

Abstract
Background
The present study investigated the antibacterial activity and underlying mechanisms of ginkgolic acid (GA) C15:1 monomer using green fluorescent protein (GFP)-labeled bacteria strains.
Results
GA presented significant antibacterial activity against Gram-positive bacteria but generally did not affect the growth of Gram-negative bacteria. The studies of the antibacterial mechanism indicated that large amounts of GA (C15:1) could penetrate GFP-labeled Bacillus amyloliquefaciens in a short period of time, and as a result, led to the quenching of GFP in bacteria. In vitro results demonstrated that GA (C15:1) could inhibit the activity of multiple proteins including DNA polymerase. In vivo results showed that GA (C15:1) could significantly inhibit the biosynthesis of DNA, RNA and B. amyloliquefaciens proteins.
Conclusion
We speculated that GA (C15:1) achieved its antibacterial effect through inhibiting the protein activity of B. amyloliquefaciens. GA (C15:1) could not penetrate Gram-negative bacteria in large amounts, and the lipid soluble components in the bacterial cell wall could intercept GA (C15:1), which was one of the primary reasons that GA (C15:1) did not have a significant antibacterial effect on Gram-negative bacteria.
Keywords: GA, Green fluorescent protein, Antibacterial activity
Background
Plants can synthesize over 200,000 compounds through various metabolic pathways [1]. Secondary metabolites in plants are derived from primary metabolites, and their categories and chemical structures are complex and diverse, including nitrogen-containing organic compounds, terpenoids, phenols and polyacetylenes, of which alkaloids, terpenoids and phenols are the most common. Secondary metabolites are widely involved in plant growth, development and defense as well as other physiological and biological processes [2]. Plant secondary metabolites provide many useful natural organic compounds for human use. Because traditional chemical pesticides contaminate soil and water, the development of environmentally friendly bio-pesticides has become a popular research focus. However, the development of synthetic pesticides has many problems, such as a low successful rate, long cycle and huge cost etc. Therefore, discovering lead compounds (plant-derived antibacterial reagents) from natural plant products with improved biological activity has become an effective method to develop new biological pesticides. Self-defense mechanisms have been evolved in plants, and many secondary plant metabolites are natural antibacterial agents [3–5]. Wilkins et al. [6] reported that 1389 plants could be used as sources of plant antibacterial agents including ingredients that could kill or inhibit bacteria, such as antibiotics, flavonoids, organic acids, polyphenols and specific proteins. Wilson et al. [7] studied the inhibition of Botrytis cinerea by 345 crude plant extracts and 49 essential oils, found that 13 crude extracts and 4 essential oils provided antibacterial activities.
Resorcinolic lipids are widely distributed plant secondary metabolites produced in large numbers. Recent studies have shown that they have extraordinarily high antibacterial activity. Resorcinolic lipids produced by Pseudomonas carboxydoflava can inhibit the growth of many bacteria species, such as Micrococcus lysodeictius and Bacillus subtilis [8, 9]. Resorcinolic lipids isolated from cashew apple have strong antibacterial effects on Gram-positive bacteria, including methicillin-resistant S. aureus strains [10, 11]. Sixteen phenolic compounds have been isolated from the cashew Anacardium occidentale (Anacardiaceae) nut shell oil, including various C15 phenolic compounds. Their antimicrobial activity has been tested against four typical microorganisms, Bacillus subtilis, a Gram-positive bacterium; Escherichia coli, a Gram-negative bacterium; Saccharomyces cereuisiae, a yeast; and Penicillium chrysogenum, a mold. Most of them exhibited potent antibacterial activity against only Gram-positive bacteria [12].
Ginkgo is a Chinese-specific rare relict species that is well known as a “living fossil of gymnosperms” [13]. The fruit and leaves of ginkgo have relatively high economic and medicinal values. However, its sarcotestas is usually discarded, causing secondary pollution of the environment [14]. GA, which is in high level in sarcotestas, is a natural plant-derived active substance contained in ginkgo, and it belongs to long-chain phenolic compounds that are derivatives of sumac acid [15]. Current studies have shown that the biological activities of GA include anti-tumor activity, neuroprotective activity, anxiolytic and antibacterial activity [16–20]. These biological activities may make a possibility that increases the utilization of ginkgo sarcotestas and reduces environmental pollution. The potential uses of Ginkgo have been attracted many concern. Studies of GA antibacterial activity have found that although GA could inhibit the activity of bacteria and plant pathogens, it just showed selective antibacterial activity, with strong inhibition towards to Gram-positive bacteria and almost no inhibition to Gram-negative bacteria [21–24].
The present study employed GFP-labeled strains and analyzed the antibacterial activity and mechanisms of GA C15:1 monomer, high amounts of which was in ginkgo sarcotestas and had relatively high antibacterial activity. Investigations of the selective antibacterial activity of GA could provide a scientific and theoretical basis for the development of new plant-derived pesticides using ginkgo sarcotestas as the raw material.
Results
Antibacterial activity of GA (C15: 1)
The antibacterial activity of GA (C15:1) is shown in Table 1. GA (C15:1) had strong antibacterial activity against Gram-positive bacteria, the MIC values of all of the tested Gram-positive bacteria were not greater than 10 μg mL−1..In this study, all of the tested Gram-negative bacteria could grow well after the addition of large doses of GA (C15:1) (final concentration 500 μg mL−1), and no differences were observed compared with the controls supplemented with salicylic acid, indicating that GA (C15:1) did not have significant antibacterial action against Gram-negative bacteria.
Table 1.
Antibacterial activities of GA (C15:1) and salicylic acid
| Strains | Control | GA (C15:1) | Salicylic acid | ||||
|---|---|---|---|---|---|---|---|
| MIC (μg mL−1) | MBC (μg mL−1) | MBC (μg mL−1) | MBC (μg mL−1) | MIC (μg mL−1) | MBC (μg mL−1) | ||
| G− | E. coli DH5α | >500 | - | >500 | - | >500 | - |
| E. coli O157:H7 | >500 | - | >500 | - | >500 | - | |
| P. putida KT2440 | >500 | - | >500 | - | >500 | - | |
| P. aeruginosa PAO1 | >500 | - | >500 | - | >500 | - | |
| R. solanacearum | >500 | - | >500 | - | >500 | - | |
| G+ | B. amyloliquefaciens SQR9 | >500 | - | 5 | 60 | 500 | >500 |
| R. jostii RHA1 | >500 | - | 10 | 20 | 500 | >500 | |
| S. thermophilus ND03 | >500 | - | 10 | 20 | 500 | >500 | |
| S. aureus | >500 | - | 10 | 20 | 500 | >500 | |
- Not measured, GA ginkgolic acid, MIC the minimum inhibitory concentration, MBC the minimum bactericidal concentration
The effect of GA (C15:1) on GFP in bacteria
Using a GFP-labeled strain as the target, we studied effect of GA (C15:1) on GFP fluorescence in bacteria, and the results are show in Fig. 1-a). GA (C15:1) could significantly affect GFP fluorescence in the Gram-positive bacteria B. amyloliquefaciens SQR9-gfp within 1 min. Compared with the results for the CK (bacteria only containing DMSO), GA (C15:1) at the concentration of 5 μg mL−1 could reduce GFP fluorescence intensity in SQR9 bacteria by more than 50% within 1 min, and GA (C15:1) at higher concentrations could almost completely quench GFP fluorescence in SQR9 bacteria within 1 min.
Fig. 1.
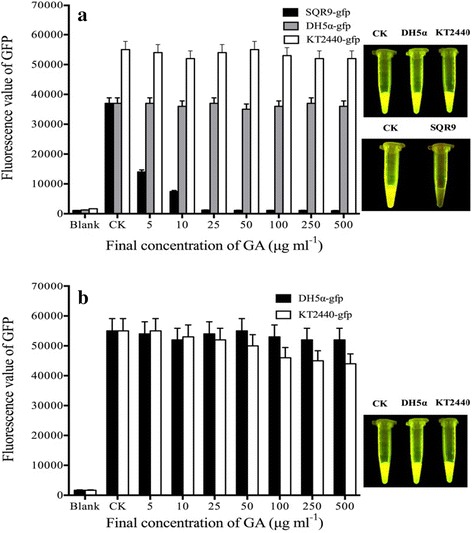
Effect of GA (C15:1) on GFP fluorescence in bacteria. Three independent experiments were conducted (n = 3); the error bars indicate one standard error. Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.). a: Bacteria were incubated at 30 °C for 1 min before GFP fluorescence was measured.10 μL of DMSO without the drug was used as a control. The blank was the E. coli bacteria solution without GFP. One-way ANOVA was used for analyzing the data (F7,16 = 656.9 P < 0.001(SQR9-gfp); F7,16 = 0.208 P > 0.05(DH5α-gfp); F7,16 = 0.357 P > 0.05 (KT2440-gfp)); (b) Bacteria were incubated at 30 °C for 4 h before GFP fluorescence was measured. 10 μL of DMSO without the drug was used as a control. The blank was the E. coli bacteria solution without GFP. One-way ANOVA was used for analyzing the data (F7,16 = 0.178 P > 0.05(DH5α-gfp); F7,16 = 1.412 P > 0.05 (KT2440-gfp))
Although GA (C15:1) could significantly affect GFP fluorescence in B. amyloliquefaciens SQR9-gfp bacteria within 1 min, it did not have a significant effect on GFP fluorescence in Gram-negative bacteria E. coli DH5α-gfp and P. putida KT2440-gfp. Within 1 min, a significant decrease of fluorescence intensity was not detected in the studied Gram-negative bacteria, and fluorescence intensity values in the CK were close to the fluorescence intensity value in bacteria supplemented with GA.
We extended the contact time of Gram-negative bacteria E. coli DH5α-gfp and P. putida KT2440-gfp with GA (C15:1) to 4 h. The results (Fig. 1-b) showed that even with longer incubation times, GA could only reduce GFP fluorescence in Gram-negative bacteria by a small amount. GA (C15:1) at the concentration of 500 μg mL−1 had the most significant effect on fluorescence in E. coli DH5α-gfp, causing approximately 30% fluorescence reduction. The fluorescence reduction values at other concentrations were all less than 25%.
The scanning electron microscopy examination showed that after the addition of GA (C15:1), the cells of the three bacteria still remained intact without apparent cell lysis (Fig. 2). Because GFP protein was only present in the bacteria, we speculated that GFP fluorescence decay in Gram-positive bacteria B. amyloliquefaciens SQR9-gfp was caused by a large amount of GA that entered the bacteria within a short time, whereas the reason that GFP fluorescence in both Gram-negative bacteria did not show decay was that GA (C15:1) did not enter these bacteria in a large amount. The lack of a significant reduction in GFP fluorescence in the two Gram-negative bacteria was caused by a limited amount of GA entering the cells.
Fig. 2.

SEM observation of bacteria cell morphology. respectively, (a, b, c) represent Bacteria cell morphology before the addition of GA; (d, e, f) represent Bacteria cell morphology after the addition of GA at a final concentration of 100 μg mL−1 for 1 min
Effect of GA (C15:1) on GFP in bacteria crude extracts
To verify the hypothesis that “GFP fluorescence decay was related to GA entering the bacteria cells”, the bacteria cells of Gram-negative bacteria E. coli DH5α-gfp and P. putida KT2440-gfp were lysed and centrifuged, and the crude lysate supernatants which contained GFP, were collected. GA (C15:1) was directly added to the supernatant, and the GFP fluorescence intensity was examined. The same procedure was performed on Gram-positive bacteria B. amyloliquefaciens SQR9-gfp. The results showed that GA (C15:1) could significantly affect GFP fluorescence in the crude lysates of E. coli DH5α-gfp and P. putida KT2440-gfp within 1 min (Fig. 3). Compared with the results for the CK, GA (C15:1) at a final concentration > 25 μg mL−1 could completely quench GFP fluorescence in the crude lysates of E. coli DH5α-gfp and P. putida KT2440-gfp within 1 min.
Fig. 3.

Effect of GA (C15:1) on GFP fluorescence in bacteria crude lysates (1 min). Three independent experiments were conducted (n = 3). The error bars indicate one standard error. All of the tests were incubated at 30 °C for 1 min before GFP fluorescence was measured. 10 μL of DMSO without the drug was used as a control. The blank was the E. coli bacteria solution without GFP. One-way ANOVA was used for analyzing the data (F7,16 = 281.4 P < 0.001 (SQR9-gfp); F7,16 = 246.3 P < 0.001 (DH5α-gfp); F7,16 = 304.0 P < 0.001 (KT2440-gfp)). Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.)
Similar fluorescence decay results were also showed in the crude cell lysates of Gram-positive bacteria B. amyloliquefaciens SQR9-gfp (Fig. 3). When the concentration of GA (C15:1) was > > 10 μg mL−1, the fluorescence in the crude lysates of B. amyloliquefaciens SQR9-gfp was completely quenched within 1 min. This results were consistent with the results of the GFP fluorescence decay experiment in B. amyloliquefaciens SQR9-gfp cells, and suggested that GA could enter the cells of Gram-positive bacteria B. amyloliquefaciens SQR9-gfp GFP within a short period of time.
Effect of GA on the activity of a variety of proteins
According to the PCR results (Fig. 4-a), GA (C15:1) significantly inhibited the PCR reaction. The addition of 1 μg mL−1 and 5 μg mL−1 GA (C15:1) could interfere with PCR reactions, resulting in decreased specific bands, although these concentrations would not completely inhibit PCR reactions. However, when the concentration of GA was ≥ 10 μg mL−1, the PCR reaction was completely inhibited, and electrophoresis could not detect specific target bands. This result suggested that lower concentration of GA (C15:1) could inhibit Taq DNA polymerase activity and interfere with DNA replication.
Fig. 4.
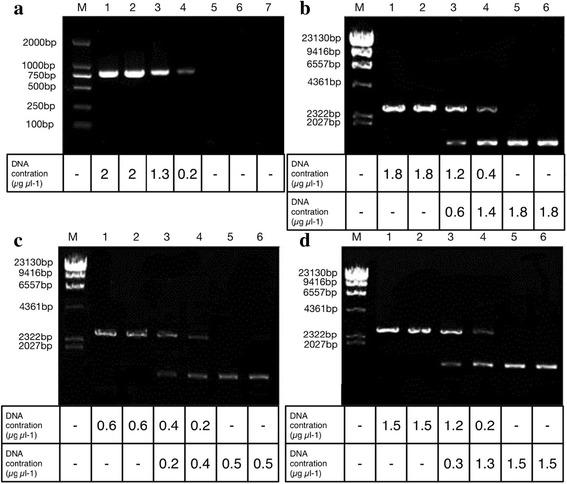
Effect of GA (C15:1) on a variety of proteins. respectively :(a) represents the effect of GA (C15:1) on the activity of Taq DNA Polymerase. The DNA contration at the bottom of fig represents the PCR reaction was inhibited by GA (C15:1). b represents the effect of GA (C15:1) on the activity of Kpn I. The DNA contration at the bottom of fig represents the enzymatic activity Kpn I was inhibited by GA (C15:1) (c) represents the effect of GA (C15:1) on the activity of Hind III. The DNA contration at the bottom of fig represents the enzymatic activity Hind III was inhibited by GA (C15:1) (d) represents the effect of GA (C15:1) on the activity of EcoR I. The DNA contration at the bottom of fig represents the enzymatic activity EcoR I was inhibited by GA (C15:1) lane 1, CK; lane 2, addition of DMSO into PCR or digestion system; lane 3, addition of 1 μg mL−1 GA (C15:1) into PCR or digestion system; lane 4, addition of 5 μg mL−1 GA (C15:1) into PCR or digestion system; lane 5, addition of 10 μg mL−1 GA (C15:1) into PCR or digestion system; lane 6, addition of 25 μg mL−1 GA (C15:1) into PCR or digestion system; lane 7, addition of 50 μg mL−1 GA (C15:1) into PCR or digestion system
Restriction digestion electrophoresis results showed that GA (C15:1) could significantly inhibit the enzymatic activity of Kpn I, Hind III and EcoR I (Fig. 4-b, c, d). GA (C15:1) at a final concentration of 1 μg mL−1 could partially inhibit the enzymatic activity of the three restriction enzymes. When the concentration of GA (C15:1) was > 5 μg mL−1, the enzymatic activity of the three restriction enzymes were inhibited almost completely, and the super coiled pUC19 plasmid was barely digested.
GA (C15:1) could significantly inhibit SOD enzyme activity and β-galactosidase activity (Fig. 5-a, b). When the final concentration of GA (C15:1) was 1 μg mL−1, both SOD enzyme activity and β-galactosidase activity were decreased by 50% compared with that of the control. When the final concentration of GA was > 5 μg mL−1, SOD enzyme activity and β-galactosidase enzyme activity were almost undetectable.
Fig. 5.
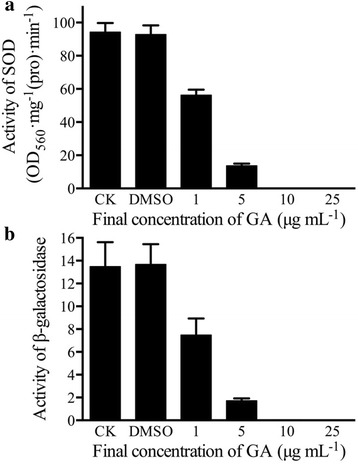
Effect of GA (C15:1) on the activity of SOD and β-galactosidase. The error bars indicate one standard error. a: Effect of GA (C15:1) on the activity of SOD.The optical density (OD)560 values were measured. 5 μL of DMSO (without the drug) was used as a control. One-way ANOVA was used for analyzing the data (F5,12 = 161.6 P < 0.001);(b) : Effect of GA (C15:1) on the activity of β-galactosidase. The OD value at 420 nm was read. 5 μL of DMSO (without the drug) was used as a control. One-way ANOVA was used for analyzing the data (F5,12 = 25.15 P < 0.001). Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.)
These proteins had different sources and were selected randomly. Thus, the results of this study suggested that the inhibition of GA (C15:1) on protein activities was non-selective.
Inhibition of isotope incorporation experiments
The aforementioned experiments showed that GA could inhibit DNA polymerase function in vitro. However, it was unclear whether GA had a similar function in bacteria, including whether GA (C15:1) could inhibit DNA polymerase activity in vivo, which would inhibit DNA replication. In addition, it is unclear whether GA could inhibit RNA polymerase and ribosome activities, which would inhibit transcription and translation. To further clarify the mechanism of GA (C15:1), we used the method of the inhibition of isotope incorporation to verify the effects of GA (C15:1) in vivo. (Methy-3H) thymine ([3H] TdR), 3H-uridine ([3H] UR) and 3H-tyrosine ([3H] Tyr) were used as precursors to determine effect of GA (C15:1) on the biosynthesis of DNA, RNA and B. amyloliquefaciens SQR9 proteins. The results are showed in Fig. 6. Compared with that of the control, GA (C15:1) could inhibit DNA replication, RNA synthesis and protein synthesis to different extents under all three concentrations (25 μg mL−1, 10 μg mL−1 and 5 μg mL−1). When the concentration of GA (C15:1) reached 25 μg mL−1, the inhibition of [3H] TdR incorporation was approximately 99%, of [3H] UR incorporation was approximately 90%, and of protein precursor [3H] tyrosine was approximately 85%. These data indicated that GA (C15:1) could inhibit DNA replication in vivo as well as RNA transcription and protein synthesis.
Fig. 6.
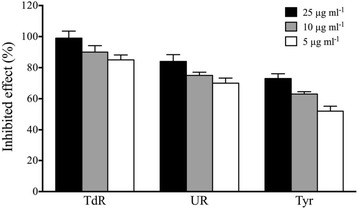
Effect of GA (C15:1) on the incorporation of precursors for the synthesis of macromolecules in B. amyloliquefaciens SQR9. The error bars indicate one standard error. The final concentrations of GA in the reaction system were 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All of the treatments were conducted at 37 °C in a shaker. One-way ANOVA was used for analyzing the data (F2,6 = 8.72 P = 0.017 (TdR); F2,6 = 19.54 P = 0.002 (UR); F2,6 = 28.59 P < 0.001 (Tyr)). Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.)
The interception of GA by Gram-negative bacteria cell walls
Using E. coli as the target, lysozyme was used to destroy the peptidoglycan structure in the cell wall and thus E. coli DH5α-gfp protoplasts were obtained. Effect of GA (C15:1) on GFP fluorescence in the protoplast within 1 minute was measured. The results (Fig. 7) showed that when the final concentration of C15:1 was lower than 25 μg mL−1, it did not have significant effect on GFP fluorescence in protoplasts. When the final concentration of GA was ≥ 25 μg mL−1, the decreased GFP fluorescence intensity in the protoplasts was found. It was positively correlated with increasing concentration of GA. When the final concentration of GA reached 500 μg mL−1, the GFP fluorescence intensity in the protoplasts decreased to approximately 80% of the control. This result suggested that after the peptidoglycan structure in the Gram-negative bacteria cell wall was destroyed, a small amount of high concentration GA could enter the Gram-negative bacteria cell and produce a low level of GFP fluorescence decay.
Fig. 7.
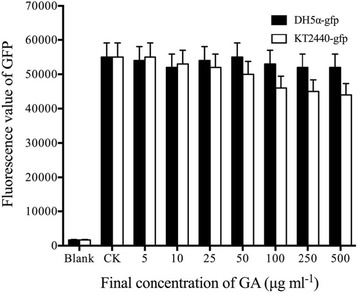
Effect of GA (C15: 1) on GFP fluorescence in the protoplasts of E. coli DH5α-gfp (1 min). Three independent experiments were conducted (n = 3); The error bars indicate one standard error. 5 μL of DMSO (without the drug) was used as a control. The blank was E. coli protoplast solution without GFP. All the tests were incubated at 30 °C for 1 min. One-way ANOVA was used for analyzing the data (F7,16 = 0.106 P > 0.05 (DH5α-gfp); F7,16 = 1.412 P > 0.05 (KT2440-gfp)). Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.)
By soaking E. coli cells in ethanol solution for a short period of time, the lipid-soluble components (mainly included lipopolysaccharide and phospholipids) in the cell wall were removed/partially removed. Effect of GA (C15:1) on GFP fluorescence in the E. coli cells that did not have lipid-soluble components in their cell walls, was measured. The results (Fig. 8) showed that when the final concentration of GA reaches was 5 μg mL−1, a large degree decrease of GFP fluorescence in E. coli occurred. When the final concentration of GA was above 10 μg mL−1, it could completely quench the GFP fluorescence in E. coli. However, if the lipid-soluble components were not removed from the E. coli bacteria cell wall, even the final concentration of GA at 500 μg mL−1 could not quench the GFP fluorescence (Fig. 1-a). These results showed that the lipid-soluble components in Gram-negative bacteria cell walls could intercept GA.
Fig. 8.
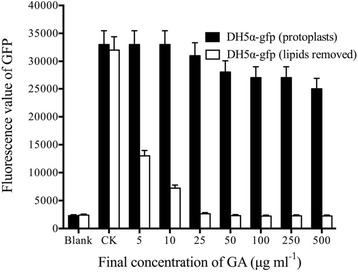
Effect of GA (C15: 1) on GFP fluorescence in E. coli DH5α-gfp (1 min), in which the lipids have been removed from the strains’ cell wall. Three independent experiments were conducted (n = 3). The error bars indicate one standard error. 5 μL of DMSO (without the drug) was used as a control. The blank was E. coli bacteria solution without GFP (after ethanol solubilization). All the tests were incubated at 30 °C for 1 min. One-way ANOVA was used for analyzing the data (F7,16 = 2.116 P > 0.05 (Black); F7,16 = 121.6 P < 0.001 (White)). Three individual tubes were collected from LB plates, and each tube was performed in triplicate. Each bar in the gram represents means of three individual tubes (mean + − non-log transfoemed SE.)
Discussion
In this study, E. coli DH5α, E. coli O157: H7, P. putida KT2440, P. aeruginosa PAO1, R. solanacearum, Rhodococcus RHA1, S. thermophilus ND03, S. aureus and other common strains were used to study the antibacterial activity of GA (C15:1), and GA was found to have significant antibacterial activity against Gram-positive bacteria but little effect on the growth of Gram-negative bacteria. A relatively strong selective antibacterial mechanism of GA was observed. The MIC value of B. amyloliquefaciens SQR9 was the smallest among all of the tested Gram-positive bacteria. However, its MBC value (60 μg mL−1) was the largest among all of the tested Gram-positive bacteria. These results might be caused by small amount of endospores that were generated when B. amyloliquefaciens SQR9 was cultured. Endospores had relatively strong resistance, and could withstand higher concentrations of GA without being killed. Therefore, B. amyloliquefaciens SQR9 had significantly higher MBC values than other Gram-positive bacteria. The antibacterial activity of GA has been reported these years. Himejima and Kubo [12] found that 2-hydroxy-6-(8-pentadecenyl) salicylic (another name of ginkgolic acid C15:1) showed lower MICs (about 10 μg mL−1) against Gram-positive bacteria and higher MICs (>100 μg mL−1) against Gram-negative bacteria. Choi et al. [23] also showed that GA (C15:1) had significant antibacterial activity against 18 g-positive vancomycin-resistant. The results of the present study are consistent with above studies.
Additional studies on antibacterial mechanisms using GFP fluorescence-labeled Gram-positive bacteria B. amyloliquefaciens SQR9 and GFP-labeled Gram-negative bacteria E. coli DH5α and P. putida KT2440 showed that GA (C15:1) could significantly affect GFP fluorescence in the cells of Gram-positive B. amyloliquefaciens SQR9-gfp, whereas it had no significant effect on GFP fluorescence in the cells of Gram-negative bacteria E. coli DH5α-gfp and P. putida KT2440-gfp. The green fluorescent protein (GFP) has been widely used as a highly useful tool in the fluorescence studies of living cells, which is found in cell cytoplasm of jellyfish and is an extremely stable protein with 238 amino acids [25, 26]. The fluorescence produced by GFP was caused by its protein conformation. In general, as long as the protein conformation of GFP did not change, the fluorescence would not decay or disappear. Previous reports showed that GA and sumac acids, which had a similar structure, could affect the activity of numerous enzymes, including protein phosphatase, lipoxygenase and histone acetyltransferase [27–29]. In addition, GA affected in vivo regulation mechanism of small ubiquitin-related modifier (SUMO) and altered protein conformation, thereby affecting protein expression [30].
According to the above test, we suggested that the mechanism by which GA (C15:1) decayed GFP fluorescence was through conformation changes in the GFP protein. In addition, the mechanism by which GA promoted antibacterial activity against Gram-positive bacteria was through conformational changes of the proteins in the bacteria that inactivated the proteins and inhibited the growth of Gram-positive bacteria.
The results of crude cell lysate experiments showed that GFP fluorescence decay might be related to the interaction between GFP and GA. The GFP fluorescence in both Gram-negative and Gram-positive bacteria crude lysates was quenched by GA in a short period of time, which indicated that GFP fluorescence would be quenched as long as it had contact with GA and was not related to the microorganism tagged with GFP. Because the structure between Gram-negative and Gram-positive bacteria was similar and results showed that peptidoglycan in Gram-positive bacteria could not prevent GA (C15:1) from entering the cell, we suggested that the peptidoglycan structure of Gram-negative bacteria also could not block GA (C15:1) from entering the cell. In the protoplast experiment, a small amount of GA molecules could enter the cells after the peptidoglycan structure in E. coli cell wall was destroyed by lysozyme, which might be the result of the action of lysozyme. After the peptidoglycan structure was destroyed by lysozyme, pores might be present on the surface of the peptidoglycan layer that allowed GA molecules to pass through. However, only a small amount of GA molecules could enter the cells because the number of pores generated on the surface of the peptidoglycan layer was low, and the surface of Gram-negative bacteria was still covered by a large amount of lipids (including lipopolysaccharides and phospholipids), which could intercept a large amount of GA molecules. In order to further confirm lipid-soluble components in the cell wall of Gram-negative bacteria intercept the majority of GA molecules, the studies use high resolution electron microscopy to observe membrane change or other methods to study transport of GA through membrane will be carried out.
Some studies demonstrated that GA markedly inhibited the biofilm formation of S. mutans and Escherichia coli O157:H7, and disrupted biofilm integrity [24, 31]. Therefore, we speculate that the GA may affect the secondary metabolism of Gram-positive and Gram-negative bacteria. Due to the secondary metabolism of bacteria, such as the formation of biofilm, fluorescence formation and synthesis of antibiotics are regulated by quorum-sensing, further studies on this section will be investigated.
Conclusions
GA (C15:1) has a relatively strong selective antibacterial mechanism, which significant antibacterial activity against Gram-positive bacteria but little effect on the growth of Gram-negative bacteria. Additional studies on antibacterial mechanisms showed that GA (C15:1) could inhibit the activities of the selected proteins to a certain degree, and non-selectively induce protein conformational changes. GA (C15:1) also inhibit DNA replication in vivo as well as RNA transcription and protein synthesis. Thus, we suggested that the mechanism by which GA (C15:1) promoted antibacterial activity against Gram-positive bacteria was through conformational changes of the proteins in the bacteria that inactivated the proteins and inhibited the growth of Gram-positive bacteria. The research results indicated that lipid-soluble components (including lipopolysaccharide and phospholipids) in the cell wall of Gram-negative bacteria intercepted the majority of GA molecules, whereas the peptidoglycan layer in the cell wall showed a reduced capacity to intercept GA molecules.
Methods
Media and reagents
Lysogeny broth (LB) medium (g L−1) was composed as follows: peptone 10.0 g L−1, yeast extract 5.0 g L−1, NaCl 10.0 g L−1, pH 7.2, (solid, addition of 1.5% agar), and deionized water 1000 mL, which was sterilized at 121 °C for 20 min.
Proteinase K, lysozyme, ampicillin (Amp), kanamycin (Km), gentamicin (Gm), isopropyl-β-D-thiogalactopyranoside (IPTG), o-nitrophenyl β-D-galactopyranoside (ONPG), and o-nitrophenol (ONP) were purchased from Shanghai Sangon Biotech (Sangon Biotech, Shanghai, China). GA C15:1 standard was purchased from Shanghai Tauto Biotechnology (Shanghai, China). Other chemical reagents were analytical grade.
Strains
Escherichia coli DH5α (E. coli DH5α) (ATCC53338), E. coli O157: H7 (E. coli O157: H7) (ATCC43895), Pseudomonas putida KT2440 (ATTC47054), Pseudomonas aeruginosa PAO1 (ATCC15692), Ralstoniasolanacearum (ATCC11696), Rhodococcusjostii RHA1 [32], Streptococcus thermophilus ND03 [33], and S. aureus (ATCC25923) were from our laboratory.
Bacillus amyloliquefaciens SQR9 (CGMCC 5808; China General Microbiology Culture Collection Center) [34], E. coli-gfp (E. coli DH5α-gfp), P. putida KT2440-gfp, and B. amyloliquefaciens SQR9-gfp were provided by the Environmental Microbiology Lab at the College of Resources and Environment, Nanjing Agricultural University.
Determination of antibacterial activity of GA
A conventional broth-dilution method was adopted [35]. GA and salicylic acid were dissolved into dimethylsulfoxide (DMSO) to prepare stock solutions with different concentrations, respectively. The final concentrations of the drug (GA or salicylic acid) in the medium (500 μg mL−1, 250 μg mL−1, 100 μg mL−1, 80 μg mL−1, 60 μg mL−1, 40 μg mL−1, 20 μg mL−1, 10 μg mL−1, 5 μg mL−1, 2 μg mL−1, 1 μg mL−1, 0.5 μg mL−1 and 0.1 μg mL−1) were obtained. 10 μL stock solutions were added to 3 mL liquid LB medium that was inoculated with bacteria. All of the tests were incubated at 200 rpm. E. coli was cultured at 37 °C for 2 d, whereas other bacteria were cultured at 30 °C for 2 d. The lowest concentration without turbidity was defined as the minimum inhibitory concentration (MIC) of that substance. The medium without turbidity (50 μL) was inoculated with 3 mL of fresh LB liquid medium, and cultured in a shaker. E. coli was cultured at 37 °C for 2 d, and all other bacteria were cultured at 30 °C for 2 d. The lowest concentration without turbidity was defined as the minimum bactericidal concentration (MBC) of that substance. Same concentration of DMSO without GA and salicylic acid was added in control groups.
Measurement of fluorescence decay of GFP in bacteria
Single colonies of E. coli DH5α-gfp, P. Putida KT2440-gfp and B. amyloliquefaciens SQR9-gfp were extracted from solid LB plates, inoculated in liquid LB medium, and then 50 μg mL−1 Amp, Gm and Km was added to the medium to maintain the normal replication of plasmids in each strain. The medium was then centrifuged, and the bacteria were collected. The supernatant was discarded, and phosphate buffer was added to the collected bacteria to obtain the bacteria concentration of 108 CFU mL−1. GA was then dissolved in DMSO to prepare stocks with different concentrations.
10 μL of GA stock solution at different concentrations was added to 1 mL of bacteria solution. The final concentrations of GA in the solution were 500 μg mL−1, 250 μg mL−1, 100 μg mL−1, 50 μg mL−1, 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 10 μL of DMSO without the drug was used as a control. All of the tests were incubated at 30 °C for 1 min before GFP fluorescence was measured. The morphology of the bacteria was monitored under a scanning electron microscopy. For strains of E. coli DH5α-gfp and P. Putida KT2440-gfp, the incubation was extended to 4 h and additional samples were collected to measure GFP fluorescence. The morphology of the bacteria was measured using a scanning electron microscopy. Bacteria GFP fluorescence was initially observed by the naked eye using an LB16 Maestrogen UltraSlim no-damage blue LED transilluminator. Measurements of bacterial GFP fluorescence were performed using a Spectra ax M5 multifunctional microplate reader. The samples’ fluorescence intensity was measured when the excitation wavelength was 488 nm and the emission wavelength was 509 nm. The blank was the E. coli bacteria solution without GFP.
1 mL of bacteria solution was lysed by sonication. The broken bacteria were centrifuged at 12,000 × g, 4 °C for 20 min, and then the supernatant was collected. GA stock solution was added to the supernatant. The final concentrations of GA in the bacteria solution were 500 μg mL−1, 250 μg mL−1, 100 μg mL−1, 50 μg mL−1, 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 10 μL of DMSO (without the drug) was used as a control. All of the tests were incubated at 30 °C for 1 min before GFP fluorescence was measured. The control was the crude enzyme solution of the corresponding bacteria without GFP.
Effect of GA on the activities of a variety of proteins
Effect of GA on the activity of Taq DNA polymerase
Stock solutions at different concentrations were prepared by dissolving GA in DMSO. GA stock solutions (0.5 μL) at different concentrations were then added into each PCR reaction system. The final concentrations of GA in the reaction system were 25 μg mL−1, 10 μg mL−1, 5 μg mL−1 and 1 μg mL−1, respectively. 0.5 μL of DMSO (without the drug) was as a control. The PCR reaction conditions were adopted the reference of Chester and Marshak [36]. After the reaction was finished, 3 μL of PCR product was analyzed on an appropriate agarose gel. GelRed nucleic acid dye was used to stain the gel.
Effect of GA on the activity of restriction enzymes
Three common restriction enzymes (Kpn I, Hind III and EcoR I) were selected as the targets. GA was dissolved in DMSO to prepare stock solutions with different concentrations. A single colony of E. coli that carried the pUC19 plasmid, was extracted and inoculated in 3 mL of LB medium containing Amp, which was then cultured at 37 °C overnight with vigorous shaking. 41.5 μL of pUC19 (200 ng μg−1), 5 μL of 10 × restriction enzyme buffer, 3 μL of corresponding restriction enzyme, and 0.5 μL of GA stock solution with different concentrations were mixed. The final concentrations of GA in the reaction system were 25 μg mL−1, 10 μg mL−1, 5 μg mL−1 and 1 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All of the components in the reaction systems were digested at 37 °C for 4 h, and then the temperature was increased to 75 °C for 15 min to stop the enzyme digestion. Electrophoresis was then carried out to examine the enzyme digestion.
Effect of GA on the activity of superoxide dismutase
According to the method by Beauchamp et al. [37], GA was dissolved in DMSO to prepare stock solutions with different concentrations. 80 μmol L -1riboflavin, 77 μmol L -1 nitro blue tetrazolium (NBT), 13 mmol L−1 methionine, 0.1 mmol L−1 EDTA, 20 μL superoxide dismutase (SOD) enzyme solution (1 μg mL−1), and 5 μL of the different concentrations of GA stock solution (final concentrations of GA in the reaction system of 25 μg mL−1, 10 μg mL−1, 5 μg mL−1 and 1 μg mL−1) were mixed. 5 μL of DMSO (without the drug) was used as a control. After the samples were exposed to light at 4500 Lux light intensity for 15 min, the reaction was stopped by shielding the light. The optical density (OD)560 values were measured. One active unit (U) occurred when NBT was inhibited by 50%, and enzyme activity = (△ A × N × 60)/(W × T × V × 50%), where △ A represented the difference in OD values between the control and sample, N represented the total volume of enzyme solution, W represented the protein mass, T represented the light reaction time and V represented the volume of enzyme solution added. Enzyme activity was represented as OD560 • mg−1 (pro) • min−1.
Effect of GA on the activity of β-galactosidase
2 μg mL−1 the enzyme β-galactosidase was prepared in 10 mM pH 7.0 phosphate buffer. GA was dissolved in DMSO to prepare the stock solutions with different concentrations. Enzyme solution (1 mL) was incubated at 37 °C for 5 min, then 1 mL of phosphate buffer (pH 7.0) containing 20 mM ONPG preheated to 37 °C was added, finally 5 μL of the different concentrations of GA stock solution were added. The final concentrations of GA in the reaction system were 25 μg mL−1, 10 μg mL−1, 5 μg mL−1 and 1 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All of the samples were incubated in a 37 °C for 10 min, and then 3 mL of 0.5 mol L−1 Na2CO3 was added to stop the reaction. The OD value at 420 nm was read. One enzyme unit was defined as the amount of enzyme required to release 1 μmol of ONP per minute at 37 °C.
Inhibition of isotopic precursor incorporation
According to the method by Aspedon and Groisman [21], [3H] TdR, [3H] UR and [3H] Tyr were incorporated into precursors to investigate effect of GA (C15:1) on the biosynthesis of DNA, RNA and B. amyloliquefaciens SQR9 protein. The logarithmic growth phase SQR9 culture was diluted with sterile water. In a 96-well plate, 0.9 mL of bacterial suspension (OD600 = 0.1) was added to each well. The isotope-labeled precursor (final concentration was 0.5 μCi mL−1) and 5 μL of the different concentration of GA (C15: 1) were also added into the well. The final concentrations of GA in the reaction system were 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All of the treatments were conducted at 37 °C in a shaker. When examining effect of GA (C15:1) on the synthesis of DNA and RNA, culture time for bacteria was limited to one generation. After growing the culture for 30 min, the culture was centrifuged at 12,000 × g, 4 °C for 5 min to harvest the bacteria pellet. Because protein synthesis was relatively slow, the bacteria culture time was extended to 2 h. OD600 was also adjusted so that the cultures had the same bacteria concentration. The bacteria pellet was washed three times with phosphate buffer and then placed in an oven overnight to dry it. Scintillation solution was directly added to the Eppendorf tubes containing bacteria, and the tubes were then transferred to a scintillation counter (LS3801, Beckman) to determine the counts per minute (CPM) values. The average CPM values of the experimental group and control group were compared. The incorporation inhibition rate was calculated. Incorporation inhibition rate = control group CPM - experimental group CPM/control group CPM × 100%.
Effect of GA (C15:1) on GFP fluorescence in the protoplasts of E. coli
E. coli protoplasts were prepared according to the method described by Weiss [38]. GA was dissolved in DMSO, and its stock solutions with different concentrations were prepared. E. coli protoplasts (1 mL) were diluted with sucrose-magnesium-maleate (SMM) buffer (protoplast number > 107 CFU mL−1), and 5 μL of the different concentration GA stock solutions were added. The final concentrations of GA in the reaction system were 500 μg mL−1, 250 μg mL−1, 100 μg mL−1, 50 μg mL−1, 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All the tests were incubated at 30 °C for 1 min, and then the samples were analyzed for GFP fluorescence. GFP fluorescence was initially observed by the naked eye using an LB-16 Maestrogen UltraSlim no-damage blue LED transilluminator. GFP fluorescence was measured with a Spectra ax M5 multifunctional microplate reader. The samples’ fluorescence intensity was measured when the excitation wavelength was 488 nm and the emission wavelength was 509 nm. The blank was E. coli protoplast solution without GFP.
The interception of GA by the lipid-soluble component in E. coli cell walls
E. coli DH5α-gfp was cultured in 100 mL of liquid LB medium at 37 °C, 200 rpm overnight. After centrifugation at 12,000 × g for 5 min, the supernatant was discarded to collect the bacteria. The bacteria pellet was then re-suspended in the same volume of phosphate buffer. The half was retained for the experiments and the left half was centrifuged at 12,000 × g for 5 min. The supernatant was then discarded. A small volume of phosphate buffer was used to re-suspend the bacteria pellet, and then 10 mL of 70% ethanol solution was added, mixed well, and kept at room temperature for 30 s before centrifuging at 12,000 × g for 1 min. The supernatant ethanol solution was then discarded. Phosphate buffer (50 mL) was used to re-suspend the bacteria pellet, which was centrifuged at 12,000 × g for 5 min. The supernatant was then discarded. Finally, 50 mL of phosphate buffer was used to re-suspend the bacteria pellet for the experiments.
GA was dissolved in DMSO, and its stock solutions with different concentrations were prepared. GA stock solutions (5 μL) at different concentrations were added to 1 mL of bacteria solution before/after ethanol solubilization of the lipids. The final concentrations of GA in the reaction system were 500 μg mL−1, 250 μg mL−1, 100 μg mL−1, 50 μg mL−1, 25 μg mL−1, 10 μg mL−1 and 5 μg mL−1, respectively. 5 μL of DMSO (without the drug) was used as a control. All the tests were incubated at 30 °C for 1 min, and then the samples were examined for GFP fluorescence. GFP fluorescence was initially observed by the naked eye using an LB-16 Maestrogen UltraSlim no-damage blue LED transilluminator. GFP fluorescence was measured using a Spectra ax M5 multifunctional microplate reader. The samples’ fluorescence intensity was measured when the excitation wavelength was 488 nm and the emission wavelength was 509 nm. The blank was E. coli bacteria solution without GFP (after ethanol solubilization).
Statistical analyses
For all the experiments throughout the study, we collected three individual tubes from LB plates, and each tube was performed in triplicate. In analysis, comparisons were carried out using the fluorescent means of each triplicate, then, finally each bar in the gram represents means of three individual tubes. The data of each strain was analyzed using one-way ANOVA (i.e., B. amyloliquefaciens SQR9-gfp E. coli DH5α-gfp and P. putida KT2440-gfp), with final concentration of GA as the fixed factors (P < 0.05). The experimental data were log transformed to meet the homogeneity of variance or a normal distribution of residuals. All statistical analyses were conducted using SPSS 13.0 (SPSS, Chicago, IL, USA).
Acknowledgements
It is a great pleasure to thank research fund for key projects in the national science & technology pillar program (2012BAD21B04) and the science and technology project of jiangsu province (BE2015315). The authors would like to acknowledge the support of the project funded by the priority academic program development (PAPD) of jiangsu higher education institutions and co-innovation entre for sustainable forestry in southern china. The authors also would like to thank Dr. Xie lulu of University of Rochester, Dr. Xiedong of Nanjing Forestry University for guidance in the data analysis.
Funding
This work was supported by research fund for key projects in the national science & technology pillar program (2012BAD21B04) and the science and technology project of jiangsu province (BE2015315).
Availability of data and materials
All data generated/analysed during the current study that are not already included in this published article, are available from the corresponding author on reasonable request.
Authors’ contributions
HZB did the experimental, analyses of experimental data, figures drawing, critical reading of the manuscript and writing of the manuscript; WCE and CFL were responsible for study conception and design, analysis and interpretation of data work; FGJ contributed to study conception, to analysis of experimental data and to the writing of the manuscript; TZX analyzed the data, supervised the statistical analyses and contributed to the writing of the manuscript. All authors discussed and commented the results and gave their final approval for submission.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
N/A.
Abbreviations
- [3H] TdR
(Methy-3H) thymine
- [3H] Tyr
3H-tyrosine
- [3H] UR
3H-uridine
- Amp
Ampicillin
- CFU
Colony-Forming Units
- CK
Control check
- CPM
Counts per minute
- DMSO
Dimethylsulfoxide
- DNA
Deoxyribonucleic acid
- EDTA
Ethylenediaminetetraacetic acid
- GA
Ginkgo acid
- GFP
Green fluorescent protein
- Gm
Gentamicin
- IPTG
Isopropyl-β-D-thiogalactopyranoside
- Km
Kanamycin
- LB
Lysogeny broth
- LED
Light emitting diode
- MBC
Minimum bactericidal concentration
- MIC
Minimum inhibitory concentration
- NBT
Nitro blue tetrazolium
- OD
Optical density
- ONP
O-nitrophenol
- ONPG
O-nitrophenyl-β-D-galactopyranoside
- PCR
Polymerase chain reaction
- RNA
Ribonucleic acid
- SMM
Sucrose-magnesium-maleate
- SOD
Superoxide dismutase
Contributor Information
Zhebin Hua, Email: huazhebin@126.com.
Caie Wu, Phone: +86-25-85427844, Email: wucaie@njfu.edu.cn.
Gongjian Fan, Email: fangongjian@njfu.edu.cn.
Zhenxing Tang, Email: tangzhenxing@126.com.
Fuliang Cao, Email: fuliancaonjfu@163.com.
References
- 1.Dixon RA, Strack D. Phytochemistry meets genome analysis, and beyond. Phytochemistry. 2003;62(6):815–6. doi: 10.1016/S0031-9422(02)00712-4. [DOI] [PubMed] [Google Scholar]
- 2.Toni M, Kutchan Ecological arsenal and developmental dispatcher. The paradigm of secondary metabolism. Plant Physiol. 2001;125(1):58–60. doi: 10.1104/pp.125.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowan MM. Plant products as antimicrobial agents. Clin Microbiol Rev. 1999;12(4):564–82. doi: 10.1128/cmr.12.4.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li JWH, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325(5937):161–5. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 5.Zhao J, Davis LC, Verpoorte R. Elicitor signal transduction leading to production of plant secondary metabolites. Biotechnol Adv. 2005;23(4):283–333. doi: 10.1016/j.biotechadv.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Wilkins KM, Board RG, Gould GW: Mechanisms of action of food preservation procedures. London: Elservierapplied Science EA; 1989.
- 7.Wilson CL, Solar JM, EI-Ghaouth A, Wisniewski ME. Rapid evaluation of plant extracts and essential oils for antifungal activity against Botrytis cinerea. Plant Dis. 1997;81(2):204–10. doi: 10.1094/PDIS.1997.81.2.204. [DOI] [PubMed] [Google Scholar]
- 8.Erin AN, Davitashvili NG, Prilipko LL, Boldyrev AA, Lushchak VI, Batrakov SG, Pridachina NN, Serbinova AE, Kagan VE. Influence of alkylresorcin on biological membranes during activation of lipid peroxidation. Biokhimiia. 1987;52:1180–5. [PubMed] [Google Scholar]
- 9.Kaprelyants AS, Suleimenov MK, Sorokina AD, Deborin GA, El-Registan GI, Stoyanovich FM, Lille YE, Ostrovsky DN. Structural functional changes in bacterial and model membranes induced by phenolic lipids. Biol Membr (Moscow) 1987;4:254–61. [Google Scholar]
- 10.Kubo I, Komatsu S, Ochi M. Molluscicides from the cashew Anacardium occidentale and their large-scale isolation. J Agr Food Chem. 1986;34(6):970–3. doi: 10.1021/jf00072a010. [DOI] [Google Scholar]
- 11.Muroi H, Kubo I. Antibacterial activity of anacardic acid and totarol, alone and in combination with methicillin, against methicillin-resistant Staphylococcus aureus. J Appl Microbiol. 1996;80(4):387–94. doi: 10.1111/j.1365-2672.1996.tb03233.x. [DOI] [PubMed] [Google Scholar]
- 12.Himejima M, Kubo I. Antibacterial Agents from the Cashew Anacardium occidentale (Anacardiaceae) Nut Shell Oil. J Agric Food Chem. 1991;39:418–21. doi: 10.1021/jf00002a039. [DOI] [Google Scholar]
- 13.Boonkaew T, Camper ND. Biological activities of Ginkgo extracts. Phytomedicine. 2005;12(4):318–23. doi: 10.1016/j.phymed.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Chen JJ, Zhang T, Jiang B, Mu WM, Miao M. Characterization and antioxidant activity of Ginkgo biloba exocarp polysaccharides. Carbohydr Polym. 2012;87(1):40–5. doi: 10.1016/j.carbpol.2011.06.083. [DOI] [PubMed] [Google Scholar]
- 15.van Beek TA, Montoro P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J Chromatogr A. 2009;1216(11):2002–32. doi: 10.1016/j.chroma.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Ahlemeyer B, Krieglstein J. Neuroprotective effects of Ginkgo biloba extract. Cell Mol Life Sci. 2003;60(9):1779–92. doi: 10.1007/s00018-003-3080-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satyan KS, Jaiswal AK, Ghosal S, Bhattacharya SK. Anxiolytic activity of ginkgolic acid conjugates from Indian Ginkgo biloba. Psychopharmacology (Berl) 1998;136(2):148–52. doi: 10.1007/s002130050550. [DOI] [PubMed] [Google Scholar]
- 18.Yang XM, Ye YR, Wang P, Chen J, Guo T. Study on anti-bacterium activities of extract of Ginkgo biloba leaves (EGbs) and Ginkgolic Acids (GAs) Food Sci. 2004;25(4):68–71. [Google Scholar]
- 19.Itokawa H, Totsuka N, Nakahara K, Takeya K, Lepoittevin JP, Asakawa Y. Antitumor principles from Ginkgo biloba L. Tokyo: JAACC; 1987. [DOI] [PubMed] [Google Scholar]
- 20.Ni XW, Wu MC. Study on isolation identification and the antibacterial activity of ginkgolic acids. Food Sci. 2004;25(9):59–63. [Google Scholar]
- 21.Aspedon A, Groisman EA. The antibacterial action of protamine: evidence for disruption of cytoplasmic membrane energization in Salmonella typhimurium. Microbiology. 1996;142(pt12):3389–97. doi: 10.1099/13500872-142-12-3389. [DOI] [PubMed] [Google Scholar]
- 22.Begum P, Hashidoko Y, Islam MT, Ogawa Y, Tahara S. Zoosporicidal activities of anacardic acids against Aphanomyces cochlioides. Z Naturforsch C: Biosci. 2002;57(9–10):874–82. doi: 10.1515/znc-2002-9-1020. [DOI] [PubMed] [Google Scholar]
- 23.Choi JG, et al. Antibacterial activity of hydroxyalkenyl salicylic acids from sarcotesta of Ginkgo biloba against vancomycin-resistant Enterococcus. Fitoterapia. 2009;80:18–20. doi: 10.1016/j.fitote.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 24.He J, Wang S, Wu T, Cao Y, Xu X, Zhou X. Effects of ginkgoneolic acid on the growth, acidogenicity, adherence, and biofilm of Streptococcus mutans in vitro. Folia Microbiol (Praha) 2013;58:147–53. doi: 10.1007/s12223-012-0191-9. [DOI] [PubMed] [Google Scholar]
- 25.Jefferson RA, Kavanagh TA, Bevan MW. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987;20:3901–7. doi: 10.1002/j.1460-2075.1987.tb02730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S, Hazelrigg T. Implications for bcd mRNA localization from spatial distribution of exu protein in Drosophila oogenesis. Nature. 1994;369:400–3. doi: 10.1038/369400a0. [DOI] [PubMed] [Google Scholar]
- 27.Ahlemeyer B, Selke D, Schaper C, Klumpp S, Krieglstein J. Ginkgolic acids induce neuronal death and activate protein phosphatase type-2C. Eur J Pharmacol. 2001;430(1):1–7. doi: 10.1016/S0014-2999(01)01237-7. [DOI] [PubMed] [Google Scholar]
- 28.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278(19):134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 29.Grazzini R, Hesk D, Heininger E, Hildenbrandt G, Reddy CC, Cox-Foster D, Medford J, Craig R, Mumma RO. Inhibition of lipoxygenase and prostaglandin endoperoxide synthase by anacardic acids. Biochem Bioph Res Co. 1991;176(2):775–80. doi: 10.1016/S0006-291X(05)80252-9. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, Kimura K, Sodeoka M, Yoshida M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol. 2009;16(2):133–40. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Lee JH, Kim YG, Ryu SY, Cho MH, Lee J. Ginkgolic acids and Ginkgo biloba extract inhibit Escherichia coli O157:H7 and Staphylococcus aureus biofilm formation. Int J Food Microbiol. 2014;174:47–55. doi: 10.1016/j.ijfoodmicro.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Seto M, Kimbara K, Shimura M, Hatta T, Fukuda M, Yano K. A novel transformation of polychlorinated biphenyls by Rhodococcus sp. strain RHA1. Appl Environ Microbiol. 1995;61:3353–8. doi: 10.1128/aem.61.9.3353-3358.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Z, et al. Identification and characterization of the dominant lactic acid bacteria from kurut: the naturally fermented yak milk in Qinghai, China. J Gen Appl Microbiol. 2010;56:1–10. doi: 10.2323/jgam.56.1. [DOI] [PubMed] [Google Scholar]
- 34.Rosado A, Duarte GF, Seldin L. Optimization of electroporation procedure to transform B. polymyxa SCE2 and other nitrogen-fixing Bacillus. J Microbiol Methods. 1994; 19(1–11). doi: 10.1016/0167-7012(94)90020-5.
- 35.Kubo I, Muroi H, Kubo A. Structural functions of antimicrobial long-chain alcohols and phenols. Bioorgan Med Chem. 1995;3(7):873–80. doi: 10.1016/0968-0896(95)00081-Q. [DOI] [PubMed] [Google Scholar]
- 36.Chester N, Marshak DR. Dimethyl sulfoxide-mediated primer Tm reduction: a method for analyzing the role of renaturation temperature in the polymerase chain reaction. Anal Biochem. 1993;209(2):284–90. doi: 10.1006/abio.1993.1121. [DOI] [PubMed] [Google Scholar]
- 37.Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44(1):276–87. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 38.Weiss RL. Protoplast formation in Escherichia coli. J Bacteriol. 1976;128(2):668–70. doi: 10.1128/jb.128.2.668-670.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated/analysed during the current study that are not already included in this published article, are available from the corresponding author on reasonable request.