

Abstract
Aim
This open‐label study investigated the effect of belatacept on cytokine levels and on the pharmacokinetics of caffeine, losartan, omeprazole, dextromethorphan and midazolam, as CYP probe substrates after oral administration of the Inje cocktail in healthy volunteers.
Methods
Twenty‐two evaluable subjects received the Inje cocktail on Days 1, 4, 7 and 11 and belatacept infusion on Day 4.
Results
Since belatacept caused no major alterations to cytokine levels, there were no major effects on CYP‐substrate pharmacokinetics, except for a slight (16–30%) increase in omeprazole exposure, which was probably due to omeprazole‐mediated, time‐dependent CYP inhibition. Belatacept did not cause major alterations in the pharmacokinetics, as measured by the geometric mean ratios and associated 90% confidence interval for area under the plasma concentration ‐time curve from time zero to infinity on Day 7 comparing administration with and without belatacept for caffeine (1.002 [0.914, 1.098]), dextromethorphan (1.031 [0.885, 1.200]), losartan (1.016 [0.938, 1.101)], midazolam (0.968 [0.892, 1.049]) or their respective metabolites.
Conclusions
Therefore, no dose adjustments of CYP substrates are indicated with belatacept coadministration.
Keywords: Biologics, cytochrome P450, interaction, pharmacokinetics, transplantation
What is Already Known about this Subject
The effect of therapeutic proteins on the pharmacokinetics of cytochrome P450 (CYP)‐substrate drugs is poorly understood and has not been evaluated for belatacept.
The effect of drugs (including therapeutic proteins) on CYP activities can be studied in vivo using a cocktail probe approach.
What this Study Adds
Since belatacept caused no major alterations to cytokine levels, there were no clinically relevant pharmacokinetic effects on CYP substrates.
The self‐mediated, time‐dependent CYP2C19 inhibition by omeprazole suggests that multiple doses of this agent are not recommended in drug–drug interaction studies, unless there is sufficient washout time.
SimCYP pharmacokinetic modelling can be used to facilitate data interpretation.
Coadministration of belatacept and the Inje cocktail is safe and well tolerated, supporting the feasibility of applying this methodology for assessing the potential drug interactions of therapeutic proteins.
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets 2 | Enzymes 3 |
| CD80 | CYP1A2 |
| CD86 | CYP2C9 |
| CTLA‐4 | CYP2C19 |
| Tumour necrosis factor | CYP2D6 |
| CYP3A4 | |
| Cytochrome P450 | |
| LIGANDS | |
|---|---|
| Belatacept | IL‐6 |
| Caffeine | IL‐10 |
| Dextromethorphan | Losartan |
| Dextrorphan | Midazolam |
| EXP3174 | Omeprazole |
| IFN‐gamma | Tocilizumab |
| IL‐2 | |
These Tables lists key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Belatacept (Nulojix®; Bristol‐Myers Squibb, Princeton, New Jersey) is a recombinant fusion protein and selective T‐cell costimulation blocker indicated for the prevention of organ rejection in Epstein–Barr virus‐positive kidney transplant recipients 4. The protein consists of the modified extracellular domain of human cytotoxic T‐lymphocyte antigen 4 fused to a portion of the constant‐region fragment of human immunoglobulin G1, and binds to CD80 and CD86 4.
Drug interactions are an important consideration in the transplant setting due to widespread polypharmacy and the presence of age‐related changes that increase the likelihood of drug‐related adverse events 5. The potential for drug interactions involving therapeutic proteins was not initially recognized, as therapeutic proteins, unlike many small‐molecule immunosuppressant drugs, are not metabolized by cytochrome P450 (CYP) enzymes 6. However, it is now known that therapeutic proteins, which are proinflammatory cytokines or cytokine modulators (e.g. monoclonal antibodies) can alter CYP expression or function and thereby interact with drugs that are CYP substrates 7, 8. Endogenous cytokines produced in response to infection and disease can downregulate transcription of specific CYP genes, and clinically relevant interactions have been reported when therapeutic cytokines such as interferon (IFN)‐α are administered with drugs that are CYP substrates 6. Conversely, therapeutic monoclonal antibodies may reverse cytokine‐mediated suppression of CYP in inflammatory diseases, leading to increased clearance of CYP substrates 6. This has been demonstrated by reduced exposure to omeprazole and simvastatin in patients with rheumatoid arthritis treated with tocilizumab, an antibody that targets interleukin (IL)‐6 6, 9.
The current Food and Drug Administration guidance on drug interaction studies recommends that therapeutic proteins that are cytokines or cytokine modulators be investigated to determine their effect on CYP enzymes or transporters 10. Belatacept has been shown to inhibit production of certain cytokines during an in vitro alloimmune response, and modulation of cytokine pathways responsible for regulating CYP expression is a common means by which therapeutic proteins may influence CYP activity 8. As the half‐life of belatacept in healthy individuals approaches 10 days (t1/2 = 9.8) 4, a single administered dose could potentially affect CYP pharmacokinetics during this time period. However, the effect of therapeutic proteins on the pharmacokinetics of CYP‐substrate drugs remains difficult to predict, and the mechanisms involved are poorly understood 6. Assessing the risk of drug interactions requires consideration of the potential for either a direct mechanistic impact on CYP or an indirect interaction with CYP substrates which occurs as part of the disease 11. During an in vitro alloimmune response, belatacept was found to inhibit the production of certain cytokines 4, but the potential for belatacept to alter in vivo exposure to drugs that are CYP substrates has not been evaluated.
The effect of multiple drugs on CYP activities can be simultaneously studied in vivo using a cocktail probe approach 12, but few such studies have been performed using therapeutic proteins. Here we report the novel methodology and findings of the first drug–drug interaction study to investigate the effect of belatacept on the pharmacokinetics of caffeine, losartan, omeprazole, dextromethorphan and midazolam. These substrates were administered orally and simultaneously to healthy volunteers as the Inje cocktail, a validated combination of sensitive probe substrates for identifying the in vivo enzyme activities of CYP1 A2, CYP2C9, CYP2C19, CYP2D6 and CYP3 A4, respectively 12. The effect of belatacept on the pharmacokinetics of these probe substrates was evaluated multiple times following the infusion of belatacept. In this way, it was possible to study the extent of the interaction when concentrations were at peak as well as during the elimination of the drug. The safety and tolerability of coadministration of belatacept and the Inje cocktail was a secondary objective. The impact of belatacept administration on cytokine levels was an exploratory objective.
Methods
Conduct of the study
This open‐label, nonrandomized, single‐sequence study was registered with ClinicalTrials.gov (NCT01766050). Institutional Review Board/Independent Ethics Committee approval was obtained, and the study was conducted in accordance with Good Clinical Practice principles and the Declaration of Helsinki. All participants provided written informed consent.
Subjects
Eligible subjects were men and women aged 18–45 years with a body mass index of 18–30 kg m−2, who were healthy as determined by medical history, physical examination, electrocardiogram (ECG) and clinical laboratory findings. Women of childbearing potential (and participating men with sexual partners in this category) were required to continue contraception for 90 days after the last dose of study medication. Exclusion criteria included known or suspected infection (or risk factors for developing infection), autoimmune disorders and a history or strong family history of malignancy. Prior exposure to belatacept, abatacept or leucocyte‐depleting agents (e.g. rituximab) was prohibited. The washout period was 3 months for hormonal contraceptive agents; 4 weeks (or five half‐lives, whichever was longer) for prescription drugs, St John's wort and live vaccine administration; 2 weeks for over‐the‐counter drugs and most herbal preparations; and 1 week for aspirin. Concomitant medications were not permitted during the study unless initiated by the investigator to treat adverse events. Subjects abstained from foods or beverages containing caffeine, as well as those known to inhibit or induce CYP1 A2, CYP2C9, CYP2C19, CYP2D6 or CYP3 A4 (Tables 1 and 2).
Table 1.
Study design
| Screening/ enrolment | IC 1 | IC 2 belatacept | IC 3 | IC 4 | Clinical furlough | Follow‐up | End of study | |
|---|---|---|---|---|---|---|---|---|
| Days −21 to −1 | Day 1 | Day 4 | Day 7 | Day 11 | Day 12 | Day 18 | Day 32 (± 2) | Day 46 (± 2) |
IC, Inje cocktail
Table 2.
Inje cocktail components
| Cytochrome P450 enzyme | Probe substrate in Inje cocktail | Metabolite |
|---|---|---|
| 1A2 | Caffeine (200 mg) | Paraxanthine |
| 2C9 | Losartan (50 mg) | E‐3174 |
| 2C19 | Omeprazole (40 mg) | 5‐hydroxyomeprazole |
| 2D6 | Dextromethorphan (30 mg) | Dextrorphan |
| 3A4 | Midazolam (5 mg) | 1′‐hydroxy‐midazolam |
Study design and treatment
Table 3 summarizes the study sequence. Participants were admitted to a clinical pharmacology unit on Day −1 for screening and remained at the clinic until furlough on Day 12, subsequently returning for two follow‐up visits. All subjects received the same drug regimen. In the morning on Days 1, 7 and 11, after fasting for at least 10 h, subjects were administered oral doses of the Inje cocktail, which consisted of caffeine (200‐mg tablet), losartan (50‐mg tablet), omeprazole (40‐mg delayed‐release capsule), dextromethorphan (30‐mg capsule) and midazolam (5‐mg oral syrup) as probe substrates for CYP1 A2, CYP2C9, CYP2C19, CYP2D6 and CYP3 A4, respectively. In the morning of Day 4, after fasting for at least 10 h, belatacept was administered as a single 10‐mg kg−1 intravenous infusion over 30 min, closely followed (within approximately 5 min) by the Inje cocktail (same doses listed). Belatacept 10 mg kg−1 was selected because it is the dose that is approved for use in the initial phase after kidney transplant 4. During dosing of the Inje cocktail, the oral dispenser used to administer midazolam was re‐filled twice with 10 ml of water, and then administered to the subject. In addition, at the time of the Inje cocktail dosing, 240 ml of water was administered to the subject along with the study medication. A physician was on site each day of dosing and remained on site for at least 4 h after each midazolam dose. Compliance was assessed by mouth check.
Table 3.
Disposition of subjects
| Data Set | Subjects, n (%) |
|---|---|
| Enrolled | 45 |
| Dosed | 22 |
| Completed study | 18 (81.8%) |
| Did not complete study | 4 (18.2%) |
| Adverse event | 1 (4.5%) |
| Withdrew consent | 2 (9.1%) |
| Lost to follow‐up | 1 (4.5%) |
| Safety analysis | 22 (100.0%) |
| Pharmacokinetic analysis | 22 (100.0%)a |
| Reduced pharmacokinetic data set for dextromethorphan a, a | 21 (95.5%) |
A reduced pharmacokinetic data set was defined for dextromethorphan and dextrorphan (one poor CYP2D6 metabolizer was excluded)
Table 4 details the Inje cocktail components and their metabolites, together with the relevant CYP subtype. Each drug in the Inje cocktail was sourced from a single commercially available lot.
Table 4.
Effect of belatacept on the pharmacokinetics of the Inje cocktail components
| Day 4 | Day 7 | Day 11 | |
|---|---|---|---|
| Omeprazole | |||
| C max | 1.260 (1.118, 1.421) | 1.292 (1.090, 1.531) | 1.178 (0.971, 1.429) |
| AUC 0–t | 1.159 (1.056, 1.272) | 1.228 (1.092, 1.381) | 1.215 (1.047, 1.410) |
| AUC 0–∞ | 1.193 (1.091, 1.304) | 1.227 (1.093, 1.379) | 1.300 (1.141, 1.482) |
| Caffeine | |||
| C max | 0.948 (0.880, 1.021) | 0.922 (0.857, 0.993) | 0.954 (0.885, 1.028) |
| AUC 0–t | 0.941 (0.874, 1.013) | 0.986 (0.902, 1.076) | 1.027 (0.942, 1.120) |
| AUC 0–∞ | 0.939 (0.868, 1.017) | 1.002 (0.914, 1.098) | 1.036 (0.940, 1.142) |
| Losartan | |||
| C max | 1.081 (0.914, 1.278) | 1.158 (1.003, 1.338) | 1.152 (0.988, 1.342) |
| AUC 0–t | 1.013 (0.944, 1.088) | 1.016 (0.936, 1.103) | 1.001 (0.893, 1.121) |
| AUC 0–∞ | 1.011 (0.942, 1.085) | 1.016 (0.938, 1.101) | 1.002 (0.896, 1.121) |
| Dextromethorphan | |||
| C max | 0.863 (0.746, 0.997) | 0.922 (0.793, 1.071) | 0.856 (0.709, 1.034) |
| AUC 0–t | 0.915 (0.817, 1.024) | 1.003 (0.856, 1.175) | 0.963 (0.821, 1.130) |
| AUC 0–∞ | 0.877 (0.783, 0.982) | 1.031 (0.885, 1.200) | 1.022 (0.839, 1.245) |
| Midazolam | |||
| C max | 0.991 (0.898, 1.093) | 0.911 (0.830, 1.000) | 0.994 (0.885, 1.116) |
| AUC 0–t | 1.050 (0.976, 1.130) | 0.966 (0.889, 1.049) | 1.033 (0.950, 1.123) |
| AUC 0–∞ | 1.049 (0.975, 1.127) | 0.968 (0.892, 1.049) | 1.031 (0.948, 1.121) |
Data are point estimates for the geometric mean ratio of the test day vs. Day 1 (90% confidence interval). AUC, area under the plasma concentration‐time curve; Cmax, maximum concentration
Pharmacogenomics
Using a blood sample collected on Day −1, genotyping was performed for polymorphic CYP2C9, CYP2C19 and CYP2D6 to identify any poor metabolizers of the Inje cocktail components in an effort to guide data interpretation.
Pharmacokinetic analysis and study end points
Blood samples for serum belatacept pharmacokinetics were taken pre‐ and post‐dose on Day 4 and on Days 5, 11 and 18. Blood samples for the assessment of plasma concentrations of the Inje cocktail components (caffeine, losartan, omeprazole, dextromethorphan and midazolam) and their metabolites (paraxanthine, E‐3174, 5‐hydroxyomeprazole, dextrorphan and 1′‐hydroxy‐midazolam, respectively) were collected over 24 h on Days 1, 4, 7 and 11 (0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12 and 24‐h postdosing).
The primary end point was maximum serum concentration (Cmax), area under the plasma concentration–time curve (AUC) from time zero to the time of last quantifiable concentration (AUC0–t), and AUC from time zero to infinity (AUC0–∞) of each Inje cocktail component, with and without administration of belatacept. Cmax, AUC0–t and AUC0–∞ of the metabolites, as well as the molecular weight‐corrected metabolite‐to‐drug ratio of these parameters, were secondary end points. Point estimates of the geometric mean ratios were calculated for the Inje cocktail components, the metabolites and metabolite‐to‐drug ratios to compare Days 4, 7 and 11 with Day 1, respectively, in the evaluable pharmacokinetic population.
All reported results for belatacept and the Inje cocktail data were conducted in compliance with applicable standard operating procedures in place at the time of the analysis. For the summaries of belatacept and the Inje cocktail plasma concentration–time data, concentrations that were less than the lower limit of quantification (LLOQ) were treated as missing in summary tables and plots. For pharmacokinetic calculations, predose concentrations and concentrations prior to the first quantifiable concentration that were less than the LLOQ were set to zero. All other concentrations less than LLOQ were set to missing.
Pharmacokinetics parameters were derived from plasma concentration vs. time data using noncompartmental analysis (WinNonlin® Professional Network Edition, Version 6.2.1; Pharsight Corp, St Louis, MO, USA).
Pharmacokinetic assay performance
A liquid chromatography and tandem mass spectrometry method was developed and validated to simultaneously determine the concentrations: of dextromethorphan; losartan and its major active metabolites, losartan carboxylic acid and E‐3174; midazolam and 1′‐hydroxymidazolam; omeprazole and its major active metabolite, 5‐hydroxyomeprazole; and caffeine and its major active metabolite, 1,7‐dimethylxanthine (paraxanthine) to support pharmacokinetic analyses. All reported results for caffeine, losartan, omeprazole, dextromethorphan, midazolam, paraxanthine, E‐3174, 5‐hydroxyomeprazole, dextrorphan and 1′‐hydroxy‐midazolam concentrations in plasma were generated using appropriate calibration curves and quality control samples that met pre‐established acceptance criteria and were conducted in compliance with applicable standard operating procedures in place at the time of analysis. A summary of assay performance for caffeine, losartan, omeprazole, dextromethorphan, midazolam, paraxanthine, E‐3174, 5‐hydroxyomeprazole, dextrorphan and 1′‐hydroxy‐midazolam concentrations in plasma is presented in Table 5.
Table 5.
Summary of bioanalytical assay performance for components of the Inje cocktail and metabolites
| Analyte | Matrix | LLOQ (ng ml −1 ) | ULOQ (ng ml −1 ) | Between‐run %CV a | Within‐run %CV a | Mean % deviation from nominal concentration a |
|---|---|---|---|---|---|---|
| Caffeine | Plasma | 25.0 | 25 000 | ≤8.17 | ≤35.1 | ±2.07 |
| Losartan | Plasma | 0.500 | 500 | ≤8.60 | ≤20.6 | ±5.89 |
| Omeprazole | Plasma | 1.00 | 1000 | ≤7.74 | ≤17.0 | ±1.78 |
| Dextromethorphan | Plasma | 0.0500 | 50.0 | ≤9.50 | ≤22.9 | ±8.70 |
| Midazolam | Plasma | 0.100 | 100 | ≤3.65 | ≤4.90 | ±2.90 |
| Paraxanthine | Plasma | 25.0 | 25 000 | ≤8.14 | ≤36.1 | ±9.45 |
| E‐3174 | Plasma | 0.500 | 500 | ≤8.37 | ≤20.9 | ±6.03 |
| 5‐hydroxyomeprazole | Plasma | 1.0 | 1000 | ≤6.41 | ≤15.5 | ±2.70 |
| Dextrorphan | Plasma | 0.800 | 800 | ≤11.5 | ≤23.8 | ±3.19 |
| 1′‐hydroxymidazolam | Plasma | 0.100 | 50.0 | ≤6.47 | ≤11.2 | ±3.99 |
Maximum value from analytical quality controls
CV, coefficient of variation; LLOQ, lower limit of quantitation; ULOQ, upper limit of quantitation.
Exploratory biomarker analyses
Serum samples for measuring cytokine levels were collected on Days −1, 1, 4, 7, 11, 18, 32 and 46; samples were taken prior to dosing on Days 1, 4, 7 and 11. Tumour necrosis factor‐α, IFN‐α, IFN‐γ, IL‐2, IL‐6 and IL‐10 were quantified by a validated multiplex immunoassay on the Luminex platform. Whole‐blood samples were collected on Days 1 (predose), 7 (predose) and 18.
Pharmacokinetic modelling of omeprazole accumulation
A physiologically based pharmacokinetic model of omeprazole and 5‐hydroxyomeprazole was developed using SimCYP (v.13; Certara, St Louis, MO, USA). This model was developed after the initial clinical study and was used to evaluate the hypothesis that the observed increase in omeprazole AUC and observed decrease in 5‐hydroxyomeprazole AUCm/AUCp were caused by omeprazole‐mediated, time‐dependent inhibition of its own CYP‐mediated elimination rather than by belatacept. The same regimen of omeprazole used in the healthy volunteer study was used for the model, but excluded belatacept; this model predicted accumulation after repeated doses of omeprazole 40 mg alone on Days 1, 4, 7 and 11 for the study population. Full model development methodology and model parameter values are provided in the supplemental material.
Safety
Safety and tolerability were secondary objectives. Blood and urine for haematology, chemistry and urinalysis assessment were collected at the screening; Days −1, 3, 6 and 10; and study discharge (Day 46 ± 2) visits. Subjects were required to fast (nothing to eat or drink except for water) for at least 10 h prior to specimen collection for clinical laboratory testing. All laboratory assessments were reviewed by the investigator prior to dosing on Day 1. The incidence of adverse events and significant laboratory abnormalities were recorded during the study, and changes in ECGs, vital signs and physical examinations were monitored.
Statistical methods
The sample size was not based on statistical power considerations. Data from 20 evaluable subjects were required to provide at least 90% confidence that the estimated ratios of the geometric mean values for AUC0–∞ for each Inje cocktail component with and without belatacept for were within 20% of the true ratios (i.e. the 90% confidence interval [CI] for test/reference ratio would be between 0.80 and 1.25, given a true ratio of 1).
To estimate the effect of concomitant belatacept on each of the Inje component drugs, linear mixed models were fitted to log‐transformed pharmacokinetic parameters. Point estimates and 90% CIs for treatment differences on the log scale were exponentiated to obtain estimates for Cmax, AUC0–t and AUC0–∞; ratios were calculated using the geometric means of the Inje cocktail ± belatacept (Days 4, 7 and 11) vs. the reference value (Day 1). No adjustments were made for multiple comparisons. Statistically significant differences were defined by the 90% CI for the geometric mean ratio falling below 0.80 or above 1.25.
Results
Study population
Table 1 shows the disposition of study participants. The study was conducted between January and April 2013 at a single centre in San Antonio, Texas. A total of 45 subjects were screened, 22 of whom met eligibility criteria and received at least one dose of study drugs. Eighteen subjects completed the study.
Of the 22 subjects who received at least one dose of study drugs, 21 were male. The mean age was 31.5 years (range, 20–43), and the mean body mass index was 24.6 (range, 19.7–29.9). The majority was white (n = 15); seven were black/African American.
Pharmacogenomics analysis identified one subject who was a poor metabolizer of CYP2D6; this subject was excluded from the pharmacokinetic analyses of dextromethorphan and its metabolite, dextrorphan.
Pharmacokinetics
Figure 1 presents the effect of belatacept on the pharmacokinetics of the Inje cocktail component drugs and their metabolites. Of the Inje cocktail drugs, only omeprazole showed a consistent increase in exposure following coadministration of belatacept. The point estimates for the geometric mean ratios for maximum concentration (Cmax) and AUC of omeprazole were approximately 16–30% higher following coadministration of belatacept and the Inje cocktail vs. the Inje cocktail alone; the upper bounds of the 90% CIs were all above 1.25 on Days 4, 7 and 11 (Table 2). The pharmacokinetics of the metabolite 5‐hydroxyomeprazole were unchanged by belatacept coadministration, but there was a statistically significant reduction in the metabolite‐to‐parent ratios as a consequence of the increased exposure to omeprazole.
Figure 1.

Effect of belatacept on the pharmacokinetics of (A) the Inje cocktail components, (B) the Inje cocktail metabolites and (C) metabolite‐to‐parent ratios. Belatacept was administered on Day 4 and the Inje cocktail was administered on Days 1, 4, 7 and 11. Data represent point estimates of the geometric mean ratios (test vs. Day 1) with 90% CIs. AUC, area under the plasma concentration–time curve; CI, confidence interval; Cmax, maximum concentration; MR, metabolite‐to‐drug ratio
Belatacept did not alter the pharmacokinetics of caffeine (Table 2) or its metabolite, paraxanthine. Geometric mean ratios for Cmax, AUC0–t and AUC0–∞, were close to 1, and the 90% CIs were all within the equivalence range of 0.80 to 1.25 on Days 4, 7 and 11.
The pharmacokinetics of losartan, dextromethorphan, midazolam and their respective metabolites were generally comparable when the drugs were given with and without belatacept: geometric mean ratios approximated 1, and most 90% CIs were within 0.80 to 1.25, although in some instances the 90% CIs were slightly outside these boundaries (Table 2). Exposure to losartan was comparable when administered with and without belatacept in terms of AUC, but Cmax increased by 8% to 16% in the presence of belatacept. Point estimates for the geometric mean ratios for Cmax were slightly above 1, and the upper bounds of the 90% CIs were above 1.25, which was consistent across Day 4 (1.081 [0.914, 1.278]), Day 7 (1.158 [1.003, 1.338]) and Day 11 (1.152 [0.988, 1.342]). The apparent increase in losartan Cmax with belatacept administration may reflect the large variability associated with this parameter (56–68% coefficient of variation). Belatacept had no effect on the pharmacokinetics of the losartan metabolite, E‐3174.
Geometric mean AUC ratios for dextromethorphan were close to 1, and 90% CIs were within the 0.80–1.25 range, except for the AUC0–∞ ratio on Day 4, which was 0.877 (0.783, 0.982; Table 2). For Cmax, the geometric mean ratios showed a decrease of 8% to 14%; all ratios were slightly below 1, and the lower bounds of the 90% CIs were below 0.80: Day 4, 0.863 (0.746, 0.997); Day 7, 0.922 (0.793, 1.071); and Day 11, 0.856 (0.709, 1.034). The pharmacokinetics of the metabolite dextrorphan showed minor deviations from equivalence; for the metabolite‐to‐parent ratios, the deviations in the 90% CIs from the limits of equivalence were <16%.
No clinically relevant drug interaction was observed for midazolam. The point estimates for the geometric mean ratios for Cmax and AUC of midazolam approximated 1, and the respective 90% CIs fell within the 0.8–1.25 limits. Minor deviations from equivalence criteria were observed for its metabolite, 1′‐hydroxy‐midazolam; these deviations from the limits of equivalence were <10%. The increases of 14% to 19% observed for the point estimates for Cmax and AUC of the metabolite on Day 11 were not considered clinically relevant. In addition, for the metabolite‐to‐parent AUC0–t ratio, the upper bound of the 90% CIs exceeded 1.25 on Day 7 (1.160 [1.059, 1.272]) and Day 11 (1.153 [1.033, 1.287]); for the metabolite‐to‐parent AUC0–∞ ratio, the upper bound of the 90% CIs also exceeded 1.25 on Day 7 (1.150 [1.048, 1.261]) and Day 11 (1.145 [1.026, 1.278]). These deviations were considered minor and not clinically relevant.
Exploratory analysis of cytokine levels
Therapeutic serum concentrations of belatacept were achieved and were similar to concentrations previously observed in healthy subjects and in patients from previous belatacept clinical trials. An exploratory analysis of the impact of belatacept administration on cytokine levels showed that cytokine levels were below the LLOQ in the majority of study participants, an observation consistent with other studies. There were no significant changes following belatacept administration in the levels of tumour necrosis factor‐α, IFN‐γ, IL‐2, IL‐6 or IL‐10. IFN‐α showed a significant change from baseline to Day 4, with a mean decrease of 15% (P = 0.0178). However, this result was affected by two outlier values that were only present on Day 4 (Figure 2). Absolute levels of IFN‐α remained within the normal range throughout the study period, and the observed change on Day 4 was not considered clinically meaningful. There were no clinically relevant correlations between the change in cytokine levels and the pharmacokinetics of the Inje cocktail drugs or their metabolites.
Figure 2.
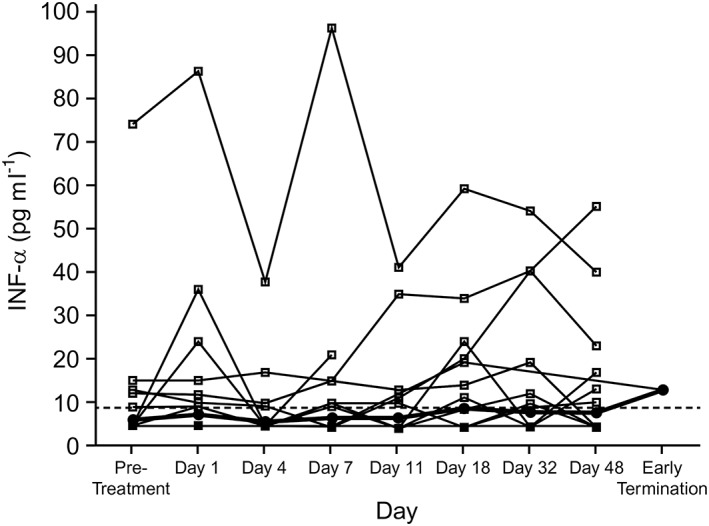
Individual serum IFN‐α levels by day (all treated subjects). Dotted line represents the LLOQ (8.6 pg ml−1). Values below LLOQ were imputed as 1/2 × LLOQ. Open circles represent individual values and closed circles represent geometric mean values. IFN, interferon; LLOQ, lower limit of quantification
Physiologically based pharmacokinetic modelling of omeprazole
A physiologically based pharmacokinetic model of omeprazole accumulation that excluded the administration of belatacept (see Methods and Supplemental Material) indicated that the observed increase in omeprazole exposure was caused by omeprazole‐mediated, time‐dependent inhibition of its own CYP‐mediated elimination. Using the same dosing regimen of omeprazole used in the present clinical study, the predicted accumulation after repeated doses of omeprazole 40 mg alone was consistent with the results obtained clinically. The model accurately predicted mean changes in AUC for omeprazole and 5‐hydroxyomeprazole, as well as the metabolite‐to‐parent AUC ratio, with predicted values falling within the 90% CIs for the observed values (Figure 3). The predicted AUC (μmol l–1 × h) of omeprazole relative to Day 1 was 1.14 on Day 4, 1.18 on Day 7, and 1.13 on Day 11; the corresponding observed values were 1.16, 1.23 and 1.22, respectively.
Figure 3.
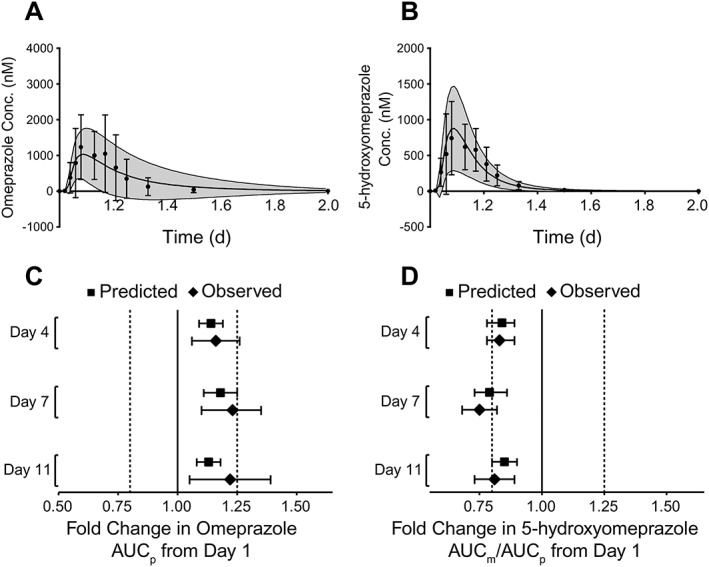
Predicted and observed plasma concentration vs. time curves of (A) omeprazole and (B) 5‐hydroxyomeprazole on Day 1 for all subjects (n = 22). The predicted data are depicted as mean (solid line) and standard deviation (grey area), whereas the observed data are depicted as mean (dots) and standard deviation (error bars). Predicted vs. observed change in (C) omeprazole AUC and (D) AUC ratio of 5‐hydroxyomeprazole to omeprazole on Days 4, 7 and 11 relative to Day 1 for all subjects (n = 22). Geometric mean data with 90% CIs are shown. AUC, area under the curve; AUCm/AUCp, metabolite‐to‐parent ratio; CI, confidence interval
Safety
No deaths or serious adverse events occurred that were attributed to treatment during the study. Five of 22 subjects (23%) had a total of 15 adverse events which were mild (n = 9), moderate (n = 5) or severe (n = 1). This included one subject who discontinued the study due to severe, exercise‐induced creatine phosphokinase elevation that was unrelated to treatment. The most common adverse event was headache (three subjects), with diarrhoea, anxiety and erythema each occurring in one subject. Diarrhoea was the only adverse event considered related to the study drug and was reported on each of the 4 treatment days. All adverse events resolved without sequelae during the study. Apart from the subject with creatine phosphokinase elevation, there were no clinically relevant laboratory results, vital signs or ECGs.
Discussion
The potential for drug interactions involving therapeutic proteins has gained attention as their clinical use has increased 7, 10, 11, 13, 14. A cocktail probe approach, whereby substrates for multiple CYP enzymes are simultaneously administered, is a convenient way to assess the potential for CYP induction or inhibition. This study used the validated, orally administered Inje cocktail to assess the effect of belatacept on the pharmacokinetics of drugs metabolized by CYP1 A2 (caffeine), CYP2C9 (losartan), CYP2C19 (omeprazole), CYP2D6 (dextromethorphan) and CYP3 A4 (midazolam). Belatacept has been shown to inhibit the production of certain cytokines during an in vitro alloimmune response, and modulation of cytokine pathways responsible for regulating CYP expression is a common means by which therapeutic proteins may influence CYP activity 8. As the half‐life of belatacept in healthy individuals approaches 10 days (t1/2 = 9.8) 4, a single dose on Day 4 could potentially affect CYP‐substrate pharmacokinetics up to Day 11. However, as belatacept had no effect on cytokine modulation in this study, no clinically relevant drug–drug interactions were identified.
Furthermore, belatacept did not alter CYP450 activity either at high or intermediate concentrations. A 10 mg kg−1 dose of belatacept represents the loading dose for initiating therapy in transplant patients, for which they continue to receive this dose every 2 weeks for the first month and then monthly for 2 additional months following transplantation, after which patients step down to a lower dose of 5 mg kg−1. As is permissible and ethical in a normal healthy volunteer population, the highest approved dose of 10 mg kg−1 was administered as a single dose to healthy subjects to be able to safely test higher exposures. While this study was not performed under multiple‐dose conditions, the results from this single‐dose evaluation suggest that as drug concentrations of belatacept declined from peak through elimination, the pharmacokinetics of the probe substrates remained consistent.
Exposure to omeprazole did increase slightly (16–30%) when administered with belatacept, but this occurred without alterations in the pharmacokinetics of its metabolite 5‐hydroxyomeprazole. Given the favourable safety profile of omeprazole 15, the observed increase in exposure is unlikely to increase adverse event risk. This increase is suggestive of weak in vivo CYP2C19 inhibition over the study period, which is unlikely to be related to belatacept administration, but rather to omeprazole‐mediated, time‐dependent inhibition of its own CYP‐mediated elimination 16, 17. In support of this, model simulations of omeprazole 40 mg administered orally on Days 1, 4, 7 and 11 and without belatacept predicted considerable accumulation of omeprazole. In a prior study of healthy volunteers, daily administration of omeprazole 40 mg resulted in accumulated exposures after 5 days (Day 5 to Day 1 AUC ratio of 2.4) 18, despite its short half‐life (t1/2 = 0.5–1 h) relative to its 24‐h dosing interval 19. No accumulation has been observed with CYP2C19‐poor metabolizers, indicating that this phenomenon is dependent on CYP2C19 activity 17. More recent in vitro studies have established that omeprazole and its metabolites are CYP2C19 time‐dependent inhibitors (also known as mechanism‐based or metabolism‐dependent inhibitors) 16, 20, 21. Our exploratory pharmacokinetic model simulations support the idea that omeprazole is inhibiting its own elimination under multiple dosing independently of belatacept coadministration. Sufficient dose reductions (≤20 mg) or increasing dosing intervals (to ≥5 days) would be needed to avoid false‐positive drug–drug interaction results due to autoinhibition.
Kidney transplant recipients, particularly the elderly, often take multiple medications under multidrug immunosuppressive regimens, as well as for the treatment of comorbidities, which leaves them at risk for drug interactions 5, 22, 23, 24, 25. Most of the commonly used immunosuppressants are metabolized by CYP and are substrates for various transporters, which may lead to interactions with commonly prescribed drugs and other immunosuppressants 5, 26, 27. Adverse events, including those resulting from the concurrent prescription of agents with potential drug interactions, are not uncommon among kidney transplant recipients, and an increased incidence of such events has been associated with a history of cardiovascular disease or diabetes 22. This emphasizes the importance of understanding potential drug interactions in belatacept‐treated patients.
Overall, the results presented herein suggest that belatacept has no clinically relevant drug interactions with the CYP substrates included in the Inje cocktail or their metabolites, despite an increase in omeprazole exposure. Furthermore, the results from this investigation reassure prescribing physicians that belatacept can be safely prescribed to renal transplant patients administered other CYP substrates, without the need for dosage adjustments 4. Coadministration of belatacept with the Inje cocktail was safe and well tolerated, supporting the feasibility of this methodology for assessing the potential drug interactions of therapeutic proteins.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: D.W., L.Z., M.S., S.Z., Z.T., S.L. and B.M. are salaried employees of Bristol‐Myers Squibb. D.W., X.T., S.Z., Z.T., B.G., S.L. and B.M. currently hold stock in Bristol‐Myers Squibb. X.T., J.D.L., E.M. and B.G. were salaried employees of Bristol‐Myers Squibb at the time these data were collected.
The authors would like to acknowledge PPD (Richmond, Virginia & Middleton, Wisconsin, USA), the analysing laboratory for all samples. Professional and medical writing and editorial assistance was provided by Meredith Kalish of CodonMedical, an Ashfield Company, part of UDG Healthcare plc, and was funded by Bristol‐Myers Squibb.
Contributors
All authors made significant contributions to the research and design of the study, data analysis and writing or review of this manuscript. As this manuscript is based on the body of data generated by a Bristol‐Myers Squibb‐sponsored study and executed by a Contract Research Organization (CRO), none of the authors cited on this manuscript is identified as a principal investigator.
Supporting information
Table S1 Final physiologically based pharmacokinetic model parameter values for omeprazole and 5‐hydroxyomeprazole.
Table S2 Final physiologically based pharmacokinetic model CYP2C19 population and expression parameter values.
Supporting info item
Williams, D. , Tao, X. , Zhu, L. , Stonier, M. , Lutz, J. D. , Masson, E. , Zhang, S. , Ganguly, B. , Tzogas, Z. , Lubin, S. , and Murthy, B. (2017) Use of a cocktail probe to assess potential drug interactions with cytochrome P450 after administration of belatacept, a costimulatory immunomodulator. Br J Clin Pharmacol, 83: 370–380. doi: 10.1111/bcp.13097.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bristol‐Myers Squibb Company . Nulojix® US prescribing information. April 2014. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125288s059lbl.pdf (last accessed 16 January 2015).
- 5. Kuypers DR. Immunotherapy in elderly transplant recipients: a guide to clinically significant drug interactions. Drugs Aging 2009; 26: 715–737. [DOI] [PubMed] [Google Scholar]
- 6. Christensen H, Hermann M. Immunological response as a source to variability in drug metabolism and transport. Front Pharmacol 2012; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang SM, Zhao H, Lee JI, Reynolds K, Zhang L, Temple R, et al. Therapeutic protein‐drug interactions and implications for drug development. Clin Pharmacol Ther 2010; 87: 497–503. [DOI] [PubMed] [Google Scholar]
- 8. Lee JI, Zhang L, Men AY, Kenna LA, Huang SM. CYP‐mediated therapeutic protein‐drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet 2010; 49: 295–310. [DOI] [PubMed] [Google Scholar]
- 9. Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease‐drug–drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther 2011; 89: 735–740. [DOI] [PubMed] [Google Scholar]
- 10. Food and Drug Administration . Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Draft Guidance for Industry. 2012. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf (last accessed 16 January 2015).
- 11. Kenny JR, Liu MM, Chow AT, Earp JC, Evers R, Slatter JG, et al. Therapeutic protein drug–drug interactions: navigating the knowledge gaps‐highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA workshop. AAPS J 2013; 15: 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ryu JY, Song IS, Sunwoo YE, Shon JH, Liu KH, Cha IJ, et al. Development of the “Inje cocktail” for high‐throughput evaluation of five human cytochrome P450 isoforms in vivo . Clin Pharmacol Ther 2007; 82: 531–540. [DOI] [PubMed] [Google Scholar]
- 13. Girish S, Martin SW, Peterson MC, Zhang LK, Zhao H, Balthasar J, et al. AAPS workshop report: strategies to address therapeutic protein‐drug interactions during clinical development. AAPS J 2011; 13: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prueksaritanont T, Chu X, Gibson C, Cui D, Yee KL, Ballard J, et al. Drug–drug interaction studies: regulatory guidance and an industry perspective. AAPS J 2013; 15: 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andersson T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors; focus on omeprazole, lansoprazole, and pantoprazole. Clin Pharmacokinet 1996; 31: 9–28. [DOI] [PubMed] [Google Scholar]
- 16. Chang M, Tybring G, Dahl ML, Götharson E, Sagar M, Seensalu R, et al. Interphenotype differences in disposition and effect on gastrin levels of omeprazole‐‐suitability of omeprazole as a probe for CYP2C19. Br J Clin Pharmacol 1995; 39: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shirasaka Y, Sager JE, Lutz JD, Davis C, Isoherranen N. Inhibition of CYP2C19 and CYP3 A4 by omeprazole metabolites and their contribution to drug–drug interactions. Drug Metab Dispos 2013; 41: 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hassan‐Alin M, Andersson T, Niazi M, Röhss K. A pharmacokinetic study comparing single and repeated oral doses of 20 mg and 40 mg omeprazole and its two optical isomers, S‐omeprazole (esomeprazole) and R‐omeprazole, in healthy subjects. Eur J Clin Pharmacol 2005; 60: 779–784. [DOI] [PubMed] [Google Scholar]
- 19. AstraZeneca Pharmaceuticals LP . Prilosec® US prescribing information. March 2014. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/019810s098,022056s014lbl.pdf (last accessed 16 January 2015).
- 20. Ogilvie BW, Yerino P, Kazmi F, Buckley DB, Rostami‐Hodjegan A, et al. The proton pump inhibitor, omeprazole, but not lansoprazole or pantoprazole, is a metabolism‐dependent inhibitor of CYP2C19: implications for coadministration with clopidogrel. Drug Metab Dispos 2011; 39: 2020–2033. [DOI] [PubMed] [Google Scholar]
- 21. Boulenc X, Djebli N, Shi J, Perrin L, Brian W, Van Horn R, et al. Effects of omeprazole and genetic polymorphism of CYP2C19 on the clopidogrel active metabolite. Drug Metab Dispos 2012; 40: 187–197. [DOI] [PubMed] [Google Scholar]
- 22. Weir MR, Gravens‐Muller L, Costa N, Ivanova A, Manitpisitkul W, Bostom AG, et al. Safety events in kidney transplant recipients: results from the Folic Acid for Vascular Outcome Reduction in Transplant Trial. Transplantation 2015; 99: 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bernardo JF, McCauley J. Drug therapy in transplant recipients: special considerations in the elderly with comorbid conditions. Drugs Aging 2004; 21: 323–348. [DOI] [PubMed] [Google Scholar]
- 24. Rostaing L, Christiaans MH, Kovarik JM, Pascual J. The pharmacokinetics of everolimus in de novo kidney transplant patients receiving tacrolimus: an analysis from the randomized ASSET study. Ann Transplant 2014; 19: 337–345. [DOI] [PubMed] [Google Scholar]
- 25. Kuypers DR, Claes K, Evenepoel P, Maes B, Vanrenterghem Y. Long‐term pharmacokinetic study of the novel combination of tacrolimus and sirolimus in de novo renal allograft recipients. Ther Drug Monit 2003; 25: 447–451. [DOI] [PubMed] [Google Scholar]
- 26. Kuypers DR. Influence of interactions between immunosuppressive drugs on therapeutic drug monitoring. Ann Transplant 2008; 13: 11–18. [PubMed] [Google Scholar]
- 27. Vicari‐Christensen M, Repper S, Basile S, Young D. Tacrolimus: review of pharmacokinetics, pharmacodynamics, and pharmacogenetics to facilitate practitioners' understanding and offer strategies for educating patients and promoting adherence. Prog Transplant 2009; 19: 277–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Final physiologically based pharmacokinetic model parameter values for omeprazole and 5‐hydroxyomeprazole.
Table S2 Final physiologically based pharmacokinetic model CYP2C19 population and expression parameter values.
Supporting info item