation of N-(deoxyguanosin-8-yl)-2-aminofluorene (AF-dG), N-(deoxyguanosin-8-yl)-N-acetyl-2-aminofluorene (AAF-dG), and N-[deoxyguanosine-8-yl]-1-aminopyrene.
Coordination
Heavy metal ions can form DNA-DNA intra-strand and inter-strand cross-links via coordination. Cr (VI) complexes permeate cell membranes and are reduced to form Cr (III) complexes that can then coordinate with oxygen atoms of phosphate backbone of two adjacent nucleotides within one DNA strand or between two DNA strands, yielding Cr (III)-DNA intra-strand yielding inter-strand cross-links [29].
Photoaddition
UV radiation leads to the formation of photoproducts (e.g., CPD) by cycloaddition of the C5-C6 double bonds of adjacent pyrimidine bases. Six diastereomers are generated, depending on the position of pyrimidine moieties with respect to the cyclobutane ring (cis/trans stereochemistry) and on the relative orientation of the two C5-C6 bonds (syn/anti regiochemistry) [30]. The cis-syn form is formed preferentially to the trans-syn diastereomers within double-stranded DNA. The trans-anti and trans-syn photoproducts are only present within single-strand or denatured DNA [31].
Hydrolysis
AP sites are generated via spontaneous, chemically-induced, or enzyme-catalyzed hydrolysis of the N-glycosyl bond [32], losing genetic information. In mammalian cells, it has been estimated that approximately 12,000 purines are lost spontaneously per genome per cell generation (20 h) in the absence of any protective effects of chromatin packaging. It was subsequently shown that depyrimidination occurs at a rate about 100 times more slowly than depurination [32]. Damaging chemicals, e.g. free radicals and alkylating agents, promote base release, mostly by generating base structures that destabilize the N-glycosyl linkage due to positively-charged leaving groups [32].
2.3. DNA polymerases and their crystal structures
DNA replication is performed by DNA polymerases. Mutations in DNA polymerases can directly lead to human diseases. Mutations in human Pol η result in one form of xeroderma pigmentosum type V (XP-V), mutations of human Pol β are associated with adenocarcinoma of the colon, and mutations of human Pol γ result in progressive external ophthalmoplegia [33].
DNA polymerases of several model organisms (including bacteriophage T7, Sulfolobus solfataricus, E. coli, and yeast) and humans have been studied extensively. Bacteriophage T7 has only one A-family DNA polymerase, gene 5 protein (gp5), which has high fidelity in DNA replication. The crenarcheon S. solfataricus has three B-family DNA polymerases (Dpo1, Dpo2, and Dpo3) and one Y-family DNA polymerase (Dpo4), used for translesion DNA synthesis. E. coli and some other prokaryotes have five DNA polymerases. A-family Pols I and II assist in replication (or repair). C-family Pol III is the major replicative polymerase involved in rapid and accurate DNA replication [34, 35]. The other two polymerases, Pol IV and Pol V, are Y-family members that bypass DNA damage and facilitate adaptive mutation [36]. Humans possess at least 19 enzymes involved in catalysis of DNA synthesis [37]: Pols α, δ, ε, and ζ are B-family members; mitochondrial Pol γ and Pols ν and θ belong to the A family; Pols β, λ, and μ are X-family members; Pols η, ι, and κ and REV1 belong to the Y-family; other enzymes include Pols σ1, σ2, φ, terminal deoxynucleotidyl transferase, and telomerase (apparently the only human reverse transcriptase). Yeasts have DNA polymerases similar but not identical to human.
X-ray crystal structures of a number of DNA polymerases are now available. All DNA polymerases consist of thumb, palm, and finger domains, holding DNA in a ‘right-hand’ mode. Only standard “Watson-Crick” (W-C) base pairs can fit into the tight active sites of high-fidelity DNA polymerases, such as A-family T7 DNA polymerase [38] (Fig. 3) and the DNA polymerases of Bacillus stearothermophilus and bacteriophage RB69 [38, 39]. It is accepted that Y-family DNA polymerases perform translesion DNA synthesis, and the structures of several Y-family DNA polymerases containing damaged DNA have been described in detail. Generally, the error-prone Y-family DNA polymerases have more open and flexible active sites and can accommodate bulky DNA damage [40, 41]. There are fewer specific contacts between the active site and a purine/pyrimidine base pair. Besides the above three domains, Y-family DNA polymerases also possess an additional “little finger” (or “PAD”) domain [40]. Subtle variations in the little finger domains of different Y-family members may be important for bypassing DNA damage. One typical Y-family polymerase is S. solfataricus Dpo4 (Fig. 3), which is comprised of four domains: the palm domain (containing the catalytic residues), the finger domain (playing a role in nucleotide selectivity), the thumb domain (making important contacts with DNA substrate), and the little finger domain (believed to play an important role in lesion bypass and polymerase processivity). Some Y-family DNA polymerases also have additional regions, such as the "N-digit" or "N-clasp" in Pol κ [42]. The N-clasp is comprised of two α-helical elements placed directly above the DNA substrate, allowing Pol κ to encircle DNA. Based on differences in structures, these polymerases bypass DNA damage in different ways. Compared with replicative DNA polymerases, the ample active sites allow Y-family DNA polymerases to accommodate bulky DNA damage, and the additional domains allow Y-family DNA polymerases to hold damaged DNA in some specific ways. These differences in structures result in diversity in translesion DNA synthesis.
Fig. 3.

Crystal structures of T7 DNA polymerase and Dpo4 containing DNA. (a) T7 DNA polymerase and (b) Dpo4 are shown in molecular surface representation with positive (blue) and negative (red) electrostatic potential. The DNA molecules are drawn in sticks. The minor groove adjacent to the replicating base pair is exposed to solvent for Dpo4 but completely buried for T7 DNA polymerase. These figures are from Ling et al. Cell107 (2001), 91-102 [40].Copyright © 2001 Cell Press. All rights reserved.
2.4. DNA damage leads to mutation in DNA replication
It is important to determine if and how DNA damage affects replication and leads to mutation when a polymerase encounters damage. Most DNA polymerases catalyze each single dNTP incorporation cycle according to a general mechanism [43] (Fig. 4). A DNA polymerase binds DNA to form a binary complex that selectively binds the correct dNTP based on Watson-Crick (W-C) (or other) base pairing to form a polymerase-DNA-dNTP ternary complex, then inducing a conformational change to facilitate the formation of the phosphodiester bond. After the chemical reaction (nucleotidyl transfer), pyrophosphate is released and the binary complex is relaxed to initiate a new cycle. The crystal structures of some ternary complexes have revealed nucleotide-induced conformational changes [38]. Efficient polymerization is dependent on the selection of the correct dNTP and a conformational change so that polymerase appropriately aligns the 3’-hydroxyl group at the end of the primer strand and the α-phosphate group of the incoming dNTP for phosphodiester bond formation [43]. Therefore, dNTP binding, conformational change, and the nucleotidyl transfer step are considered as three checkpoints to control the fidelity of a DNA polymerase. Single-molecule fluorescence resonance energy transfer (FRET) studies have shown that the closed conformation is observed only upon correct dNTP binding that stabilizes the polymerase-DNA-dNTP ternary complex, whereas incorrect dNTPs destabilize the complex [44].
Fig. 4.

The mechanism of single dNTP incorporation. E, DNA polymerase; Dn, DNA substrate; E*, conformationally changed DNA polymerase; Dn+1, DNA substrate extended by one base (product); PPi, pyrophosphate [43]. The “chemical step” is also termed phosphodiester bond formation or nucleotidyl transfer.
For normal DNA replication, only the correct dNTP is selected to make standard W-C base pairing, and it is rapidly incorporated while the wrong dNTPs are repelled outside the active site or incorporated very inefficiently. These check points maintain high fidelity in DNA replication, especially for A-family DNA polymerases, due to the tight contact between the active site and DNA. However, DNA adducts alter base structures [15] that may lead to deviation from the standard W-C base pairing and changes in binding affinity, conformational change, and/or nucleotidyl transfer. These changes may result in the attenuation of dNTP incorporation efficiency, blocking DNA replication during DNA incorporation. All of these changes may produce miscoding in replication, leading to disease. It is necessary to analyze the mechanisms of how DNA adducts lead to mutations.
3. DNA damage produces mutations in three major ways
It has been well-established that Y-family DNA pols are mainly responsible for TLS, and their crystal structures have therefore been discussed in greater detail in this review. Some recent reviews have described various types of DNA damage, individual DNA polymerases, and important information about how these polymerases bypass DNA damage [45-47]. Herein, based on our own work and recent progress in this area, we consider these results and propose that DNA damage leads to mutation during DNA replication through three major pathways: decreasing incorporation fidelity, blocking DNA replication, and producing frameshifts.
3.1. Mutation due to reduced replication fidelity
Replication fidelity is dependent on the efficiency of correct dNTP incorporation relative to incorrect incorporation. For normal DNA replication by most DNA polymerases, correct incorporation is much faster than incorrect. However, due to changes in DNA structure caused by adducts, the efficiency for correct incorporation may decrease and/or the incorrect incorporation efficiency may increase, leading to a reduction in replication fidelity.
3.1.1. Attenuation of correct incorporation efficiency
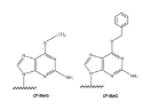
Many DNA adducts reduce only correct incorporation efficiency but do not affect incorrect incorporation. Two typical examples are some N2-alkylG and O6-alkylG adducts.
Our group has studied the bypass of a series of N2-alkylG adducts by S. solfataricus Y-family DNA polymerase Dpo4 [48]. Dpo4 preferentially incorporates dCTP opposite all of these adducts, but incorporation efficiencies (kcat/Km) were decreased 3- to 125-fold compared to unmodified G. The misincorporation efficiencies remained almost unchanged. Thus, the incorporation fidelities were increased 100-fold compared with unmodified G. In pre-steady-state kinetic analysis, the maximal rates of incorporation of dCTP opposite N2-alkylG adducts (kpol) were decreased 4- to 32-fold compared with unmodified G, but all the nucleoside triphosphate binding affinities (Kd,dCTP) were similar (varying from 8 to 29 μM). The conformational changes during dCTP incorporation have been further studied [43]. Compared with the smaller N2-MeG, bulkier alkyl groups at the N2 position of G strongly perturbed the fast conformation change, as evidenced by the disappearance of the fast fluorescence signal phase [43]. X-ray crystal structures of the complexes of Dpo4 and DNA containing N2-methylnaphthyl (Naph) G showed that the incoming dCTP was severely buckled and located outside the active site if the N2-NaphG residue is in the trans- form, indicating a non-productive complex; dCTP can also pair with the cis- form of the N2-NaphG residue via a Hoogsteen mode to continue DNA polymerization [48]. Only partial complexes were active, providing structural reasons for the decrease in incorporation efficiency.
Alkylating agents can produce N2,N2-dialkyl guanines, at least in model systems [49]. The efficiency (kcat/Km) of dCTP incorporation opposite N2,N2-Me2G was drastically decreased, 16,000-fold compared with G. The misincorporation frequencies were almost unchanged. Therefore, the misincorporation ratios were increased (to 0.36 to 2.3), causing random misincorporation opposite N2,N2-Me2G and completely eliminating the incorporation fidelity.
In order to discover the reasons for the decrease in incorporation efficiency, single-atom substituted purines were used as template bases to study single dNTP incorporation by Y-family Dpo4 or A-family T7 DNA polymerase. The decrease in efficiency cannot be simply attributed to steric influences of these substitutions that had often been considered as the sole reason, but should be attributed to both steric and electrostatic factors of these substituted groups, providing a new perspective in translesion DNA synthesis [50].
We also studied mutagenic O6-MeG and its bulkier analog O6-BzG using Dpo4. Compared with unmodified G, incorporation of dCTP opposite O6-MeG was inhibited by three orders of magnitude but the misincorporation efficiencies remained almost unchanged, decreasing the incorporation fidelity by three orders of magnitude compared with unmodified G. Finally, ~70% dCTP, 20% dTTP, and 10% dATP were incorporated opposite O6-MeG. Pre-steady-state catalytic efficiency (kpol/Kd.dCTP) was decreased about 14-fold for the insertion of dCTP opposite O6-MeG compared with G, due to a 6-fold decrease in rate (kpol) and a 2-fold increase in dissociation constant (Kd,dCTP) [51].
Bypass of the bulkier lesion O6-BzG was similar to O6-MeG except for a greater decrease in incorporation efficiency. The correct incorporation efficiency (kcat/Km) was strongly inhibited (5000-fold by O6-BzG compared with G) while the misincorporation efficiencies were reduced only 10-fold, decreasing the incorporation fidelity about three orders of magnitude compared with unmodified G. Pre-steady-state kinetic analysis showed that the value of kpol was decreased 6-fold and Kd,dNTP was increased 11-fold for the incorporation of dCTP opposite O6-BzG, relative to undamaged G [52]. In the crystal structures, both O6-MeG:CandO6-BzG:C structures showed a wobble base pair with two hydrogen bonds between O6-alkylG lesions and dCTP, shifting the base-pairing orientation from the standard W-C mode to the Wobble mode and thus decreasing the correct incorporation efficiency [51, 52].
3.1.2. Increased misincorporation efficiency
Some DNA adducts only partially affect the correct incorporation efficiency but drastically increase the incorrect incorporation efficiency, thus reducing incorporation fidelity. One typical example is 8-oxoG.
8-oxoG is a major lesion produced from oxidation stress in both lower organisms and eukaryotes. The mechanism of human DNA polymerase κ (hPolκ) bypassing 8-oxoG has been studied [53]. Based on steady-state kinetic analysis, the efficiency (kcat/Km) for incorporation of dCTP opposite 8-oxoG was decreased 80-fold compared with unmodified G. The efficiency for misincorporation of dATP opposite 8-oxoG, however, was increased 100-fold compared with unmodified G. Therefore, the incorporation fidelity opposite 8-oxoG was decreased 8,000-fold compared with unmodified G. hPolκ prefers to insert dATP rather than dCTP opposite 8-oxoG, leading to a conversion from G:C pairing to T:A pairing, i.e. transversion [53].
Pre-steady-state kinetic analysis yielded results similar to the steady-state results. The pre-steady-state catalytic efficiency (kpol/Kd,dCTP) for the insertion of dCTP opposite 8-oxoG was decreased 65-fold compared with G, due to a 25-fold decrease in the maximal rate (kpol) and a 2.5-foldincrease in the apparent nucleotide dissociation constant (Kd,dCTP). Unexpectedly, incorporation of dATP opposite 8-oxodG showed a fast burst, different from the lack of burst for the incorporation of dATP opposite G. The catalytic efficiency (kpol/Kd, dCTP) for insertion of dATP opposite 8-oxoG was increased up to 6.3 × 105M−1s−1, 20-fold higher than that for incorporation of dCTP opposite 8-oxoG [53]. Crystal structures of a complex of hPol κ and DNA containing 8-oxoG showed that the N-terminal extension of hPol κ stabilized its little finger domain that surrounds the Hoogsteen base pair of 8-oxoG and incoming dATP, explaining the increase in efficiency for the incorporation of dATP opposite 8-oxoG[53].
Bypass of the 8-oxoG lesion by S. solfataricus Dpo4 has also been studied. Steady-state kinetic analysis showed that the insertion of dCTP opposite G or 8-oxoG has similar catalytic efficiency, and the efficiency for incorporation of dATP opposite 8-oxoG was increased 250-fold compared with G, thus decreasing fidelity 200-fold opposite 8-oxoG relative to unmodified G [54]. Crystal structures of the ternary complex of Dpo4, 8-oxoG, and dCTP show that the Arg-332 residue in the little finger domain of Dpo4 can form a hydrogen bond with the oxygen atom at the C8 position of 8-oxoG and stabilizes the Watson-Crick base pair of incoming dCTP and 8-oxoG (Fig. 5). Notably, no stabilization was observed for W-C pairing of 8-oxoG and dCTP at the active site of hPolκ. Hoogsteen pairing of 8-oxoG:dATP is favorable relative to 8-oxoG:dCTP pair in hPolκ, leading to structurally different bypass of 8-oxoG by Dpo4 and hPol κ [55].
Fig. 5.

Hydrogen bonding interaction between Arg-332 and the oxygen atom at the C8 position of 8-oxodG was observed in an 8-oxodG:dCTP pair but not in a 8-oxodG:dATP pair in the Dpo4 active site. The protein is shown with schematic helices and strands. The DNA duplex and selected Dpo4 residues are shown in a stick mode. Ca2+ ions and water molecules are shown as yellow and red spheres, respectively, and hydrogen bonds as dashed lines. These figures are cited from Eoff et al., J. Biol. Chem. 282 (2007) 19831-19843 [55].
The catalytic core of yeast DNA polymerase η (residues 1-513) showed similar efficiency for incorporation of dCTP opposite G or 8-oxoG, but 23-fold higher efficiency in incorporation of dATP opposite 8-oxoG than that opposite G (Biochimie 2016 xue). Such a high priority in incorporation of dATP opposite 8-oxoG results in 57% correct dCTP incorporation and 43% misincorporation of dATP opposite 8-oxoG in full length extension as determined by LC-MS/MS.
3.2. Mutation due to replication blockage
In addition to reducing the fidelity of DNA incorporation, DNA damage can also block DNA replication. A blocking lesion may possibly lead to cell death. However, various translesion DNA polymerases are present in cells. If a translesion polymerase replaces the normal polymerase and misincorporates opposite the lesion, mutations are produced. Chemically, the essential point for blocking DNA replication is that the incoming dNTP cannot readily form the phosphodiester bond with the 3′-OH at the primer end due to misalignment of dNTP at the active site. There are at least three reasons for these misalignments: (i) the incoming dNTP is accommodated at the active site but paired with the template base via non-standard W-C pairing modes, thus misaligning dNTP configuration, e.g. in the case of N2,3-εG [21]; (ii) the incoming dNTP is repulsed outside of the active site and is far away from 3′-OH at the primer end, as in the case of N2,N2-diMeG [49]; (iii) the DNA adduct is too bulky to be accommodated and/or disordered in the active site, misaligning dNTP and 3′-OH, e.g. PAH-DNA adducts [56], CPD [57], DNA-protein cross-link adducts [58], and AAF [27]. All of these misalignments lead to the blockage of DNA replication.
3.2.1. Non-standard W-C paring modes
The exocyclic adduct N2,3-εG is produced by lipid peroxidation or from epoxide oxidation products of vinyl monomers [21]. Human DNA Pol ι and REV1 are blocked by N2,3-εG, yielding only 1-base incorporation opposite the adduct. dCTP incorporation is preferred over dTTP. In the crystal structure, Pol ι can accommodate an N2,3-εG:dCTP base pair rather well at the active site without significant conformational changes of protein or DNA, but the phosphate group of the incoming dCTP and 3′-OH at the primer end are misaligned. Two hydrogen bonds were observed in the N2,3-εG:dCTP base pair, whereas only one appears present in the N2,3-εG:dTTP pair. The crystal structures explain the slightly favored dCTP insertion compared with dTTP for hPol ι in steady-state kinetic analysis [21].
3.2.2. Repulsion of dNTP outside the active site
Excess alkylating agents can form N2,N2-dialkylG, at least in model systems, and even N2,N2-Me2G strongly blocks DNA replication by various DNA polymerases, including T7 DNA polymerase, HIV reverse transcriptase, Pol κ, Pol ι, Pol η, and Dpo4 [59-62]. For Dpo4, dCTP incorporation efficiency was decreased 160,000-fold compared with unmodified G, blocking DNA replication [49]. Pre-steady-state kinetic analysis showed almost no burst for the incorporation of dCTP opposite N2,N2-Me2G, indicating that only little (< 5%) of the polymerase-DNA-dCTP ternary complex is active. No obvious fast conformational change was observed, indicating that N2,N2-Me2G strongly perturbs the conformational change [43]. All of these kinetic analyses indicate strong blockage of DNA replication upon encountering this DNA damage. Analysis of the crystal structure showed that the 3′-terminal dideoxycytidine of the primer that should pair with the template N2,N2-Me2G is repulsed outside of the active site and folded back into the minor groove, as a catalytically incompetent complex, explaining the blockage of dNTP incorporation [49].
3.2.3. Bulkier adducts and disorder of the active site
PAHs are toxic environmental mutagens [63]. The reactive diastereoisomeric7,8-diol-9,10-epoxides of benzo[a]pyrene form N2-B[a]P guanine and N6-B[a]P adenine adducts, which strongly block dNTP incorporation. Among the four dNTPs, dATP is preferentially incorporated by T7 DNA polymerase, but the catalytic efficiency is decreased by at least four orders of magnitude. The lack of a burst for dNTP incorporation opposite all of these DNA adducts indicates that the rate-limiting step is at or before phosphodiester bond formation [63]. Dpo4 yielded the same results as T7 DNA polymerase in bypassing N6-B[a]P A, in that incorporation was blocked and dATP was preferably inserted by Dpo4 [56]. A crystal structure of the adduct residues in the Dpo4 active site showed an intercalation mode, in which reaction of the incoming dNTP with the 3′-OH at the primer end is inhibited because the distance us too long for an efficient chemical reaction (Fig. 6) [64].
Fig. 6.

Intercalation of N6-BPDE A adduct in the Dpo4 active site (BP-1), blocking dNTP incorporation. (A) The N6-BPDE-dA:dTTP ternary complex and the surrounding base pairs are shown as ball-and-stick models. (B) Looking down the DNA helical axis, two layers of base pairs and the BPDE adduct are shown. The figures are cited from Ling et al. Proc. Natl. Acad. Sci. U S A, 101 (2004) 2265-2269 [64].Copyright (2004) National Academy of Sciences, U.S.A
Nitropolycyclic aromatic hydrocarbons (NPAHs) lead to the formation of hydrophobic NPAH bulky lesions, blocking DNA replication by Dpo4, because the conformations of the lesions inhibit DNA translocation through the polymerase active site [28]. Mouse Pol κ could bypass N2-B[a]P G efficiently and accurately, but a mutant with reduced gap size (Pol κ gap mutant, PGM) was strongly blocked by this lesion, suggesting that the presence of this gap is essential for the DNA adduct bypass. From the crystal structure, the gap physically accommodates the bulky aromatic adduct and keeps the active site ordered, explaining crucial functions of the gap in Pol κ in maintenance of the active site for translesion DNA synthesis [65].
UV and ionizing radiation lead to the formation of CPDs by covalently cross-linking two adjacent pyrimidines. hPol κ can insert dATP opposite the 5′-T of a cis-syn T-T dimer but cannot insert nucleotides opposite the 3′-T of the dimer, leading to the blockage of DNA replication[57]. In a crystal structure, the active site of Pol κ was not spacious enough for two unpaired nucleotides but could only accommodate the incoming dATP that W-C pairs with the 5′-T of the T-T dimer, leaving the 3′-T misaligned in the active site. Met-135, in the active-site cleft, makes extensive van der Waals contacts with the 5′-T of the T-T dimer but appears to impede the binding of dNTP to the 3′-T of the dimer [57].
DNA-protein and DNA-DNA cross-links adducts are formed after exposure to α,β-unsaturated aldehydes or acrolein. Generally, these adducts block replicative polymerases, e.g. A-family DNA polymerases. Some TLS DNA polymerases, such as E. coli Pol IV and Pol V, are also incapable of bypassing these cross-links. Crystal structures of these complexes have not been reported yet, but the generally held opinion is that these adducts are too bulky to be accommodated by the active sites of DNA polymerases. However, hPolκ can efficiently bypass DNA-peptide cross-links if the peptide is N-substituted and linked to the N2 position of G [58], which can be considered an N2-alkylG adduct. The crystal structure of the complex containing N2-alkylG shows that hPol κ completely encircles the DNA duplex using its unique N-terminal extension. This kind of structure may be flexible enough in the hinge domain to allow hPolκ to pass by these kinds of cross-linked bulky lesions [42].
AAF-dG almost completely stalls DNA synthesis. Single-molecule fluorescence resonance energy transfer (smFRET) and protein-induced fluorescence enhancement (smPIFE) experiments show that E. coli DNA polymerase I (Klenow fragment) binds to this adduct in an ′orientation′, that is very unstable and then rapidly transfers DNA from the active site to a more stable exonuclease site, blocking DNA replication [44].
3.3. Frameshift mutations
DNA damage not only reduces polymerization fidelity (i.e. increased misincorporation ratio) and blocks DNA replication but can also lead to frameshifts during DNA replication. A frameshift is a serious mutation that adds or subtracts nucleotides in the newly synthesized DNA strand, disturbing the DNA sequence, genetic information, RNA transcription, and protein expression. A −1 frameshift means one nucleotide is missing after DNA replication. Four different mechanisms have been proposed to explain how −1 deletion frameshifts are produced (Fig. 12).
3.3.1. dNTP-stabilized misalignment
dNTP-stabilized misalignment, or a "Type II complex" mechanism (Fig. 7A), has been proposed to explain the formation of −1 frameshifts in some cases [40]. Dpo4 can produce −1 frameshifts, and we have found stacking interactions between bases at the template incorporation position; the nature of the incoming dNTP is crucial for the formation of −1 frameshifts. Few frameshifts (≤2%) are produced with thymine or cytosine at the incorporation position, 9-12% with adenine or guanine, and 25-50% with larger planar cyclic DNA adducts [66], e.g. 1,N2-εG [67], which has the strongest stacking interaction with the incoming dNTP. In crystal structures, purines and larger planar cyclic DNA adducts at the incorporation position can stack with the incoming dNTP that has paired with the next template base, forming a Type II complex. As judged by kinetic analysis, the conformational change of Dpo4 upon forming the Type II complex is very fast, followed by a slow step for the formation of phosphodiester bond [66]. Therefore, in this mechanism, the nucleotidyl transfer step is rate-limiting for frameshift formation.
Fig. 7.

Four mechanisms proposed for −1 deletion frameshifts.
3.3.2. Template-slippage
The template-slippage mechanism (Fig. 7B) was proposed for the replication of repetitive DNA sequences. The primer misaligns on a repetitive template DNA strand, forming an unpaired template base in the newly synthesized DNA that is one base shorter than the original template [68]. One typical example is Y-family DNA polymerase Dbh, which generates −1 deletion frameshifts in repetitive sequences with error frequencies up to 50% [68]. In the crystal structure of Dbh [68], a cleft between the polymerase domain and the C-terminal domain provides ample space to accommodate extrahelical template bases. Two residues, Tyr-249 and Arg-333 in the C-terminal domain, stabilize the extrahelical base at the −3 position of template, and residues in the flexible loop of Dbh interact with the bulged base at the −2 position of template. Therefore, Dbh does not appear to strictly regulate the entry of template bases into the active site, allowing template misalignment to occur more readily.
3.3.3. Misincorporation-misalignment
In the misincorporation-misalignment mechanism (Fig. 7C), a dNTP is firstly misincorporated and then the template misaligns so that the next template base can pair with this dNTP correctly [69]. One example is E. coli Klenow fragment (DNA polymerase I) in the catalysis of DNA replication on M13 double-stranded DNA containing a 361-nucleotide single-stranded DNA gap, using an imbalanced dNTP pool [70, 71]. Frameshifts (−1) are preferentially formed when the template sequence has a 5′-neighbor nucleotide that is complementary to the dNTP provided in excess. When a dNTP complementary to the 5′ next nucleotide in the template is first misincorporated opposite a template nucleotide, this misincorporated nucleotide then misaligns and correctly pairs with the next complementary template nucleotide, forming a correct terminal base pair and −1 frameshift. If the mispaired nucleotide at the end of primer is complementary to the 5′- next template nucleotide, the in vivo frequency of −1 frameshift deletion was 58-fold higher than if the nucleotide at the primer end was non-complementary to the 5′-template nucleotide, reflecting the misincorporation-misalignment mechanism[70, 71].
3.3.4. Loop-out
Another mechanism to generate a −1 frameshift is a loop-out mechanism (Fig. 7D) [72]. One typical example is the bypass of an abasic site by S. solfataricus Dpo4. The abasic site in the template cannot base stack with an incoming dNTP and then loops out from the duplex; the incoming dNTP pairs with the next template base [73]. In crystal structures, the abasic lesion that loops out can be accommodated in the cavity between the fingers and little finger domains [73]. The catalytic core of yeast DNA polymerase η (residues 1-513) also showed −1 frameshift during bypass of abasic site (Yang MR-2015).
The four frameshift mechanisms were proposed for normal and damaged DNA. Additionally, frameshifts can also be produced by other approaches. For example, planar molecules (e.g. acridine or proflavin) insert into favorable DNA sequences and produce frameshifts [74]. These chemicals are physically intercalated between base pairs in duplex DNA but not covalently bound to DNA. This kind of frameshift is out of the scope of the article and not discussed in this review.
4. Bypassing DNA damage by the DNA replisome
We have discussed how DNA damage leads to mutation during DNA replication performed by a DNA polymerase. Actual DNA replication includes both leading- and lagging-strand DNA synthesis in a coordinated mode. This process is not performed by a single DNA polymerase but by the complex DNA replisome, a protein complex containing DNA polymerase(s), helicase, primase, single-stranded DNA binding protein, and other accessory proteins. Of course, DNA damage will affect the DNA replisome during both leading- and lagging-strand DNA synthesis, increasing the frequency of gene mutation and genomic instability.
4.1. DNA replisomes
DNA replisomes are protein assemblies that function as molecular motors to mediate assembly of proteins at the replication fork, unwinding of DNA, template-directed polymerization of nucleotides, and synthesis of RNA primers. A major role of the replisome is to coordinate DNA polymerization mediated by its protein constituents, so that leading- and lagging-strand DNA synthesis proceed at the same rates. The replisome is a dynamic structure, involving the release and recruitment of proteins as the replisome proceeds [75]. The complexity of the DNA replisome results from protein interactions and various functions of accessory proteins within the complex.
DNA replication machinery constantly encounters DNA lesions under normal growth conditions. Cox et al. [76] have estimated that 10-50% of all replication forks may be subject to collapse in one generation of a single cell. TLS DNA synthesis by single DNA polymerases may not reflect the actual situation in vivo. Therefore, studies of TLS DNA synthesis using DNA replisomes are needed.
The DNA replisomes of E. coli and bacteriophages T7 and T4 have been constructed in vitro. A mammalian DNA replisome is considered too complicated to be constructed at this time, and only simple replication complexes have been reported in vitro. DNA replisomes have shown different patterns of DNA synthesis and damage bypass from systems using a single DNA polymerase. The differences mainly result from two major reasons: protein-protein interactions and accessory proteins in DNA replisome that facilitate DNA polymerase bypass of damage, as described below.
4.2. Protein interactions in the T7 DNA replisome facilitate nick bypass by DNA polymerase
The T7 DNA replisome consists of gene 5 DNA polymerase (gp5), the E. coli processivity factor thioredoxin (trx), gene 4 helicase-primase (gp4), and gene 2.5 single-stranded DNA binding protein (gp2.5) [77] (Fig. 8). Gp5 and trx form a high-affinity complex (gp5/trx) to increase the processivity of nucleotide polymerization [78]. The helicase at the C-terminal domain of gp4 assembles as a hexamer and unwinds double-stranded DNA to yield two single-stranded DNA templates for leading- and lagging-strand DNA synthesis. Gp2.5 coats single-stranded DNA to remove secondary structures and also physically interacts with gp5/trx, interactions essential for coordination of leading- and lagging-strand DNA synthesis [79].
Fig. 8.

Model of the bacteriophage T7 replisome.
Single phosphodiester bond interruptions (nicks) are the most frequent DNA roadblock that a replisome encounters. Nicks can be introduced by endonucleases, recombination, or the presence of two adjacent Okazaki fragments[80]. In addition, the repair of DNA damage (e.g., the excision repair of thymine dimers) also produces nicks. Either T7 helicase or DNA polymerase alone terminates its movement upon encountering a nick in duplex DNA. However, the helicase-polymerase complex can bypass this nick [81]. In double-stranded DNA unwinding, the helicase has contacts with both DNA strands [82]. However, a nick does not provide these contacts and the helicase itself cannot unwind a nick. When the helicase is associated with the DNA polymerase, strong protein interactions [77] allow the helicase to bind to the template rather than to dissociate, encircling helicase onto the 5′-end single-stranded DNA of a nick, thus bypassing the nick. Addition of single-stranded DNA binding protein gp2.5 to the complex further facilitates nick bypass by ~ 2-fold [81].
Another important finding is that gp4 and gp5/trx cannot initiate strand-displacement DNA synthesis from a nick in duplex DNA without gp2.5 [83]. gp2.5 binds to the displaced single-stranded DNA, and the acidic C-terminal tail of gp2.5 also interacts with gp5. The helicase replaces gp2.5 and catalyzes strand-displacement DNA synthesis with gp5/trx at this nick. Due to the presence of gp2.5, the gp5/trx-gp4 complex can efficiently initiate strand-displacement DNA synthesis from a nick. Therefore, protein interactions within the T7 DNA replisome may alter the ability of the DNA polymerase to bypass DNA damage and may produce completely different results from those obtained from isolated T7 DNA polymerase (or the trx complex).
4.3. Accessory proteins in the DNA replisome facilitate polymerase bypass of DNA damage
The T7 DNA replisome contains only four proteins but the replisomes of E. coli and bacteriophage T4 contains thirteen and eight proteins, respectively, most of which are accessory proteins. These accessory proteins have roles in protein interaction in the bacteriophage T7 system. In mammals, including humans, the DNA replisome is far more complex. These accessory proteins in the replisome assist DNA polymerase(s) in bypass of DNA damage in some specific pathways.
4.3.1. Bypass of DNA damage by the E. coli replisome
The replisome of E. coli comprises DnaB helicase, DnaG primase, single-stranded DNA binding protein, DNA polymerase III holoenzyme, and multiple accessory proteins (Fig. 9). The Pol III holoenzyme consists of a three-subunit catalytic core (α-polymerase, ε-exonuclease, and θ), a homodimeric β processivity clamp, and a δδ’τ2γχϕ-clamp-loader complex [12]. The β clamp consists of ring-shaped dimeric proteins that encircle DNA and bind to the Pol III core, thereby giving the DNA polymerase high processivity. The clamp loader complex opens and loads the β clamp onto a primed site. Each τ subunit binds to the DnaB helicase and one Pol III core polymerase, coupling the helicase and the polymerase [12].
Fig. 9.

The DNA replisome of E. coli. This figure is cited from Hamdan et al., Annu. Rev. Biochem. 78 (2009) 205-243 [12]. Copyright © 2009, Annual Reviews.
The fate of the E. coli DNA replisome was examined after leading-strand DNA synthesis was blocked by the addition of ddTTP, which selectively blocks leading-strand DNA synthesis [84]. The blocked leading-strand DNA polymerase remained stably bound to the helicase at the replication fork. The helicase continued to unwind DNA for ~ 1 kb ahead of the blocked leading-strand DNA polymerase. The lagging-strand DNA polymerase was still connected to the stalled leading-strand polymerase and remained active in converting the lagging single-stranded DNA to duplex DNA [84].
When the lagging-strand DNA polymerase was blocked by a DNA lesion on the lagging strand, the lesion did not affect the activities of helicase or leading-strand polymerase [85]. The leading- and lagging-stand DNA polymerases remained physically coupled but functionally uncoupled. The leading-strand polymerase continued unabatedly, allowing the replication fork to continue. This action caused a large loop of single-stranded DNA to accumulate on the lagging-strand template. This loop grew until the supply of single-stranded DNA binding protein was depleted. At that point, the naked single-stranded DNA triggered the release of the stalled lagging-strand polymerase from the blocked site and resumed the synthesis of a new Okazaki fragment on a newly primed site[85].
When the E. coli DNA replisome encounters a CPD DNA lesion at leading-strand template, fork progression is only transiently blocked [86]. Leading-strand DNA synthesis is then reinitiated downstream of the damage, which is dependent on the assistance of primase DnaG in the DNA replisome but not on any other known replication-restart proteins. The results show that the E. coli DNA replisome can tolerate leading-strand template lesions and synthesize the leading-strand template discontinuously [86]. However, the single E. coli DNA polymerase alone cannot bypass this DNA damage. Another approach to tolerate CPD involves polymerases transiently dissociating upon encountering this lesion and allowing repair enzymes or translesion polymerases to repair or bypass this lesion. The helicase-primase complex remains bound to the template DNA and serves to maintain the integrity of the replication fork, directing the reassembled replisome to the correct location [87].
4.3.2. Bypass of DNA damage by the bacteriophage T4 replisome
The T4 DNA replisome contains helicase, DNA polymerase, single-stranded DNA binding protein, a trimetric clamp processivity factor, a pentameric clamp-loader complex, and six monomers of the primase [12] (Fig. 10). The T4 DNA polymerase has both polymerase and exonuclease activities. The clamp-loading complex consists of four molecules of the gene 44 protein and one molecule of the gene 62 protein [12].
Fig. 10.

The bacteriophage T4 DNA replisome. This figure is cited from Hamdan et al., Annu. Rev. Biochem. 78 (2009) 205-243 [12].Copyright © 2009, Annual Reviews.
Bypass of a noncoding abasic site lesion by the T4 DNA replisome has been investigated in both leading- or lagging-strand templates[88]. The lesion in the lagging-strand template blocked the lagging-strand DNA polymerase but did not block the helicase, primase, or leading-strand polymerase. When the primase synthesized another RNA primer and the clamp was loaded, the stalled lagging-strand polymerase recycled from the DNA lesion and initiated the synthesis of a new Okazaki fragment. Therefore, this lesion does not affect the progression of the replication fork, with only a single-stranded DNA gap left behind [88]. In contrast, when a blocking lesion was located in the leading-strand template, the leading-strand polymerase was blocked but the bound helicase continued to travel, causing the leading-strand template to loop out. The leading-strand DNA template was then rapidly coated with single-stranded DNA binding protein. The replication fork traveled approximately 1 kb beyond the DNA lesion before the replication fork completely collapsed. The primase and lagging-strand polymerase remained active during this period, where Okazaki fragments were synthesized beyond the leading-strand lesion [88].
The mechanism of restarting the replication fork blocked by damaged DNA has also been investigated using single-molecule magnetic tweezers [89]. The T4 DNA holoenzyme, in cooperation with UvsW helicase, can overcome the presence of a leading-strand lesion by periodical formation and migration of a four-way Holliday junction. The initiation of the repair process required partial disassembly of the replisome through the departure of the replicative helicase [89]. With the assistance of other accessory proteins, T4 DNA holoenzyme could bypass this leading-strand lesion.
5. Conclusions and perspectives
DNA damage is produced from various environmental mutagens through different chemical pathways. DNA damage leads to mutations through three major ways: reducing incorporation fidelity, blocking DNA replication, and forming frameshifts. Notably, in vivo DNA replication is performed by DNA replisomes and not single DNA polymerases, and the actions of the E. coli, T4, and T7 DNA replisomes and a partial human DNA replication complex have been investigated with regard to DNA damage. Due to protein-protein interactions or the functions of some other accessory proteins, DNA replisomes can tolerate some DNA damage, producing results different from those observed using single DNA polymerases. However, research in this area is still very limited. More details and functions of protein interactions or accessory proteins in DNA replication and TLS DNA synthesis should be further investigated. The use of DNA replisomes and not only single DNA polymerases will be very important in advancing our understanding of TLS DNA synthesis in vivo. Additionally, in this review, we mainly focused on the roles of DNA damage in gene mutation for some model bacteria and phages but not in chromosomal mutation of mammalian cells.
Following are some perspectives on future research directions in this area:
(1) Recent studies have discovered that protein interactions and components in DNA replisomes play important roles in DNA damage bypass. Variant proteins that lack protein interactions should be used to further study the functions of protein interactions in DNA damage bypass. Each component in an DNA replisome should be further studied to clarify its function in DNA damage bypass.
(2) DNA replication is very fast, and dNTP incorporation rates can be as high as 300 nucleotides/s. Bypass of DNA damage by the T4 DNA replisome is also too fast to be detected by many normal biochemical methods..Fast kinetic analysis methods (e.g., rapid-quench or stopped-flow instruments) should be used here to study how DNA polymerases and DNA replisomes handle DNA damage. These methods give more information on a millisecond time scale.
(3) General overall reactions give average results, e.g. an average rate of 1012-1015molecules. However, DNA replication is very complicated and the overall reaction cannot give information on bypass of DNA damage by a single replisome. Single molecule techniques (based on force or fluorescence) should be used to investigate single molecule mechanisms of a DNA replisome bypass of DNA damage, giving real information for a single DNA replication event.
(4) Until now, only the E. coli, T4, and T7 DNA replisomes have been reconstructed in vitro, providing opportunities to study bypass of DNA damage by DNA replisomes. a major aim is to determine how DNA damage affects DNA replication in a human system. However, construction of a mammalian DNA replisome in vitro is far more complicated. One possible way is to use extracts of human cells that contain the proteins involved in DNA replication, to study how DNA damage affects human DNA replication. Another way is to use the DNA replisomes of certain other organisms, e.g. S. solfataricus, whose replisome is expected to be similar to a eukaryotic system and is easier to construct in vitro. Such a DNA replisome could provide more important information for human systems, compared with the E. coli, T4, and T7 systems.
(5) In addition to the DNA polymerases and replisomes, other proteins, e.g. E. coli replicative helicases DnaB, DNA repair helicase UvrA2B protein complex, and replicative DNA helicases of the simian virus 40 are also involved in DNA damage bypass. More proteins should be explored. The mutations induced by DNA damage due to the action of other proteins (that do not belong to the replisome) should be further considered. .
Acknowledgements
We acknowledge financial support by the Natural Science Foundation of China (NSFC Nos. 31370793 and 814220410) and U. S. National Institutes of Health grants R01 ES010375 and R01 ES010546.
Footnotes
Conflict of interest
The authors declare that no conflict of interest exists regarding the manuscript.
References
- [1].Yamagiwa K, Ichikawa K. Experimentelle studie über die pathogenese der epithelialgeschwülste. Mitt. Med. Fak. Tokio. 1915;15:295–344. [Google Scholar]
- [2].Cook JW, Hewett CL, Hieger I. The isolation of a cancer-producing hydrocarbon from coal tar. Parts I, II, and III. J. Chem. Soc. 1933:395–405. [Google Scholar]
- [3].Bechtel DH. Molecular dosimetry of hepatic aflatoxin B1-DNA adducts: linear correlation with hepatic cancer risk. Regul. Toxicol. Pharmacol. 1989;10:74–81. doi: 10.1016/0273-2300(89)90014-7. [DOI] [PubMed] [Google Scholar]
- [4].Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hot spot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- [5].Vahakangas KH, Samet JM, Metcalf RA, Welsh JA, Bennett WP, Lane DP, Harris CC. Mutations of p53 and ras genes in radon-associated lung cancer from uranium miners. Lancet. 1992;339:576–580. doi: 10.1016/0140-6736(92)90866-2. [DOI] [PubMed] [Google Scholar]
- [6].Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in p53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- [7].Miller JA. Carcinogenesis by chemicals: an overview. Cancer Res. 1970;30:559–576. [PubMed] [Google Scholar]
- [8].Hemminki K, Dipple A, Shuker DEG, Kadlubar FF, Segerbäck D, Bartsch H. DNA Adducts: Identification and biological significance. IARC Publ. 1994;135:1–478. [Google Scholar]
- [9].Kadlubar FF, Hammons GJ. The role of cytochrome P450 in the metabolism of chemical carcinogens. Curr. Top. Med. Chem. 1987;2:81–130. [Google Scholar]
- [10].Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- [11].Hamdan SM, Richardson CC. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009;78:205–243. doi: 10.1146/annurev.biochem.78.072407.103248. [DOI] [PubMed] [Google Scholar]
- [12].Furukohri A, Goodman MF, Maki H. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J. Biol. Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Villani G, Le Gac NT. Interactions of DNA helicases with damaged DNA: Possible biological consequences. J. Biol. Chem. 2000;275:33185–33188. doi: 10.1074/jbc.R000011200. [DOI] [PubMed] [Google Scholar]
- [14].Yardimci H, Wang XD, Loveland AB, Tappin I, Rudner DZ, Hurwitz J, van Oijen AM, Walter JC. Bypass of a protein barrier by a replicative DNA helicase. Nature. 2012;492:205–211. doi: 10.1038/nature11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guengerich FP. Interactions of carcinogen-bound DNA with individual DNA polymerases. Chem. Rev. 2006;106:420–452. doi: 10.1021/cr0404693. [DOI] [PubMed] [Google Scholar]
- [16].Yasui M, Matsui S, Ihara M, Laxmi YR, Shibutani S, Matsuda T. Translesional synthesis on a DNA template containing N2-methyl-2'-deoxyguanosine catalyzed by the Klenow fragment of Escherichia coli DNA polymerase I. Nucleic Acids Res. 2001;29:1994–2001. doi: 10.1093/nar/29.9.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng TF, Hu XP, Gnatt A, Brooks PJ. Differential blocking effects of the acetaldehyde-derived DNA lesion N2-ethyl-2'-deoxyguanosine on transcription by multisubunit and single subunit RNA polymerases. J. Biol. Chem. 2008;283:27820–27828. doi: 10.1074/jbc.M804086200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Georgiadis P, Samoli E, Kaila S, Katsouyanni K, Kyrtopoulos SA. Ubiquitous presence of O6-methylguanine in human peripheral and cord blood DNA. Cancer Epidem. Biomar. 2000;9:299–305. [PubMed] [Google Scholar]
- [19].Margison GP, Koref MFS, Povey AC. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis. 2002;17:483–487. doi: 10.1093/mutage/17.6.483. [DOI] [PubMed] [Google Scholar]
- [20].Cheng SC, Hilton BD, Roman JM, Dipple A. DNA adducts from carcinogenic and noncarcinogenic enantiomers of benzo[a]pyrene dihydrodiol epoxide. Chem. Res. Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- [21].Zhao L, Pence MG, Christov PP, Wawrzak Z, Choi JY, Rizzo CJ, Egli M, Guengerich FP. Basis of miscoding of the DNA adduct N2,3-ethenoguanine by human Y-family DNA polymerases. J. Biol. Chem. 2012;287:35516–35526. doi: 10.1074/jbc.M112.403253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lawley P, Brookes P. Interstrand cross-linking of DNA by difunctional alkylating agents. J. Mol. Biol. 1967;25:143–160. doi: 10.1016/0022-2836(67)90285-9. [DOI] [PubMed] [Google Scholar]
- [23].Michaelson-Richie ED, Loeber RL, Codreanu SG, Ming X, Liebler DC, Campbell C, Tretyakova NY. DNA-protein cross-linking by 1,2,3,4-diepoxybutane. J. Proteome Res. 2010;9:4356–4367. doi: 10.1021/pr1000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jelitto B, Vangala R, Laib R. Biological monitoring of exposure and the response at the subcellular level to toxic substances. Arch. Toxicol. 1989:246–249. [Google Scholar]
- [25].Degan P, Shigenaga MK, Park EM, Alperin PE, Ames BN. Immunoaffinity isolation of urinary 8-hydroxy-2-deoxyguanosine and 8-hydroxyguanine and quantitation of 8-hydroxy-2'-deoxyguanosine in DNA by polyclonal antibodies. Carcinogenesis. 1991;12:865–871. doi: 10.1093/carcin/12.5.865. [DOI] [PubMed] [Google Scholar]
- [26].Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2'-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vrtis KB, Markiewicz RP, Romano LJ, Rueda D. Carcinogenic adducts induce distinct DNA polymerase binding orientations. Nucleic Acids Res. 2013;41:7843–7853. doi: 10.1093/nar/gkt554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kirouac KN, Basu AK, Ling H. Structural mechanism of replication stalling on a bulky amino-polycyclic aromatic hydrocarbon DNA adduct by a Y family DNA polymerase. J. Mol. Biol. 2013;425:4167–4176. doi: 10.1016/j.jmb.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].O'Brien T, Mandel HG, Pritchard DE, Patierno SR. Critical role of chromium (Cr)-DNA interactions in the formation of Cr-induced polymerase arresting lesions. Biochemistry. 2002;41:12529–12537. doi: 10.1021/bi020452j. [DOI] [PubMed] [Google Scholar]
- [30].Cadet J, Voituriez L, Hruska FE, Kan L-S, Leeuw F.A.d., Altona C. Characterization of thymidine ultraviolet photoproducts. Cyclobutane dimers and 5, 6-dihydrothymidines. Can. J. Chem. 1985;63:2861–2868. [Google Scholar]
- [31].Ravanat J-L, Douki T, Cadet J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. 2001;63:88–102. doi: 10.1016/s1011-1344(01)00206-8. [DOI] [PubMed] [Google Scholar]
- [32].Wilson DM, III, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res/DNA Repair. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- [33].Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kelman Z, O'Donnell M. DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- [35].McHenry CS. DNA polymerase III holoenzyme of Escherichia coli. Annu. Rev. Biochem. 1988;57:519–550. doi: 10.1146/annurev.bi.57.070188.002511. [DOI] [PubMed] [Google Scholar]
- [36].Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- [37].Loeb LA, Monnat RJ. DNA polymerases and human disease. Nat. Rev. Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- [38].Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 angstrom resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- [39].Wang J, Sattar AA, Wang C, Karam J, Konigsberg W, Steitz T. Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- [40].Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- [41].Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- [42].Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase kapa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- [43].Zhang H, Guengerich FP. Effect of N2-guanyl modifications on early steps in catalysis of polymerization by Sulfolobus solfataricus P2 DNA polymerase Dpo4 T239W. J. Mol. Biol. 2010;395:1007–1018. doi: 10.1016/j.jmb.2009.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brenlla A, Markiewicz RP, Rueda D, Romano LJ. Nucleotide selection by the Y-family DNA polymerase Dpo4 involves template translocation and misalignment. Nucleic Acids. Res. 2013;42:2555–2563. doi: 10.1093/nar/gkt1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pata JD. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta, Proteins Proteomics. 2010;1804:1124–1135. doi: 10.1016/j.bbapap.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sharma S, Helchowski CM, Canman CE. The roles of DNA polymerase zeta and the Y family DNA polymerases in promoting or preventing genome instability. Mutat. Res. 2013;743-744:97–110. doi: 10.1016/j.mrfmmm.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee MY, Zhang S, Lin SH, Chea J, Wang X, LeRoy C, Wong A, Zhang Z, Lee EY. Regulation of human DNA polymerase delta in the cellular responses to DNA damage. Environ. Mol. Mutagen. 2012;53:683–698. doi: 10.1002/em.21743. [DOI] [PubMed] [Google Scholar]
- [48].Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Versatility of Y-family Sulfolobus solfataricus DNA polymerase Dpo4 in translesion synthesis past bulky N2-alkylguanine adducts. J. Biol. Chem. 2009;284:3563–3576. doi: 10.1074/jbc.M807778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang H, Eoff RL, Kozekov ID, Rizzo CJ, Egli M, Guengerich FP. Structure-function relationships in miscoding by Sulfolobus solfataricus DNA polymerase Dpo4 guanine N2,N2-dimethyl substiton produces inactive and miscoding polymerase complexes. J. Biol. Chem. 2009;284:17687–17699. doi: 10.1074/jbc.M109014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang H, Bren U, Kozekov ID, Rizzo CJ, Stec DF, Guengerich FP. Steric and electrostatic effects at the C2 atom substituent influence replication and miscoding of the DNA deamination product deoxyxanthosine and analogs by DNA polymerases. J. Mol. Biol. 2009;392:251–269. doi: 10.1016/j.jmb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Eoff RL, Irimia A, Egli M, Guengerich FP. Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “Wobble” pairing between O6-methylguanine and cytosine but accurate bypass is preferred. J. Biol. Chem. 2007;282:1456–1467. doi: 10.1074/jbc.M609661200. [DOI] [PubMed] [Google Scholar]
- [52].Eoff RL, Angel KC, Egli M, Guengerich FP. Molecular basis of selectivity of nucleoside triphosphate incorporation opposite O6-benzylguanine by Sulfolobus solfataricus DNA polymerase: Steady-state and pre-steady-state kinetics and x-ray crystallography of correct and incorrect pairing. J. Biol. Chem. 2007;282:13573–13584. doi: 10.1074/jbc.M700656200. [DOI] [PubMed] [Google Scholar]
- [53].Irimia A, Eoff RL, Guengerich FP, Egli M. Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase κopposite the 7,8-dihydro-8-oxo-2'-deoxyguanosine adduct. J. Biol. Chem. 2009;284:22467–22480. doi: 10.1074/jbc.M109.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zang H, Chowdhury G, Angel KC, Harris TM, Guengerich FP. Translesion synthesis across polycyclic aromatic hydrocarbon diol epoxide adducts of deoxyadenosine by Sulfolobus solfataricus DNA polymerase Dpo4. Chem. Res. Toxicol. 2006;19:859–867. doi: 10.1021/tx060056s. [DOI] [PubMed] [Google Scholar]
- [55].Eoff RL, Irimia A, Angel KC, Egli M, Guengerich P. Hydrogen bonding of 7,8-dihydro-8-oxodeoxyguanosine with a charged residue in the little finger domain determines miscoding events in Sulfolobus solfataricus DNA polymerase Dpo4. J. Biol. Chem. 2007;282:19831–19843. doi: 10.1074/jbc.M702290200. [DOI] [PubMed] [Google Scholar]
- [56].Zang H, Chowdhury G, Angel KC, Harris TM, Guengerich FP. Translesion synthesis across polycyclic aromatic hydrocarbon diol epoxide adducts of deoxyadenosine by Sulfolobus solfataricus DNA polymerase Dpo4. Chem Res Toxicol. 2006;19:859–867. doi: 10.1021/tx060056s. [DOI] [PubMed] [Google Scholar]
- [57].Vasquez-Del Carpio R, Silverstein TD, Lone S, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Role of human DNA polymerase κ in extension opposite from a cis-syn thymine dimer. J. Mol. Biol. 2011;408:252–261. doi: 10.1016/j.jmb.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Minko IG, Yamanaka K, Kozekov ID, Kozekova A, Indiani C, O'Donnell ME, Jiang Q, Goodman MF, Rizzo CJ, Lloyd RS. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008;21:1983–1990. doi: 10.1021/tx800174a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Choi J-Y, Guengerich FP. Kinetic evidence for efficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase i. J. Biol. Chem. 2006;281:12315–12324. doi: 10.1074/jbc.M600112200. [DOI] [PubMed] [Google Scholar]
- [60].Choi J-Y, Angel KC, Guengerich FP. Translesion synthesis across bulky N2-alkylguanine DNA adducts by human DNA polymerase i. J. Biol. Chem. 2006;281:21062–21072. doi: 10.1074/jbc.M602246200. [DOI] [PubMed] [Google Scholar]
- [61].Choi J-Y, Guengerich FP. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J. Mol. Biol. 2005;352:72–90. doi: 10.1016/j.jmb.2005.06.079. [DOI] [PubMed] [Google Scholar]
- [62].Choi J-Y, Guengerich FP. Analysis of the effect of bulk at N2-alkylguanine DNA adducts on catalytic efficiency and fidelity of the processive DNA polymerase T7 exonuclease− and HIV-1 reverse transcriptase. J. Biol. Chem. 2004;279:19217–19229. doi: 10.1074/jbc.M313759200. [DOI] [PubMed] [Google Scholar]
- [63].Zang H, Harris TM, Guengerich FP. Kinetics of nucleotide incorporation opposite polycyclic aromatic hydrocarbon- DNA adducts by processive bacteriophage T7 DNA polymerase. Chem. Res. Toxicol. 2005;18:389–400. doi: 10.1021/tx049683c. [DOI] [PubMed] [Google Scholar]
- [64].Ling H, Sayer JM, Plosky BS, Yagi H, Boudsocq F, Woodgate R, Jerina DM, Yang W. Crystal structure of a benzo[a]pyrene diol epoxide adduct in a ternary complex with a DNA polymerase. Proc. Natl. Acad. Sci.U S A. 2004;101:2265–2269. doi: 10.1073/pnas.0308332100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liu Y, Yang Y, Tang TS, Zhang H, Wang Z, Friedberg E, Yang W, Guo C. Variants of mouse DNA polymerase reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. Proc. Natl. Acad. Sci.U S A. 2014;111:1789–1794. doi: 10.1073/pnas.1324168111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang H, Beckman JW, Guengerich FP. Frameshift deletion by Sulfolobus solfataricus P2 DNA polymerase Dpo4 T239W is selective for purines and involves normal conformational change followed by slow phosphodiester bond formation. J. Biol. Chem. 2009;284:35144–35153. doi: 10.1074/jbc.M109.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4 - Analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J. Biol. Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- [68].Wilson RC, Pata JD. Structural insights into the generation of single-base deletions by the Y family DNA polymerase dbh. Mol. Cell. 2008;29:767–779. doi: 10.1016/j.molcel.2008.01.014. [DOI] [PubMed] [Google Scholar]
- [69].Kunkel TA, Alexander PS. The base substitution fidelity of eucaryotic DNA polymerases: mispairing frequencies, site preferences, insertion preferences, and base substitution by dislocation. J. Biol. Chem. 1986;261:160–166. [PubMed] [Google Scholar]
- [70].Bebenek K, Kunkel TA. Framshift errors initiated by nucleotide misincorporation. Proc. Natl. Acad. Sci. U S A. 1990;87:4946–4950. doi: 10.1073/pnas.87.13.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kunkel TA, Soni A. Mutagenesis by transient mislignment. J. Biol. Chem. 1988;263:14784–14789. [PubMed] [Google Scholar]
- [72].Fiala KA, Hypes CD, Suo Z. Mechanism of abasic lesion bypass catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 2007;282:8188–8198. doi: 10.1074/jbc.M610718200. [DOI] [PubMed] [Google Scholar]
- [73].Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol. Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- [74].Ripley LS, Dubins JS, deBoer JG, DeMarini DM, Bogerd AM, Kreuzer KN. Hotspot sites for acridine-induced frameshift mutations in bacteriophage T4 correspond to sites of action of the T4 type II topoisomerasen. J. Mol. Bio. 1988;200:665–680. doi: 10.1016/0022-2836(88)90479-2. [DOI] [PubMed] [Google Scholar]
- [75].Zhang H, Lee SJ, Richardson CC. Essential protein interactions within the replisome regulate DNA replication. Cell Cycle. 2011;10:3413–3414. doi: 10.4161/cc.10.20.17523. [DOI] [PubMed] [Google Scholar]
- [76].Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- [77].Zhang H, Lee SJ, Zhu B, Tran NQ, Tabor S, Richardson CC. Helicase-DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. U S A. 2011;108:9372–9377. doi: 10.1073/pnas.1106678108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tabor S, Huber HE, Richardson CC. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 1987;262:16212–16223. [PubMed] [Google Scholar]
- [79].Marintcheva B, Hamdan SM, Lee SJ, Richardson CC. Essential residues in the C terminus of the bacteriophage T7 gene 2.5 single-stranded DNA-binding protein. J. Biol. Chem. 2006;281:25831–25840. doi: 10.1074/jbc.M604601200. [DOI] [PubMed] [Google Scholar]
- [80].Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- [81].Zhu B, Lee SJ, Richardson CC. Bypass of a nick by the replisome of bacteriophage t7. J. Biol. Chem. 2011;286:28488–28497. doi: 10.1074/jbc.M111.252023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kaplan DL. The 3′-tail of a forked-duplex sterically determines whether one or two DNA strands pass through the central channel of a replication-fork helicase. J. Mol. Biol. 2000;301:285–299. doi: 10.1006/jmbi.2000.3965. [DOI] [PubMed] [Google Scholar]
- [83].Ghosh S, Marintcheva B, Takahashi M, Richardson CC. C-terminal phenylalanine of bacteriophage T7 single-stranded DNA-binding protein is essential for strand displacement synthesis by T7 DNA polymerase at a nick in DNA. J. Biol. Chem. 2009;284:30339–30349. doi: 10.1074/jbc.M109.024059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].McInerney P, O'Donnell M. Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J. Biol. Chem. 2007;282:25903–25916. doi: 10.1074/jbc.M703777200. [DOI] [PubMed] [Google Scholar]
- [85].McInerney P, O'Donnell M. Functional uncoupling of twin polymerases - Mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 2004;279:21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- [86].Yeeles JTP, Marians KJ. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011;334:235–238. doi: 10.1126/science.1209111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jeiranian HA, Schalow BJ, Courcelle CT, Courcelle J. Fate of the replisome following arrest by UV-induced DNA damage in Escherichia coli. Proc. Natl. Acad. Sci. U S A. 2013;110:11421–11426. doi: 10.1073/pnas.1300624110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nelson SW, Benkovic SJ. Response of the bacteriophage T4 replisome to noncoding lesions and regression of a stalled replication fork. J. Mol. Bio. 2010;401:743–756. doi: 10.1016/j.jmb.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338:1217–1220. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]