

Abstract
Background
Ankyrin repeat and SOCS box protein 3 (ASB3) is a member of ASB family and contains ankyrin repeat sequence and SOCS box domain. Previous studies indicated that it mediates the ubiquitination and degradation of tumor necrosis factor receptor 2 and is likely involved in inflammatory responses. However, its effects on oncogenesis are unclear. This study aimed to investigate the effects of ASB3 on the growth and metastasis of colorectal cancer (CRC).
Methods
We used next-generation sequencing or Sanger sequencing to detect ASB3 mutations in CRC specimens or cell lines, and used real-time quantitative polymerase chain reaction, Western blotting, and immunohistochemical or immunofluorescence assay to determine gene expression. We evaluated cell proliferation by MTT and colony formation assays, tested cell cycle distribution by flow cytometry, and assessed cell migration and invasion by transwell and wound healing assays. We also performed nude mouse experiments to evaluate tumorigenicity and hepatic metastasis potential of tumor cells.
Results
We found that ASB3 gene was frequently mutated (5.3%) and down-regulated (70.4%) in CRC cases. Knockdown of endogenous ASB3 expression promoted CRC cell proliferation, migration, and invasion in vitro and facilitated tumorigenicity and hepatic metastasis in vivo. Conversely, the ectopic overexpression of wild-type ASB3, but not that of ASB3 mutants that occurred in clinical CRC tissues, inhibited tumor growth and metastasis. Further analysis showed that ASB3 inhibited CRC metastasis likely by retarding epithelial-mesenchymal transition, which was characterized by the up-regulation of β-catenin and E-cadherin and the down-regulation of transcription factor 8, N-cadherin, and vimentin.
Conclusion
ASB3 dysfunction resulted from gene mutations or down-regulated expression frequently exists in CRC and likely plays a key role in the pathogenesis and progression of CRC.
Keywords: Ankyrin repeat and SOCS box protein 3 (ASB3), Colorectal cancer, Epithelial-mesenchymal transition, Cell proliferation, Tumor metastasis
Background
In developed countries, colorectal cancer (CRC) is the third most common cancer in men and the second most common cancer in women [1, 2]. Although the incidence and mortality of CRC are lower in developing countries, including China, than in developed countries, they are rapidly rising with increasing economic development [1, 3]. Mounting evidences have confirmed that a series of gene mutations and epigenetic changes are involved in CRC tumorigenesis and progression [4–11]. Ubiquitination is a fundamental post-translational modification, and the ubiquitin–proteasome system plays an important role in regulating cell proliferation, apoptosis, angiogenesis, and motility. Additionally, abnormal regulations of the ubiquitin–proteasome system are known to promote colorectal carcinogenesis by regulating p53, Smad4, and components of the K-ras and Wnt/β-catenin pathways [12].
The ankyrin repeat and suppressor of cytokine signaling (SOCS) box (ASB) family contains 18 proteins, which interact with Cul5-Rbx2 to form a functional E3 ubiquitin ligase [13]. ASB proteins likely function as the substrate-recognition subunits of ElonginBC–Cullin–SOCS box (ECS)-type Cullin-Ring E3 ubiquitin ligase complexes that specifically transfer ubiquitin to cellular proteins for degradation by the proteasome [14]. ASB proteins, containing a SOCS box, are involved in the negative regulation of cytokine signaling [15]. Reportedly, ASB4 inhibits JNK activity and blocks insulin signal transduction by binding and inducing the ubiquitination and degradation of insulin receptor substrate 4 [16] and G-protein pathway suppressor 1 [17], as well as confers migration and invasion properties in hepatocellular carcinoma cells [18]. Furthermore, it was found that ASB9 expressed higher in CRC tissue than in corresponding normal tissue; that knockdown of ASB9 promoted the invasion of CRC cells; and that patients who expressed low levels of ASB9 had a lower overall survival rate than those who expressed high levels of ASB9 [19].
Human ASB3 gene, another member of ASB gene family, is located on chromosome 2p16.2. It has three transcript variants that encode two isoforms. Isoform A of ASB3 contains 518 amino acid residues [20], which form 11 coterminous ankyrin (ANK) repeats followed by a SOCS box domain in the C terminal of the peptide [NCBI (The National Center for Biotechnology Information) Reference Sequence: NP_057199.1]. It has been reported that ASB3 mediates ubiquitination and degradation of tumor necrosis factor receptor 2, which plays a crucial role in several inflammatory responses [21]. In this study, we detected the mutations and expression of ASB3 gene in CRC tissues and cells, and investigated the role of ASB3 in the pathogenesis of CRC.
Methods
Tissue samples
Paraffin-embedded and fresh frozen CRC specimens were collected from patients treated at Sun Yat-sen University Cancer Center, Guangzhou, China. All specimens contained matched cancer tissues (percentage of tumor cells ≥70%) and corresponding normal mucosal tissues (>5 cm laterally from the edge of the cancerous region). The study protocol was approved by the Institutional Review Board and the Human Ethics Committee of Sun Yat-sen University Cancer Center, and informed consent was obtained from each patient.
Cell lines and cell culture
Human normal colon epithelium cell line FHC; human CRC cell lines HT-29, COLO205, LoVo, HCT116, SW620, SW480, and DLD-1; and the human embryonic kidney cell line 293T were obtained from the American Type Culture Collection. Human CRC cell line THC8307 was kindly provided by Prof. Rui-Hua Xu at Sun Yat-sen University Cancer Center [22]. The FHC cell line was cultured in Dulbecco’s Modified Eagle Medium (DMEM)/nutrient mixture F-12 media containing 100 ng/mL hydrocortisone, 10 ng/mL cholera toxin, 5 μg/mL insulin, and 5 μg/mL transferrin supplemented with 10% fetal bovine serum (FBS). COLO205 was cultured in RPMI-1640 medium supplemented with 10% FBS. All other cells were cultured in DMEM supplemented with 10% FBS. All materials for cell culture were from Invitrogen/ThermoFisher Scientific (Carlsbad, CA, USA).
ASB3 exonic sequence analysis
Genomic DNA was extracted from fresh frozen samples or cells using a Gentra Puregene Tissue Kit (Qiagen, Hilden, Germany). The exonic sequence was analyzed by next-generation sequencing at the Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China. Sequencing files were deposited in the European Genome-phenome Archive under accession number EGAS00001001088. The exon sequence of the ASB3 gene was analyzed by Sanger sequencing at Invitrogen Trading (Shanghai) Co. Ltd (Shanghai, China).
Small interfering RNAs and transient transfection
ASB3 small interfering RNAs (siRNAs) and negative control siRNA (sequences are shown in Table 1) were synthesized by Shanghai GenePharma Co. Ltd. (Shanghai, China). For transient transfection, THC8307 (2 × 105/well) or SW620 cells (4 × 105/well) were seeded in 6-well plates for 24 h and then transfected with siRNAs (100 pmol/well) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The cells were cultured for 24 h in standard media and then used for further analysis at the indicated time points.
Table 1.
The sequences of small interfering RNAs (siRNAs) involved in this study
| Target | Name | Direction | Sequences |
|---|---|---|---|
| ASB3 | siRNA1 | Sense | 5′-r(GGACUUGUUAAUACCACUU)dTdT-3′ |
| Antisense | 5′-r(AAGUGGUAUUAACAAGUCC)dTdT-3′ | ||
| siRNA2 | Sense | 5′-r(GCAUUGACACCCUUAUCUU)dTdT-3′ | |
| Antisense | 5′-r(AAGAUAAGGGUGUCAAUGC)dTdT-3′ | ||
| Negative control | siNC | Sense | 5′-r(GCGACGAUCUGCCUAAGAU)dTdT-3′ |
| Antisense | 5′-r(UAUGCUCAAGCUUUCUAGC)dTdT-3′ |
Retroviral expression vector construction, packaging, and stable cell line construction
To generate retroviral expression vectors, the fragments of human wild type (WT) ASB3 and the artificial loss-of-function ASB3 mutant ΔSOCS [21] were amplified by polymerase chain reaction (PCR) from cDNA of THC8307 cells with specific primers (Table 2) and cloned into Xho I and Not I (or Cla I) sites of pLNCX2 plasmid (Clontech, Mountain View, CA, USA). ASB3 mutant-expressing vectors were generated using the GENEART site-directed mutagenesis system (Invitrogen) based on WT ASB3-expressing vector with specific primers (Table 2). All constructs were verified by DNA sequencing analysis. The retroviruses were then packaged and tittered following the manufacturer’s instructions.
Table 2.
The sequences of primers used in this study
| Target | Direction | Sequences |
|---|---|---|
| For qPCR | ||
| ASB3 | Forward | 5′-CATACTTATTTCATCGGGTGC-3′ |
| Reverse | 5′-GGTAACTGCCAACTGTCCTC-3′ | |
| GAPDH | Forward | 5′-CTCCTCCTGTTCGACAGTCAGC-3′ |
| Reverse | 5′-CCCAATACGACCAAATCCGTT-3′ | |
| For vector construction (italics indicates restriction enzyme recognition sequence) | ||
| WT ASB3 | Forward | 5′-GCGCTCGAGATGGATTTTACAGAGGCT-3′ |
| Reverse | 5′-GGCGCGGCCGCTTATCCATCTTGAATAGCTG-3′ | |
| ΔSOCS | Forward | 5′-GCGCTCGAGATGGATTTTACAGAGGCT-3′ |
| Reverse | 5′-GGGATCGATTTACCTTTCAACAGCTGGTG-3′ | |
| For point mutation generation (capital letter indicates mutated nucleotide) | ||
| G135E | Forward | 5′- ggctgttgcttcaacacgAagcaaatgttaatggatc-3′ |
| Reverse | 5′-gatccattaacatttgctTcgtgttgaagcaacagcc-3′ | |
| K339I | Forward | 5′-gtgaacattcttttgaTatatggagcccagata-3′ |
| Reverse | 5′-tatctgggctccatatAtcaaaagaatgttcac-3′ | |
| R362C | Forward | 5′-cgagaagttttcgatatttTgctactttttgaggaaagg-3′ |
| Reverse | 5′-cctttcctcaaaaagtagcAaaatatcgaaaacttctcg-3′ | |
To construct stable ASB3-overexpressing cells, HCT116 or DLD-1 cells that endogenously expresses the mutated ASB3 at a low level were infected with each retrovirus with 8 μg/mL of polybrene (Sigma-Aldrich, Milwaukee, WI, USA) and then were selected with G418 (Calbiochem, La Jolla, CA, USA) for 2–3 weeks.
Real-time quantitative PCR assay
Real-time quantitative PCR (qPCR) analysis was conducted as described previously [23]. Briefly, total RNA was extracted with Trizol Reagent (Invitrogen), reversely transcribed into cDNA with M-MLV reverse transcriptase (Promega, Madison, WI, USA), and sequentially subjected to qPCR analysis with the SYBR Green PCR Kit (Invitrogen) using primers shown in Table 2. The threshold cycle (Ct) values were determined and normalized against that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal control. The relative mRNA levels were shown as the value of 2−ΔCt against the control group [24].
Western blotting analysis
Western blotting was performed as described previously [24, 25]. Briefly, cell pellets were lysed in RIPA lysis buffer (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) followed by centrifugation to remove insoluble materials. Protein were then separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane. Blots were probed with specific primary antibody for ASB3 (1:250, Novus Biologicals, Littleton, CO, USA); E-cadherin, N-cadherin, vimentin, transcription factor 8 (TCF8), β-catenin, and zonula occludens-1 (ZO-1) (1:1000; Cell Signaling Technology, Inc., Beverly, MA, USA); and GAPDH (1:2000; Santa Cruz Biotechnology, Inc.), followed by reaction with horseradish peroxidase-conjugated secondary antibody. Signals were visualized using the enhanced chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ, USA).
Immunohistochemical assay
Protein levels of ASB3 were detected by immunohistochemical (IHC) assay with a peroxidase kit (DAKO, Carpinteria, CA, USA) as described previously [26, 27]. Briefly, after routine deparaffinization, rehydration, and blocking with 0.3% H2O2 and antigen retrieval, the slides were incubated overnight at 4 °C with rabbit anti-ASB3 antibody (1:400, NBP1-88,812; Novus Biologicals), followed by incubation with HRP-conjugated secondary antibody and visualized with the EnVision Detection Kit (DAKO). Then, the sections were counterstained with hematoxylin. ASB3 staining intensity (I0, negative; I1, weak; I2, moderate; and I3, strong) (representative images shown in Fig. 1a) and the percentage of corresponding positive area (P1-P3) were evaluated by two pathologists who were blinded to clinical parameters. The ASB3 protein levels were presented as H score: H score = I1 × P1 + I2 × P2 + I3 × P3 [23, 28, 29].
Fig. 1.
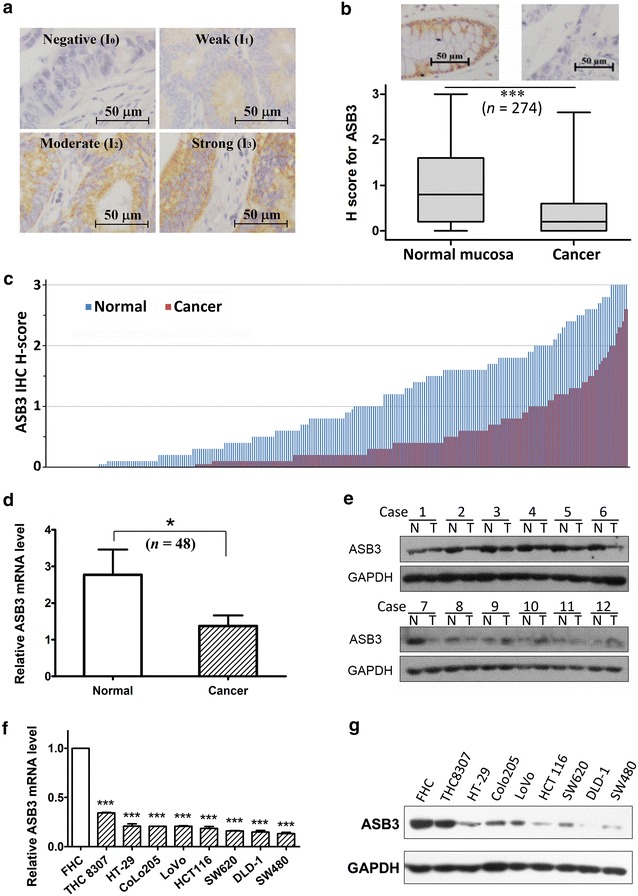
Ankyrin repeat and suppressor of cytokine signaling (SOCS) box protein 3 (ASB3) is generally down-regulated in colorectal cancer (CRC) tissues and cell lines. ASB3 expression was detected by immunohistochemical (IHC), real-time quantitative polymerase chain reaction (qPCR), and Western blotting assays in CRC tissues (a, b, c, d and e) and cell lines (f and g). a Representative images of IHC staining for ASB3 in CRC tissues. ASB3 protein expression levels were divided into 4 levels: I0, negative; I1, weak staining; I2, moderate staining; I3, strong staining. The scale bar represents 50μm. b ASB3 protein levels (H score) determined by IHC assay in 274 pairs of CRC and corresponding normal mucosal tissues (***P < 0.001). c IHC H-scores ordered from low to high respectively. d The relative mRNA levels of ASB3 in 48 pairs of CRC and corresponding normal mucosal tissues (normalized against that of glyceraldehyde-3-phosphate dehydrogenase [GAPDH] internal control; *P = 0.016). e ASB3 protein levels detected by Western blotting assay in 12 pairs of CRC tissues (T) and corresponding normal mucosal tissues (N). GAPDH was used as a loading control. f ASB3 mRNA levels detected by qPCR in normal intestinal epithelial cell line FHC and 8 CRC cell lines (***P < 0.001 compared with FHC cells). g ASB3 protein levels detected by Western blotting assay in cell lines; GAPDH was used as a loading control
Cell proliferation assay
Cell proliferation was analyzed using MTT and colony formation assays as described previously [23, 25, 30]. For the MTT assay, cells were seeded in 96-well plates with a density of 2000 cells/well and incubated for indicated times. Cells were stained with MTT, and then absorbance was determined at 490 nm. For the colony formation assay, cells were seeded in 6-well plates at 500 cells/well and maintained in standard media for 14 days. Colonies were fixed with 4% paraformaldehyde and stained with crystal violet, and the ones containing more than 50 cells were counted.
Cell cycle analysis
Cells were collected by centrifugation (1000 rpm × 10 min) after trypsinization, washed with ice-cold PBS twice, and fixed in 70% ethanol at 4 °C overnight. Cell suspensions were washed and re-suspended in PBS, treated with RNase A, and stained with propidium iodide. Finally, flow cytometry was performed to analyze cell cycle distribution [31].
Wound healing assays
After serum deprivation for 12 h, confluent monolayers were scratched using a 10-μL pipette tip and washed once with serum-free medium to create a cell-free gap. Then, cells were incubated in DMEM medium containing 10% FBS. Wound healing in the same field was monitored and photographed under a microscope every 6 h from 0 to 36 h post-scratch [32, 33]. Using the Image-J software, images were then analyzed and calculated [34] to determine the rate of cell migration.
Migration and invasion assays
Transwell assays were used to measure the migration or invasion ability of cells. In 200 μL of serum-free medium, 1 × 105 cells were seeded into a Boyden chamber without or with Matrigel (8-μm pore; BD Falcon, San Jose, CA, USA) for migration or invasion assay, respectively; then the chambers were put in 24-well plates with 600 μL of medium containing 10% FBS. After 16–24 h of incubation, cells on the underside of the polycarbonate membrane were fixed in ethanol and stained with crystal violet; the numbers of migratory or invasive cells were determined from seven independent microscopic fields (200×).
Immunofluorescence assays
Immunofluorescence assay was performed to detect the expression of epithelial-mesenchymal transition (EMT) markers. Cells cultured in glass-bottom cell culture dishes (NEST Biotechnology Co., Ltd., Wuxi, Jiangsu, China) were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 4% bovine serum albumin. Then, they were incubated with primary antibodies (Cell Signaling Technology, Inc.) for E-cadherin (1:100), vimentin (1:200), β-catenin (1:200), or N-cadherin (1:100) overnight at 4 °C, followed by incubation with Alexa fluor-594-conjugated or Alexa fluor-488-conjugated secondary antibody (Invitrogen). The samples were co-stained with DAPI and imaged by confocal laser scanning microscopy (Olympus FV1000; Olympus, Tokyo, Japan) [27].
In vivo tumorigenicity and hepatic metastasis assays
Female BABL/c nude mice (4–5 weeks old) were purchased from the Experimental Animal Center of Guangdong Province, Guangzhou, China. All animal studies were performed following the United States National Institutes of Health (NIH) animal use guidelines and the current Chinese regulations and standards on the use of laboratory animals. All animal procedures were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee.
For the tumorigenicity assay, the mice were injected subcutaneously with 1 × 106 HCT116 cells overexpressing WT ASB3, ASB3 mutants [ΔSOCS, G135E, K339I, or G135E/K339I (a natural mutant containing G135E and K339I)], or vector-transfected control cells in 100 μL PBS (eight mice per group). Xenograft formation was monitored, tumor size was measured every 4 days, and tumor volume (V) was calculated according to the following formula: V = 0.52 × width2 × length [31, 35, 36]. Four weeks after implantation, the mice were euthanized, and the tumors were removed, photographed, and weighed.
For the hepatic metastasis assay, mice were anesthetized and subjected to laparotomy. One million HCT116 cells overexpressing WT ASB3 or ASB3 mutants or control cells in 20 μL PBS were respectively injected into the distal tip of the spleen (8 mice per group). Six weeks after incubation, the mice were euthanized and the spleens and livers were removed for pathologic examination [26, 27].
Statistical analysis
All in vitro experiments were performed at least three times; all in vivo experiments were performed twice. Statistical analysis was conducted using the SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA). Differences were analyzed by one-way analysis of variance (ANOVA) or exact χ2 test. Metering data are presented as mean ± standard deviation (SD). P values < 0.05 were considered statistically significant.
Results
ASB3 gene was frequently mutated in CRC tissues and cell lines
Using next-generation sequencing (for 50 cases) and Sanger sequencing assays (for 83 new cases and 3 cases with ASB3 mutations found in next-generation sequencing assays), we analyzed the ASB3 gene mutations in tumor tissues collected from 133 CRC patients treated at Sun Yat-sen University Cancer Center, and found that ASB3 gene had a high frequency of somatic mutations (5.3%, 7/133). We also performed Sanger sequencing to analyze ASB3 gene exons in the normal intestinal epithelial cell line FHC and in CRC cell lines HCT116, HT-29, SW480, SW620, DLD-1, THC8307, CoLo205, and LoVo. We found that the ASB3 gene mutated in HCT116, HT-29, and DLD-1 cells but not in other cells. The mutation types included missense, nonsense, and splice site mutations; of these, only missense mutations K339I and G135E occurred in Chinese CRC patients (Table 3).
Table 3.
ASB3 gene mutations in colorectal cancer tissues and cell lines
| Mutation genotype | Mutation type | Exon | Codon change | Amino acid change | Case ID/Cell line |
|---|---|---|---|---|---|
| Colorectal cancer tissues | |||||
| CDS 21C→Ga | Nonsense | Exon 2 | TAC→TAG b | Y7stop | C56 |
| chr2:53810098 G→C | Splice-site | Exon 3/Intron | – | – | C53 |
| CDS 404G→R | Missense | Exon 4 | GGA→GAA | G135E | S78, S64 |
| CDS 1084C→Y | Missense | Exon 8 | CGC→TGC | R362C | S35 |
| CDS 1016A→W | Missense | Exon 8 | AAA→ATA | K339I | S12, S61, S64 |
| Colorectal cancer cell lines | |||||
| CDS 471A→T | Missense | Exon 5 | GAA→GAT | E157D | HCT116 |
| CDS 471A→W | Missense | Exon 5 | GAA→GAT | E157D | HT-29 |
| CDS 24G→R | Synonymous | Exon 2 | GCG→GCA | Non | DLD-1 |
| CDS 25G→K | Missense | Exon 2 | GAC→TAC | D9Y | DLD-1 |
| CDS 154T→Y | Missense | Exon 2 | TAT→CAT | Y52H | DLD-1 |
| CDS 922G→R | Missensee | Exon 7 | GCC→ACC | A308T | DLD-1 |
ASB3, ankyrin repeat and suppressor of cytokine signaling (SOCS) box protein 3
The nucleotides in bold indicate the one mutated
a CDS coding domain sequence; A, T, C, G, four types of nucleotides; R = A/G; Y = C/T; W = A/T; K = G/T
bamino acid codon
ASB3 was down-regulated in most CRC tissues and cell lines
We detected the expression of ASB3 by using IHC, qPCR and Western blotting assays. IHC assay for 274 CRC samples showed that average ASB3 protein levels were significantly lower in tumor tissues than in paratumor mucosa (P < 0.001) and that 70.4% (193/274) of CRC cases exhibited a down-regulated expression in tumor tissues (Fig. 1b, c). qPCR and Western blotting assays showed that ASB3 mRNA levels were significantly lower in CRC tissues than in paired normal colorectal mucous epitheliums (n = 48, P = 0.016; Fig. 1d) and that ASB3 protein levels were lower in 75.0% (9/12) of tumor tissues than in paired normal epithelial tissues (Fig. 1e). The examination of ASB3 expression in CRC cell lines revealed similar results: ASB3 expression was decreased at both mRNA and protein levels in CRC cells compared with FHC cells (Fig. 1f, g). These data suggest that ASB3 expression is down-regulated in CRC.
ASB3 inhibited the proliferation of CRC cells in vitro
Since the ASB3 gene is frequently mutated or down-regulated in CRC, we assumed that dysfunction of ASB3 likely plays a role in the pathogenesis and progression of the disease. To confirm this hypothesis, first, we analyzed the effect of ASB3 on the proliferation of CRC cells in vitro. We performed ASB3 knockdown in THC8307 and SW620 CRC cells that are with relatively high levels of WT ASB3 (Table 3; Figs. 1f, g, 2a) and then analyzed cell proliferation by MTT and colony formation assays. We found that the knockdown of ASB3 significantly promoted CRC cell proliferation (Fig. 2b, c) and colony formation (Fig. 2d).
Fig. 2.
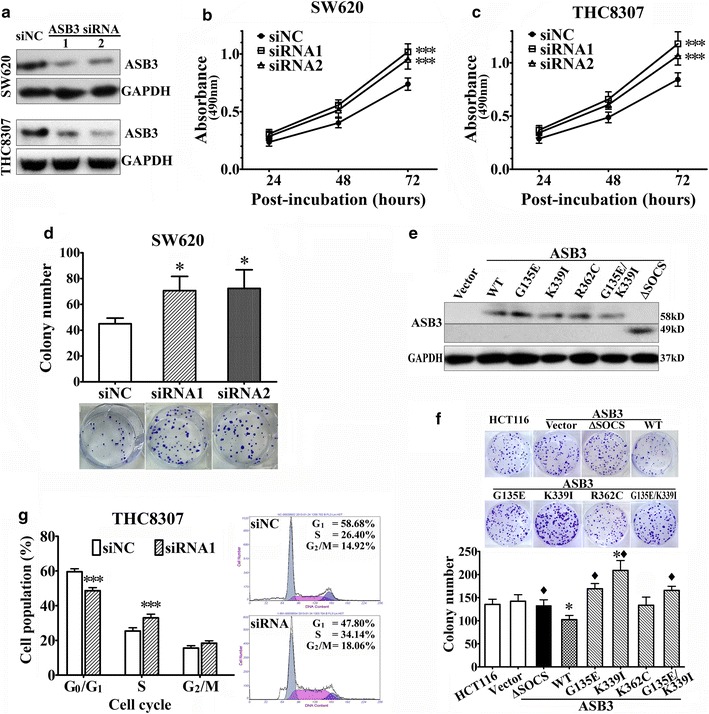
ASB3 inhibits the proliferation of CRC cells in vitro. ASB3 protein levels, cell proliferation, or cell cycle distributions were analyzed in CRC cells using Western blotting assay (a and e), MTT assay (b and c), colony formation assay (d and f), or flow cytometry (g) after ASB3 knockdown by transiently transfected with small interfering RNAs (siRNAs) or after ASB3 overexpression by stably transfected with ASB3 cDNA or its mutants. siNC, negative control siRNA; WT, wild type; G135E, K339I, R362C, and ΔSOCS are ASB3 mutants. Representative pictures are also included in d, f, and g. *P < 0.05 compared with those transfected with siNC or vector control; ♦ P < 0.05 compared with that transfected with WT ASB3; ***P < 0.001 compared with that transfected with siNC
We performed colony formation assays in HCT116 CRC cells with a mutated ASB3 gene which was expressed at a lower level (Fig. 1f, g; Table 3). We stably transfected ASB3 cDNA or its mutants G135E, K339I, or R362C that were detected in clinical CRC tissues (Table 3) or ΔSOCS into HCT116 cells. The data showed that ectopic overexpression of WT ASB3 inhibited HCT116 cell colony formation (Fig. 2e, f). However, overexpressing ASB3 mutants, including G135E, K339I, R362C, or ΔSOCS, did not inhibit HCT116 cell colony formation. On the contrary, K339I seemingly promoted colony formation (Fig. 2f). The inhibitory effects of these mutants (except R362C) on HCT116 cell colony formation were significantly different from that of WT ASB3 (Fig. 2f). This indicates that these mutants likely lose the inhibitory effect of WT ASB3 on CRC cell proliferation.
Next, we analyzed cell cycle distributions in THC8307 cells after ASB3 knockdown and found that the down-regulation of ASB3 expression promoted THC8307 cells from G1 into S phase of the cell cycle. In THC8307 cells transfected with ASB3 siRNAs, the percentage of cells at G1 phase was significantly lower, and the percentage of cells at S phase was higher than those in control cells (P < 0.001, Fig. 2g). This indicates that ASB3 inhibits CRC cell proliferation as a tumor suppressor and that dysfunctions of ASB3 resulting from mutations or down-regulation are among the possible events that lead to CRC pathogenesis or progression.
ASB3 inhibited the migration and invasion of CRC cells in vitro
After artificially regulating ASB3 expression in CRC cells as described above, we investigated the effects of ASB3 on cell migration and invasion in vitro. We found that ASB3 knockdown facilitated wound healing, transwell migration, and invasion in THC8307 and SW620 CRC cells (Fig. 3a–f), whereas the overexpression of WT ASB3 suppressed cell migration and invasion in HCT116 CRC cells (Fig. 3g–j). However, ASB3 mutants G135E, K339I, or G135E/K339I did not hinder but actually promoted HCT116 cell migration. Mutant ΔSOCS had no effect on cell migration, whereas mutant R362C acted similarly to WT ASB3 (Fig. 3g, h). The effects of ASB3 mutants G135E, K339I, G135E/K339I, and ΔSOCS (except R362C) on HCT116 cell migration were markedly different from that of WT ASB3 (Fig. 3h). Similar results were obtained from the invasion assay (Fig. 3i, j).
Fig. 3.
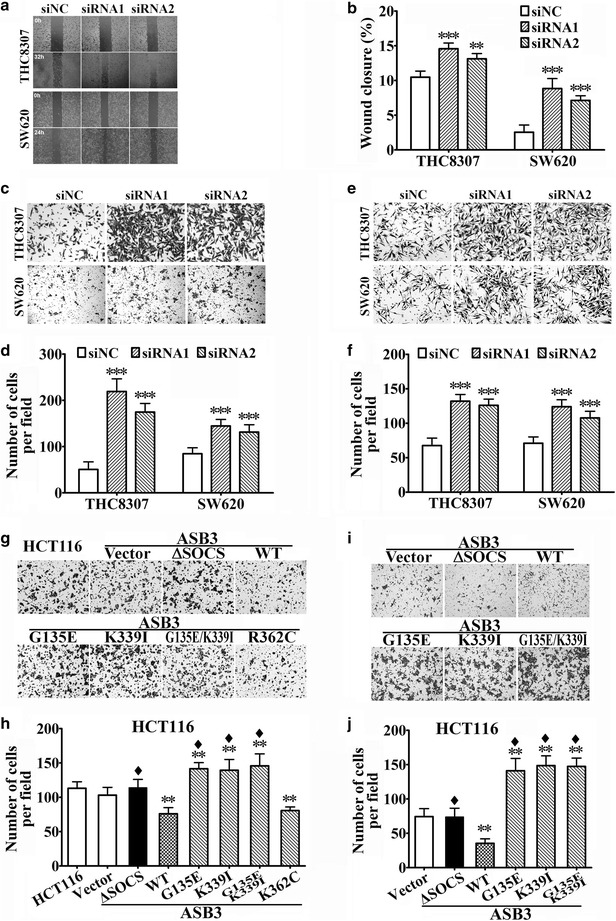
ASB3 inhibits the migration and invasion of CRC cells in vitro. The migration and invasion abilities of CRC cells were detected by wound healing and transwell assays after ASB3 knockdown by transiently transfected with siRNAs or overexpression by stably transfected with ASB3 cDNA or its mutants. siNC negative control siRNA; WT wild type; G135E, K339I, R362C, and ΔSOCS are ASB3 mutants. a and b Wound healing assay, c and d transwell migration assay, and e and f invasion assay in THC8307 and SW620 cells after ASB3 knockdown. g and h transwell migration and i and j invasion assays in HCT116 cells after ASB3 (WT or mutants) overexpression. **P < 0.01, ***P < 0.001 compared with siNC or vector control; ♦ P < 0.01 compared with that transfected with WT ASB3
ASB3 inhibited the tumorigenicity and hepatic metastasis of CRC cells in nude mice
To further elucidate the effects of ASB3 on the oncogenesis and progression of CRC, HCT116 cells stably overexpressing ASB3 cDNA or its mutants (1 × 106 cells each mouse) were subcutaneously or intrasplenically injected into nude mice, and the tumorigenicity and hepatic metastasis were investigated. We found that, in subcutaneous implant model, the tumor formation rates of subcutaneous xenografts of HCT116 cells overexpressing WT ASB3, ASB3 mutants G135E, K339I, G135E/K339I, and ΔSOCS were 37.5% (3/8), 75.0% (6/8), 87.5% (7/8), 87.5% (7/8), and 75.0% (6/8), respectively, whereas that of vector-transfected control cells was 75.0% (6/8) (Fig. 4a). The tumorigenicity of xenografts derived from HCT116 cells overexpressing WT ASB3 was lower, the volume of the tumors was smaller, and the weight of the tumors was lighter than those of xenografts from cells overexpressing ASB3 mutants or control cells (all P values < 0.05, Fig. 4b, c). In intrasplenic injection models, we found that overexpressing WT ASB3 in HCT116 cells suppressed intrasplenic tumor formation and hepatic metastasis (Table 4; Fig. 4d, e, f). However, overexpressing ASB3 mutants G135E, K339I, and G135E/K339I in turn facilitated hepatic metastasis (P < 0.05), whereas loss-of-function mutant ΔSOCS had no effects on hepatic metastasis (Table 4; Fig. 4d, e, f). These data indicate that overexpression of WT ASB3 may suppress the tumorigenicity and hepatic metastasis of HCT116 cells in nude mice, whereas ASB3 mutants G135E, K339I, and G135E/K339I lose the inhibitory functions.
Fig. 4.
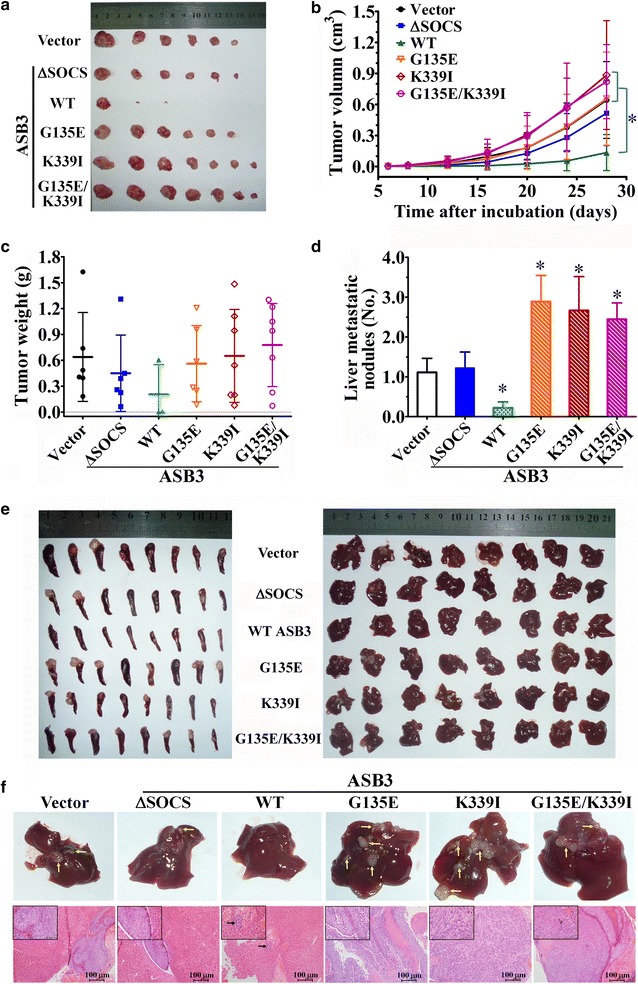
ASB3 inhibits the tumorigenicity and hepatic metastasis of CRC cells in vivo. HCT116 cells stably transfected with ASB3 cDNA or its mutants (1 × 106 cells each) were subcutaneously or intrasplenically injected into nude mice (8 in each group), and the formation and growth of subcutaneous xenografts a–c and liver metastatic tumors d and e were monitored. a The picture of subcutaneous xenografts. b Growth curves of subcutaneous xenografts (*P < 0.05). c Average weight of subcutaneous xenografts. d Numbers of liver metastatic tumor nodules (*P < 0.05 compared with the vector group). e Gross tumor growth in the spleen and liver after intrasplenic injection of HCT116 cells stably transfected with ASB3 cDNA or its mutants (1 × 106 cells each) for 8 weeks. Left general spleen images; Right general liver images. f Representative images (upper) and hematoxylin and eosin (HE) staining pictures (lower) of the livers from each group; the metastatic tumor nodules are indicated with yellow arrows. The scale bars in HE staining pictures represent 100 µm
Table 4.
The number and percentages of intrasplenic and hepatic tumor formation after intrasplenic injection of HCT116 stable cell lines (1×106 cells each) for 8 weeks
| Modified HCT116 stable cell line | Number of mice | Tumor formationa | |
|---|---|---|---|
| Intrasplenic | Liver | ||
| Vectro | 8 | 6 (75.0) | 6 (75.0) |
| ΔSOCS | 8 | 6 (75.0) | 6 (75.0) |
| WT ASB3 | 8 | 4 (50.0) | 2 (25.0)* |
| G135E | 8 | 7 (87.5) | 8 (100) |
| K339I | 8 | 7 (87.5) | 8 (100) |
| G135E/K339I | 8 | 8 (100) | 8 (100) |
* P < 0.05 compared with other groups (Students’ t test)
a The data are presented as the number of mice with tumors followed by percentage in parentheses
ASB3 inhibited the EMT of CRC cells
Since ASB3 inhibited the metastasis of CRC cells in vitro and in vivo, we investigated whether ASB3 regulates EMT or not by Western blotting and immunofluorescence assays. The results showed that ASB3 knockdown resulted in the down-regulation of β-catenin and epithelial marker E-cadherin and the up-regulation of TCF8 and mesenchymal markers N-cadherin and vimentin in THC8307 and SW620 CRC cells (Fig. 5a, b). Conversely, the ectopic overexpression of ASB3 led to the up-regulation of β-catenin and E-cadherin and down-regulation of TCF8, N-cadherin, and vimentin (Fig. 5c, d). However, we found no obvious changes of these markers’ expression after overexpressing ASB3 mutants G135E, K339I, or G135E/K339I compared with the control (Fig. 5c, d).
Fig. 5.
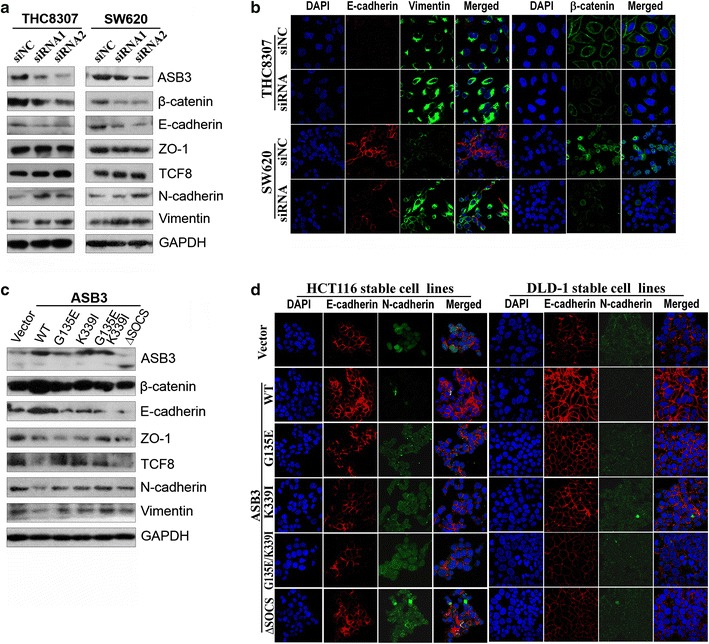
ASB3 inhibits the epithelial-mesenchymal transition (EMT) of CRC cells. After THC8307 and SW620 cells were transiently transfected with siRNAs to knockdown ASB3 expression for 48 h, EMT markers were detected with Western blotting (a) and immunofluorescence assays (b), whereas EMT markers were detected with Western blotting (c) and immunofluorescence assays (d) in HCT116 or DLD-1 cells stably transfected with ASB3 cDNA or its mutants
Discussion
ASB proteins were initially considered negative regulators of cytokine signaling because they contain SOCS box domain [15, 37]. However, mounting evidence has shown that ASB proteins are involved in many cellular processes and pathways. ASB11, an endoplasmic reticulum-associated ubiquitin ligase, interacts with and promotes the ubiquitination of ribophorin 1, which is involved in the glycosylation of nascent proteins [38]. ASB9 interacts with and promotes the ubiquitination and degradation of creatine kinase, and inhibits cell growth by negatively regulating mitochondrial energy metabolism [14, 39, 40]. ASB2α enhances adhesion of hematopoietic cells to fibronectin by degradating filamin A [41]. Furthermore, ASB2-involved ECS-type Cullin RING E3 ubiquitin ligase complex mediates mixed lineage leukemia (MLL) protein degradation during hematopoietic differentiation. One critical cause for MLL is likely that MLL fusion protein derived from chromosomal translocation is unable to be degraded due to the loss of the ASB2-binding site [42].
In the present study, we found that the ASB3 gene had a high frequency of somatic mutations: it was mutated in 5.26% (7/133) of CRC cases and in HCT116, HT-29, and DLD-1 CRC cell lines. However, we observed that there are no obvious mutation hotspots in the ASB3 gene in CRC (we consider that G135E and K339I are mutations reoccurred only in low frequency in CRC cases). Expression analysis showed that ASB3 was frequently down-regulated in CRC tissues and cell lines. Further investigations showed that the knockdown of ASB3 promoted cell proliferation, migration, and invasion in cultured CRC cells, whereas the overexpression of WT ASB3 inhibited cell proliferation, migration, and invasion in vitro and reduced tumorigenicity and hepatic metastasis of CRC xenografts in vivo. Furthermore, we found that overexpression of ASB3 inhibited EMT of CRC cells, characterized by up-regulating epithelial markers β-catenin and E-cadherin and down-regulating mesenchymal markers TCF8, N-cadherin, and vimentin [43, 44]. Conclusively, ASB3 exerts a tumor-suppressive role in the pathogenesis and progression of CRC. However, the molecular mechanisms of how ASB3 regulates the proliferation, migration, and invasion remain unknown.
Like other ASB family members, ASB3 consists of an N-terminal ANK repeat domain (ARD) and a C-terminal SOCS box. Human ASB3 ARD contains 11 ANK repeats, called ANK1 to ANK11 (NCBI Reference Sequence: NP_057199.1), and is different from murine ASB3 that contains 12 ANK repeats [20, 21]. The ANK repeats are responsible for substrate recognition, whereas the SOCS box is responsible for assembling Elongin B/C, Cul5, and Rbx2 to form the E3 ubiquitin ligase complex [45]. Our current study demonstrated that not only ASB3 ΔSOCS mutant but also G135E, K339I, and G135E/K339I mutants that were detected in CRC cases lost the tumor-suppressive function of WT ASB3. Surprisingly, in some experiments, ASB3 mutants G135E, K339I, and G135E/K339I even displayed the opposite oncogenic effects. ASB3 mutant ΔSOCS loses its function resulting from the failure to recruit and compose E3 ubiquitin ligase complex. The ASB3 G135E mutation, encoded by exon 4, leads to an additional negative charge in ANK4 in physiological condition, whereas K339I, encoded by exon 8, leads to a loss of positive charge in ANK10. Theoretically, these two mutations will affect the spatial structure of ARD of ASB3, which is verified by the altered three-dimensional structure predicted by SWISS-MODEL (Fig. 6). Thus, we speculate that these ASB3 mutants affect substrate recognization, which makes ASB3 no longer recognize its original substrates, or even recognize novel substrates. The former case will result in a loss of the functions of WT ASB3, whereas the latter will bring in a novel function.
Fig. 6.
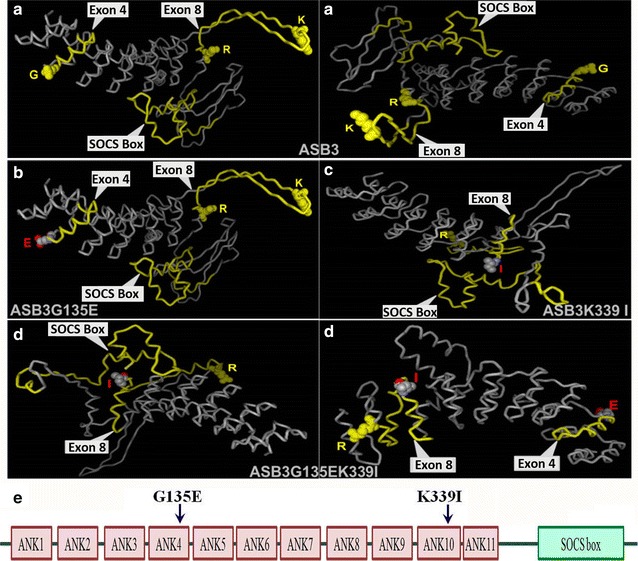
Comparison among three-dimensional (3D) models of WT ASB3 and ASB3 mutant proteins by computer homogeneity modeling and prediction with the SWISS-MODEL. The ASB3/ASB3 mutant proteins were modeled and predicted. a Images of spatial structure of ASB3 from two different angles. b Image of 3D model of G135E (ASB3G135E, on the left). c Image of 3D model of K339I (ASB3K339I, on the right). d Images of spatial structure of G135E/K339I (ASB3G135EK339I) from two different angles. e Structure sketch of ASB3 protein with G135E and K339I mutations
There are several limitations in our study. First, we did not clarify whether the ASB3 dysfunction resulted from gene mutations or down-regulated expression affects the clinical prognosis for CRC cases due to the shorter follow-up. Second, further studies are required to confirm what molecules ASB3 directly interact with to play the tumor-suppressive role in CRC tumorigenesis.
Conclusions
There are frequent mutations (5.3%) and down-regulated expression (70.4%) of the ASB3 gene in Chinese patients with CRC. WT ASB3 inhibits CRC cell proliferation, migration, and invasion in vitro and decreases the tumorigenicity and hepatic metastasis in vivo; whereas mutated ASB3 lost this tumor-suppressive role. In conclusion, dysfunctions of the ASB3 gene that result from mutations or down-regulated expression are possible events that lead to the pathogenesis or progression of CRC.
Authors’ contributions
WLH and RYL managed the research fund; WLH, RYL, SC, and WYD designed the study; WYD and CMK performed MTT and colony formation assays; WYD and XF performed animal experiments; WYD performed transwell and wound healing assays; ZHL and ZZP collected clinical specimens and performed data analysis; WYD, ZHL, and YZ performed IHC and immunofluorescence assays; WY performed DNA extraction and sequencing; WYD and JW constructed expression vectors; WYD, RYL, WLH, and SC performed data analysis and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. Xiangqi Meng, Ms. Hongyan Yu, Dr. Xiangfang Ying, and Ms. Ling Zhou for their technical assistance. This study was supported by the National Natural Science Foundation of China (No. 81472256, 81272638), the Guangdong Provincial Science and Technology Project (No. 2016A020215081, 2016A020217007) and the National High Technology Research and Development Program of China (863 Program, No. 2012AA02A204).
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- ANK
ankyrin
- ARD
ANK repeat domain
- ASB
ankyrin repeat and SOCS box protein
- ANOVA
analysis of variance
- CRC
colorectal cancer
- EMT
epithelial-mesenchymal transition
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- h
hours
- HRP
horseradish peroxidase
- IHC
immunohistochemical
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
- PBS
phosphate-buffered saline
- qPCR
real-time quantitative PCR
- SD
standard deviation
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SOCS
suppressor of cytokine signaling
- WB
western blotting
- WT
wild type
Footnotes
Wu-Ying Du and Zhen-Hai Lu contributed equally to this work
Contributor Information
Wu-Ying Du, Email: duwuying@163.com.
Zhen-Hai Lu, Email: luzhh@sysucc.org.cn.
Wen Ye, Email: yewensysu@163.com.
Xiang Fu, Email: fuxiangsysucc@163.com.
Yi Zhou, Email: z-y-b-b-c@163.com.
Chun-Mei Kuang, Email: kuangchm@sysucc.org.cn.
Jiang-Xue Wu, Email: wujx@sysucc.org.cn.
Zhi-Zhong Pan, Email: panzhzh@sysucc.org.cn.
Shuai Chen, Email: chenshuai@sysucc.org.cn.
Ran-Yi Liu, Email: liury@sysucc.org.cn.
Wen-Lin Huang, Email: hwenl@mail.sysu.edu.cn.
References
- 1.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2016 doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Zeng H, Zhang S. The updated incidences and mortalities of major cancers in China, 2011. Chin J Cancer. 2015;34(11):502–507. doi: 10.1186/s40880-015-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabeneck L, Horton S, Zauber AG, Earle C. Colorectal cancer. In: Gelband H, Jha P, Sankaranarayanan R, Horton S, editors. Cancer: Disease control priorities. 3. Washington: The International Bank for Reconstruction and Development; 2015. [Google Scholar]
- 5.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–267. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahara T, Yamamoto E, Madireddi P, Suzuki H, Maruyama R, Chung W, et al. Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology. 2014;146(2):530–538. doi: 10.1053/j.gastro.2013.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Network Cancer Genome Atlas. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(74):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia Y, Guo M. Epigenetic changes in colorectal cancer. Chin J Cancer. 2013;32(1):21–30. doi: 10.5732/cjc.011.10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xuan Y, Yang H, Zhao L, Lau WB, Lau B, Ren N, et al. MicroRNAs in colorectal cancer: small molecules with big functions. Cancer Lett. 2015;360(2):89–105. doi: 10.1016/j.canlet.2014.11.051. [DOI] [PubMed] [Google Scholar]
- 11.Yamagishi H, Kuroda H, Imai Y, Hiraishi H. Molecular pathogenesis of sporadic colorectal cancers. Chin J Cancer. 2016;35(1):4. doi: 10.1186/s40880-015-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voutsadakis IA. The ubiquitin-proteasome system in colorectal cancer. Biochim Biophys Acta. 2008;1782(12):800–808. doi: 10.1016/j.bbadis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Kohroki J, Nishiyama T, Nakamura T, Masuho Y. ASB proteins interact with Cullin5 and Rbx2 to form E3 ubiquitin ligase complexes. FEBS Lett. 2005;579(30):6796–6802. doi: 10.1016/j.febslet.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 14.Balasubramaniam D, Schiffer J, Parnell J, Mir SP, Amaro RE, Komives EA. How the ankyrin and SOCS box protein, ASB9, binds to creatine kinase. Biochemistry. 2015;54(8):1673–1680. doi: 10.1021/bi501420n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott J, Johnston JA. SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 2004;25(8):434–440. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Li JY, Chai B, Zhang W, Wu X, Zhang C, Fritze D, et al. Ankyrin repeat and SOCS box containing protein 4 (Asb-4) colocalizes with insulin receptor substrate 4 (IRS4) in the hypothalamic neurons and mediates IRS4 degradation. BMC Neurosci. 2011;12:95. doi: 10.1186/1471-2202-12-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li JY, Chai BX, Zhang W, Liu YQ, Ammori JB, Mulholland MW. Ankyrin repeat and SOCS box containing protein 4 (Asb-4) interacts with GPS1 (CSN1) and inhibits c-Jun NH2-terminal kinase activity. Cell Signal. 2007;19(6):1185–1192. doi: 10.1016/j.cellsig.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Au V, Tsang FH, Man K, Fan ST, Poon RT, Lee NP. Expression of ankyrin repeat and SOCS box containing 4 (ASB4) confers migration and invasion properties of hepatocellular carcinoma cells. Biosci Trend. 2014;8(2):101–110. doi: 10.5582/bst.8.101. [DOI] [PubMed] [Google Scholar]
- 19.Tokuoka M, Miyoshi N, Hitora T, Mimori K, Tanaka F, Shibata K, et al. Clinical significance of ASB9 in human colorectal cancer. Int J Oncol. 2010;37(5):1105–1111. doi: 10.3892/ijo_00000762. [DOI] [PubMed] [Google Scholar]
- 20.Kile BT, Viney EM, Willson TA, Brodnicki TC, Cancilla MR, Herlihy AS, et al. Cloning and characterization of the genes encoding the ankyrin repeat and SOCS box-containing proteins Asb-1, Asb-2, Asb-3 and Asb-4. Gene. 2000;258(1–2):31–41. doi: 10.1016/S0378-1119(00)00402-9. [DOI] [PubMed] [Google Scholar]
- 21.Chung AS, Guan YJ, Yuan ZL, Albina JE, Chin YE. Ankyrin repeat and SOCS box 3 (ASB3) mediates ubiquitination and degradation of tumor necrosis factor receptor II. Mol Cell Biol. 2005;25(11):4716–4726. doi: 10.1128/MCB.25.11.4716-4726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P, et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res. 2014;20(4):1042–1052. doi: 10.1158/1078-0432.CCR-13-0171. [DOI] [PubMed] [Google Scholar]
- 23.Fu X, Hu J, Han HY, Hua YJ, Zhou L, Shuai WD, et al. High expression of XPA confers poor prognosis for nasopharyngeal carcinoma patients treated with platinum-based chemoradiotherapy. Oncotarget. 2015;6(29):28478–28490. doi: 10.18632/oncotarget.4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu RY, Dong Z, Liu J, Zhou L, Huang W, Khoo SK, et al. Overexpression of asparagine synthetase and matrix metalloproteinase 19 confers cisplatin sensitivity in nasopharyngeal carcinoma cells. Mol Cancer Ther. 2013;12(10):2157–2166. doi: 10.1158/1535-7163.MCT-12-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Wu J, Meng X, Ying X, Zuo Y, Liu R, et al. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis. 2011;32(7):1033–1042. doi: 10.1093/carcin/bgr081. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y, Wu J, Fu X, Du W, Zhou L, Meng X, et al. OTUB1 promotes metastasis and serves as a marker of poor prognosis in colorectal cancer. Mol Cancer. 2014;13:258. doi: 10.1186/1476-4598-13-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X, et al. Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin Cancer Res. 2014;20(13):3434–3445. doi: 10.1158/1078-0432.CCR-13-2952. [DOI] [PubMed] [Google Scholar]
- 28.Meng X, Wu J, Pan C, Wang H, Ying X, Zhou Y, et al. Genetic and epigenetic down-regulation of microRNA-212 promotes colorectal tumor metastasis via dysregulation of MnSOD. Gastroenterology. 2013;145(2):426–436. doi: 10.1053/j.gastro.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Qiu L, Wu J, Pan C, Tan X, Lin J, Liu R, et al. Downregulation of CDC27 inhibits the proliferation of colorectal cancer cells via the accumulation of p21Cip1/Waf1. Cell death disease. 2016;7:e2074. doi: 10.1038/cddis.2015.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Wu J, Zhou L, Pan C, Zhou Y, Du W, et al. ZD6474, a new treatment strategy for human osteosarcoma, and its potential synergistic effect with celecoxib. Oncotarget. 2015;6(25):21341–21352. doi: 10.18632/oncotarget.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu RY, Zhu YH, Zhou L, Zhao P, Li HL, Zhu LC, et al. Adenovirus-mediated delivery of interferon-gamma gene inhibits the growth of nasopharyngeal carcinoma. J Transl Med. 2012;10:256. doi: 10.1186/1479-5876-10-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter MN, Wright KT, Fuller HR, MacNeil S, Johnson WE. Mesenchymal stem cell-conditioned medium accelerates skin wound healing: an in vitro study of fibroblast and keratinocyte scratch assays. Exp Cell Res. 2010;316(7):1271–1281. doi: 10.1016/j.yexcr.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 33.Cory G. Scratch-Wound Assay. In: Wells CM, Parsons M, editors. Cell Migration: developmental methods and protocols. New York: Humana Press; 2011. pp. 25–30. [Google Scholar]
- 34.Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43(1 Suppl):25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Liu RY, Huang JL, Liu QC, Li Y, Wu PH, et al. Adenovirus-mediated intra-tumoral delivery of the human endostatin gene inhibits tumor growth in nasopharyngeal carcinoma. Int J Cancer. 2006;118(8):2064–2071. doi: 10.1002/ijc.21585. [DOI] [PubMed] [Google Scholar]
- 36.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–285. doi: 10.1016/S0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 37.Zhou X, Liu Z, Cheng X, Zheng Y, Zeng F, He Y. Socs1 and Socs3 degrades Traf6 via polyubiquitination in LPS-induced acute necrotizing pancreatitis. Cell Death Disease. 2015;6:e2012. doi: 10.1038/cddis.2015.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andresen CA, Smedegaard S, Sylvestersen KB, Svensson C, Iglesias-Gato D, Cazzamali G, et al. Protein interaction screening for the ankyrin repeats and suppressor of cytokine signaling (SOCS) box (ASB) family identify Asb11 as a novel endoplasmic reticulum resident ubiquitin ligase. J Biol Chem. 2014;289(4):2043–2054. doi: 10.1074/jbc.M113.534602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon S, Kim D, Rhee JW, Park JA, Kim DW, Kim DS, et al. ASB9 interacts with ubiquitous mitochondrial creatine kinase and inhibits mitochondrial function. BMC Biol. 2010;8:23. doi: 10.1186/1741-7007-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debrincat MA, Zhang JG, Willson TA, Silke J, Connolly LM, Simpson RJ, et al. Ankyrin repeat and suppressors of cytokine signaling box protein asb-9 targets creatine kinase B for degradation. J Biol Chem. 2007;282(7):4728–4737. doi: 10.1074/jbc.M609164200. [DOI] [PubMed] [Google Scholar]
- 41.Lamsoul I, Burande CF, Razinia Z, Houles TC, Menoret D, Baldassarre M, et al. Functional and structural insights into ASB2alpha, a novel regulator of integrin-dependent adhesion of hematopoietic cells. J Biol Chem. 2011;286(35):30571–30581. doi: 10.1074/jbc.M111.220921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Muntean AG, Hess JL. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood. 2012;119(5):1151–1161. doi: 10.1182/blood-2011-06-362079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aparicio LA, Blanco M, Castosa R, Concha A, Valladares M, Calvo L, et al. Clinical implications of epithelial cell plasticity in cancer progression. Cancer Lett. 2015;366(1):1–10. doi: 10.1016/j.canlet.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Zhu S, Zhang J, Xu F, Xu E, Ruan W, Ma Y, et al. IGFBP-rP1 suppresses epithelial-mesenchymal transition and metastasis in colorectal cancer. Cell Death Dis. 2015;6:e1695. doi: 10.1038/cddis.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, et al. Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J Biol Chem. 2008;283(12):8005–8013. doi: 10.1074/jbc.M706987200. [DOI] [PubMed] [Google Scholar]