

Abstract
Apoptosis, especially the intrinsic mitochondrial cell death pathway, is regulated by the BCL-2 family of proteins. Defects in apoptotic machinery are one of the main mechanisms that cells employ to evade cell death and become cancerous. Targeting the apoptotic defects, either by direct inhibition of BCL-2 family proteins or through modulation of regulatory pathways, can restore cell sensitivity to cell death. This review will focus on the aspects of BCL-2 family proteins, their interactions with kinase pathways, and how novel targeted agents can help overcome the apoptotic blockades. Furthermore, functional assays, such as BH3 profiling, may help in predicting responses to chemotherapies and aid in the selection of combination therapies by determining the mitochondrial threshold for initiating cell death.
Abbreviations: ASH, American Society of Hematology; ATAP, amphipathic tail-anchoring peptide; BAD, BCL-2-associated death promoter protein; BAK, BCL-2 homologous antagonist killer; BAX, BCL-2-associated X protein; BCL-2, B-cell lymphoma 2; BCL-w (BCL2L2), BCL-2-like protein 2; BCL-xL, B-cell lymphoma X long; BFL-1 (BCL2A1), BCL-2-related protein A1; BCR, B-cell receptor; BH3, BCL-2 homology 3; BID, BH3 interacting domain death agonist; BIK, BCL-2-interacting killer; BIM, BCL-2-interacting mediator of cell death; BOK, BCL-2 related ovarian killer; BTK, Bruton׳s tyrosine kinase; CDK, cyclin-dependent kinase; CLL, chronic lymphocytic leukemia; CHOP, cyclophosphamide, hydroxydaunorubicin, oncovin-vincristine and prednisone; CML, chronic myelogenous leukemia; CR, complete response;EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; FDA, U. S. Food and Drug Administration; GSK-3, glycogen synthase kinase-3; ITK, interleukin-2-inducible T-cell kinase; MCL, myeloid cell leukemia; MOMP, mitochondrial outer membrane permeabilization; NHL, non-Hodgkin lymphoma; NIH, National Institutes of Health; NSCLC, non-small cell lung cancer; PI3K, phosphatidylinositol-3-kinase; PUMA, p53 up-regulated modulator of apoptosis; SLL, small lymphocytic lymphoma; T-ALL, T-acute lymphocytic leukemia
KEY WORDS: Apoptosis, BCL-2 family, Mitochondrial priming, BH3 profiling, Targeted therapy, Combination therapy
Graphical abstract
Evading apoptosis is a hallmark of cancer cells. How to attack the apoptotic defects, through direct inhibition of BCL-2 family proteins or by indirect regulation of other promoting survival signaling pathways, is the challenge for modern cancer therapies. Fortunately, a lot of progress has been made to meet that challenge.
1. Targeted therapies fueled by the discovery of cancer genome and signaling webs
In 2003, the human genome project was pronounced completed, after thirteen years of U. S. National Institutes of Health (NIH) leading multi-national research efforts. The “finish” period has continued since the final version of sequences was published in the journal Nature in 20041, and more detailed information for the human genome has yet to be revealed. With the evolutionary development of sequencing and computing technology, which lead to a drastic decrease in costs for sequencing (from 100 million to 1000 dollars per genome2), and possible interpretation of the results, the focus of current DNA sequencing is naturally oriented towards human diseases. And cancer is one of them; the discovery of important gene variants/mutations in cancer genome has changed treatment regimens and benefits many cancer patients. The most exciting examples are mutations uncovered in epidermal growth factor receptor (EGFR) and BRAF in lung cancer3 and melanoma4, respectively.
In parallel, proteomics is expanding its web to the most of the signaling networks governing our physiological functions. New nodes and loops continue to be discovered, and novel interventions are being tested and exploited in disease treatments. In 2015, the U. S. Food and Drug Administration (FDA) approved 45 novel drugs, including four used to treat multiple myeloma5. About a decade ago, most chemotherapies were limited to conventional pan-cytotoxic drugs that targeted DNA or microtubules such as the CHOP regimen (cyclophosphamide, hydroxydaunorubicin, oncovin-vincristine and prednisone), which cured millions of people of otherwise fatal diseases, but also were accompanied by severe side effects due to damage to healthy tissues. At present, targeted agents, newly approved or in clinical trials, are more precise and bring the overall response rates to much higher levels than before. This is true even for subgroups with adverse prognostic features and those who respond poorly to conventional therapies, especially in hematological cancers demonstrated at the annual meeting of American Society of Hematology—ASH 2015 (Orlando, FL, USA). In April, 2016, following promising activity in its clinical trials6, 7, the selective BCL-2 inhibitor venetoclax (AbbVie) was approved by the FDA8 for chronic lymphocytic leukemia (CLL) with chromosomal abnormalities, and it becomes the first-in-class direct inhibitor targeting BCL-2 family proteins that has got approved. With so many novel agents and biomarkers emerging, there is no doubt that cancer research and therapies have entered a new era, in which hopefully more cancers will become manageable in the near future.
Regardless of their categorization as conventional or targeted therapies, most of chemotherapies kill cancer cells via the apoptotic cell death pathway. In this review, the focus will be on research progress made on this intrinsic mitochondrial cell death pathway regulated by B-cell lymphoma 2 (BCL-2) family proteins; on targeted therapies that have been developed to intervene in its dysfunction in cancers; and how we can use BH3 profiling, a functional assay measuring mitochondrial priming, to predict patient responses and provide guiding information for potential combination therapies.
2. Apoptosis is regulated by BCL-2 family proteins
Apoptosis, which includes both extrinsic and intrinsic pathways, is one of the most important forms of cell death in multicellular organisms. The intrinsic cell death pathway is regulated mostly by BCL-2 family proteins residing in or recruited to the mitochondria after death insults imposed on cells9, 10. The BCL-2 family comprises both anti- and pro-apoptotic proteins. Anti-apoptotic proteins include at least BCL-2, BCL-xL, MCL-1, BCL-w and BFL-1. High expression of anti-apoptotic proteins, especially BCL-211, 12, 13, 14, BCL-xL15 and MCL-116, 17, 18, 19, 20, has been shown in various types of cancers, and they play important roles in tumorigenesis in different tumor models 9, 21, 22, 23, 24, 25. Pro-apoptotic proteins can be further divided into two subgroups, including multi-domain proteins, like the death effectors/executioners BAX and BAK; and BH3-only proteins, like activators BIM, BID and PUMA, or sensitizers including BAD, NOXA, HRK and BMF. Recently, BOK, a non-canonical BCL-2 family effector of apoptosis, has been shown to mediate cell death triggered by endoplasmic reticulum (ER)-associated degradation independent of BAX and BAK, or when BAX/BAK are absent and cells are overwhelmed by unfolded proteins26.
The interactions within the BCL-2 family members are complex, and the interplay of anti- and pro-apoptotic proteins determines cell fate (see Fig. 1). The activation of BAX, BAK or BOK (in some circumstances)26 can lead to their oligomerization, which forms pores in the mitochondrial outer membrane and resulting the release of cytochrome c27, 28. Thus mitochondrial outer membrane permeabilization (MOMP) is generally considered a point-of-no-return, and triggers downstream caspase activation, proteolysis and DNA fragmentation. Different apoptotic blockades, resulting from BCL-2 family protein interactions, in which pro-death signals were sequestered or counteracted by anti-apoptotic proteins, have been observed in cancer cells as a means for cells to evade apoptosis29.
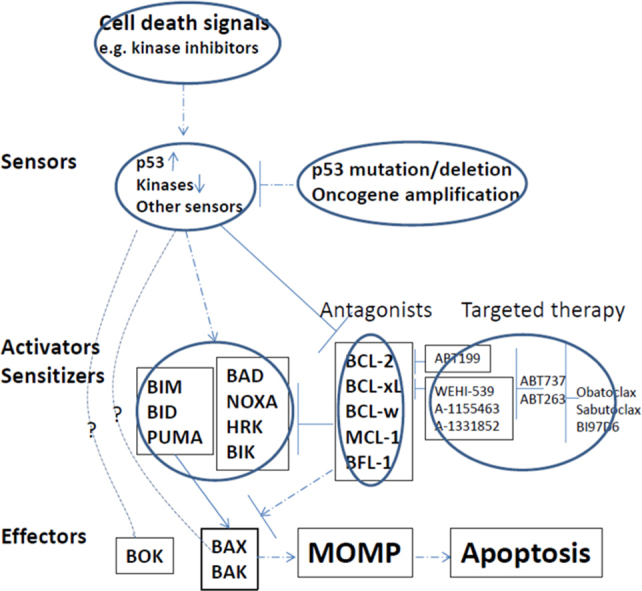
Figure 1.

The schema of the BCL-2 family proteins and their regulation by signaling pathways and targeted therapies.
Apoptosis is initiated at mitochondria; however, the regulation of BCL-2 family proteins is tightly controlled by upstream signaling networks, from receptors on cell surface to transcription factors residing in the nuclei. There are usually two sets of regulations when cells face death insults. One is to upregulate pro-death signals and the other is to downregulate anti-death factors. For example, pro-apoptotic protein BIM-EL is kept low through phosphorylation (by extracellular signal-regulated kinase—ERK30) mediated degradation when cells are stimulated with growth signals, but its protein level can be stabilized and increased when the ERK pathway is inhibited. BIM can be induced transcriptionally by transcription factor FOXO3A31, 32 when it is translocated to nuclei after PI3K/AKT inhibition. AKT can phosphorylate BAD33, 34 and BAX35, 36, and regulate their pro-apoptotic functions. BID37, PUMA38, NOXA39 and BAX40 are targets of p53 transcription factor, and their induction by p53 in response to DNA damage or other death insults keep cells in balance between cell cycle arrest and cell death. Besides p53, PUMA can also be transcriptionally regulated by FOXO3A41. On the other hand, for anti-apoptotic proteins, BCL-242, BCL-xL43 and BFL-144 are target genes of NF-κB signaling, which is consistent with the pro-survival function of the NF-κB pathway. The anti-apoptotic function of these proteins also can be modulated by phosphorylation45, 46, 47. Nevertheless, the best example of phosphorylation and its sub-sequential effects on apoptosis lies in MCL-1. This short half-life protein can be rapidly degraded via the proteasome pathway after phosphorylation by glycogen synthase kinase-3 (GSK-3) in the AKT pathway48, 49. The mutation of E3 ligase FBW7 and resulting stabilization of MCL-1 protein is critical in tumorigenesis of T- acute lymphocytic leukemia (T-ALL)50, and determines the sensitivity of cancer cells to anti-microtubule drugs51.
Thus far, the genetic mutations harbored by BCL-2 family proteins are uncommon (more indeed will be discovered when more cancer genomes are sequenced), but other chromosomal abnormalities exist, which can lead to the upregulation of BCL2 family proteins. For instance, the amplification of MCL-1 gene locus is often associated with a variety of tumors, including breast cancer and non-small cell lung cancer (NSCLC)22. Chromosomal (t14;18) translocation in follicular lymphoma39, 52, 53 and diffuse large B cell lymphoma54 is critical for BCL-2 overexpression and oncogenesis in those types of cancers. Other means that cancer cells employ to counteract cell death include microRNAs. It has been discovered that they can negatively regulate anti-apoptotic proteins. For example, miR-15a and miR-16-1 regulate BCL-2 in CLL55, let-7 family56, miR-49157 and miR-133a58 for BCL-xL (miR-133a can also target MCL-1), and miR-19559 for BFL-1. Downregulation of these inhibitory microRNAs would elevate anti-death proteins, which then initiate tumorigenesis or drug resistance.
3. Modulating the apoptotic machinery with kinase inhibitors
As illustrated above, the regulation of BCL-2 family proteins is tightly connected with pro-survival signaling networks, including NF-κB, phosphatidylinositol-3-kinase (PI3 kinase, PI3K), and other pathways which serve as barometers for the abundance of nutrients in the microenvironment. In the last decade or so, the roles of these pro-survival pathways have been illuminated by cancer genome sequencing, proteomics and other systematic approaches. As such, kinase inhibitors have been developed to inhibit those pathways. Some of these inhibitors have brought in important clinical applications.
Indeed, each type of cancers has selected unique pathways during their tumorigenesis, and specific inhibitors will be required to target the unique pathway and trigger cell death accordingly. Erlotinib and gefitinib are inhibitors targeted the activating mutations in EGFR3 discovered in some of NSCLC patients. The pro-apoptotic proteins BIM and PUMA are induced by EGFR inhibition and trigger downstream apoptotic signaling in these cells60, 61, 62. In BRAF-mutant melanoma, BIM, PUMA, and BMF are reported contributing to the apoptosis induced by BRAF and/or MEK inhibitor treatments63. BIM and BAD trigger the apoptotic response to imatinib treatments in chronic myelogenous leukemia (CML)64. Conversely, downregulation of the anti-apoptotic MCL-1 protein by PI3K-mTORC1 inhibition can act together with BIM induction in EGFR-mutant cells to induce tumor regression65.
Most recently, inhibitors against Bruton׳s tyrosine kinase (BTK), which is downstream of the B-cell receptor (BCR) pathway, have shown promising clinical activity in B-cell malignancies. Ibrutinib (developed by Pharmacyclics) is the first generation BTK inhibitor, and was approved by the FDA for mantle cell lymphoma (MCL) in 2013 and for CLL in 201466. A 71% overall response rate was reported in the original clinical trial for CLL and small lymphocytic lymphoma (SLL), which was independent of clinical and genetic risk factors such as 17p deletion67. However, ibrutinib has relatively poor selectivity. Besides BTK, it also can inhibit other kinases such as interleukin-2-inducible T-cell kinase (ITK)66. Second generation BTK inhibitors were designed for better selectivity for BTK to avoid off-target effects. A recent report for a phase I–II clinical trial for one of such second generation inhibitors, acalabrutinib (ACP-196, Acerta Pharma), has demonstrated robust clinical activities68. In that trial with acalabrutinib, a 95% overall response rate was achieved in relapsed CLL patients, and it is also efficacious in patients bearing 17p deletion (100%), a subgroup that has been associated with poor prognosis.
Despite their encouraging clinical activities, it is still unclear how ibrutinib or acalabrutinib kill cancer cells. They may trigger the PI3K-mTOR pathway and upregulate pro-apoptotic BH3-only proteins like BIM69. The real mechanism of actions for these BTK inhibitors has yet to be fully understood since these agents don׳t elicit cell death quickly in vitro66 (as opposed to the BCL-2 inhibitors described below). Lymphocytosis has been observed for ibrutinib67 and acalabrutinib68, suggesting that these agents may inhibit the interaction between cancer cells and microenvironment, and displace cancer cells from protective compartments, which in turn generates pro-death signals like BIM and kills cells.
Taken together, indirect induction of BH3-only proteins or reduction of anti-apoptotic proteins by kinase inhibitors can play a central role in inducing cell death in cancers driven by constitutively activated oncogenic kinases, such as BCR-ABL, EGFR, BRAF, KRAS, BTK, some of which are described here.
4. Direct modulation of the apoptotic pathway with inhibitors for BCL-2 family proteins
The main concern for kinase inhibitors is feedback inhibition within the same or proximal pathways, which would hinder the effectiveness of these agents in killing cancer cells. Agonists or inhibitors of BCL-2 family proteins, act directly on apoptotic machinery. Such modulators can overcome the problems of inhibiting kinase pathways, and have thus been explored and developed by pharmaceutical companies.
One new approach to modulate BCL-2 family proteins is to enhance the pro-apoptotic functions. Stapled peptides based on BH3-only proteins, constructed using non-natural amino acids designed to protect peptides from proteolysis, have been tested in the pre-clinical setting70, 71. Further clinical development of these modified peptides is necessary in order to benefit patients, as they may have difficulty entering cancer cells.
For inhibitors of the anti-apoptotic proteins, early attempts included using natural products identified from chemical library screening (like gossypol and other natural polyphenols) for their inhibitory effects on BCL-2 and BCl-xL72. However, none of these compounds have become drugs for clinical usage. Novel agents, based on medicinal chemistry and/or structure-activity relationship, have since been developed. These inhibitors include the antisense oligonucleotide oblimersen (Genasense, by Genta), the pan-inhibitor obatoclax (by Teva) and the ABT737/ABT263 (navitoclax)/ABT199 (venetoclax) series (by AbbVie). Oblimersen is a single-stranded 18-mer DNA molecule complementary to BCL-2 mRNA, and it had been shown to inhibit BCL-2 protein expression, presumably by inducing degradation of BCL-2 mRNA. Despite some evidence of benefits in phase I studies of CLL, myeloma and melanoma, oblimersen was not effective in a phase III study in myeloma, and only modestly beneficial when added to fludarabine in a phase III study of CLL73. Thus oblimersen did not receive FDA approval and further development of the drug was not pursued. Obatoclax (GX-15-070) is considered a pan-inhibitor to the BCL-2 family since it can bind to the BH3 domain of BCL-2, BCL-xL and MCL-174. Clinical trials for obatoclax showed only modest efficacies for CLL75, but no benefit for myelofibrosis76 or small cell lung cancer (SCLC)77. Furthermore, the neurological side effects limited its further clinical development77, and the drug was not approved by the FDA.
The most potent and selective BCL-2 inhibitors are those developed by AbbVie. As mentioned above, the first-in-class BCL-2 inhibitor venetoclax has just been approved by the FDA8. ABT737, the first generation BCL-2 inhibitor by AbbVie, is a BH3-mimetic of the BAD BH3-only protein78. ABT263 (navitoclax) is an orally bioavailable counterpart of ABT73779. Both molecules can inhibit BCL-2, BCL-xL and BCL-w with a binding affinity on the order of 10 to 10,000 times greater than other molecules, including obatoclax80. These are large (nearly 1000 Da) compounds that mimic the amphipathic BH3 domain׳s α-helices. As such, they compete for the BH3-binding sites on anti-apoptotic proteins, freeing pro-apoptotic proteins from sequestration. These freed pro-apoptotic proteins then activate BAX/BAK, which in turn form the outer mitochondrial membrane pores, triggering downstream caspase activation and cell death (Fig. 2). ABT737 has poor oral bioavailability and has been limited to pre-clinical research. Navitoclax, however, has been limited in progressing through clinical trials due to thrombocytopenia, an on-target effect on BCL-xL that is required for platelet function81. Even with that stated, specific BCL-xL inhibitors like WEHI-53982, A-1155463 and A-133185283, 84 are also under development. Perhaps, with better dose management, supportive care, and combination therapy, BCL-xL inhibitors can still bring benefits to patients with BCL-xL-dependent cancers85, 86, which are more common in solid tumors87.
Figure 2.

Targeted therapies can modulate the mitochondrial priming probed by BH3 profiling. MOMP, mitochondrial outer membrane permeabilization.
The problem of thrombocytopenia triggered by navitoclax led to the reengineered development of venetoclax, an inhibitor with better selectivity for BCL-2. It has reduced affinity for BCL-xL by three orders of magnitude88. Venetoclax rapidly kills malignant cells through the intrinsic mitochondrial apoptosis pathway, and is selective for cells dependent on BCL-2, but not those dependent on BCL-xL88. In preclinical models, the drug exhibited efficacy against a wide variety of tumor types, including leukemia, non-Hodgkin lymphoma (NHL), and myeloma, with no significant thrombocytopenia observed in in vivo models89.
As a single agent, venetoclax has recently demonstrated robust clinical efficacy. A paper published by Roberts and colleagues6 reports results of a phase I clinical trial for relapsed CLL, which depends on BCL-2 for survival90. Among the 116 patients who received venetoclax, 92 (79%) of them had a response. Even more encouraging is the response rate (from 71% to 79%) among patients from the subgroups with an adverse prognosis, including those with resistance to fludarabine, those with chromosome 17p deletion and those with unmutated IGHV (Immunoglobulin heavy chain variable region). In the phase-II trial reported by Stilgenbauer and colleagues7, an overall response rate of nearly 80% was also achieved among patients with 17p deletion, marking venetoclax a promising new treatment option for patients with poor prognostic features.
Neither navitoclax nor venetoclax inhibits MCL-1, the anti-apoptotic protein that not only plays critical roles in multiple myeloma91, mantle cell lymphoma16 and some solid tumors18, 24, 92, but is also involved in drug resistance to ABT73793, 94, anti-tubulin51 chemotherapy and other cancer drugs. Currently, specific MCL-1 inhibitors for clinical applications are lacking. However, its short half-life feature has been exploited in the setting of kinase and cell cycle inhibition, which can lead to either translational or transcriptional repression of MCL-1 protein or transcript, as seen following treatment with PI3K-mTOR inhibitors or cyclin-dependent kinase (CDK) inhibitors like flavopiridol95 and roscovitine96.
As described above, obatoclax is a pan-BCL-2 inhibitor that can inhibit MCL-1 as well as BCL-2, BCL-xL and BCL-w, but the clinical activity of this agent has been disappointing due to modest anti-cancer activity and neurological toxicity. Besides obatoclax, apogossypol derivative BI97C1 (sabutoclax) has been reported to inhibit BH3 peptide binding to BCL-2, BCL-xL and MCL-197. Apogossypolone is a third generation gossypol derivative, designed to effectively target MCL-1. One of the structure derivatives is BI97D6, which has displayed a higher selectivity for MCL-1 over BCL-2 or BCL-xL, and can overcome ABT737 resistance by acting on MCL-198. However, these inhibitors lacked the potency to induce similar extents of death in BCL-2, BCL-xL or MCL-1 dependent cells, indicating that they are weak inhibitors for BCL-2 family proteins87. By analogy to the development of ABT737, several groups have generated specific small molecule inhibitors for MCL-199. Advances in pre-clinical models raise the possibility that potent and selective MCL-1 inhibitors may be available for clinical examination in the near future.
The inhibitors described in this section have been developed aiming to antagonize the up-regulation of ant-apoptotic proteins during tumorigenesis, such as BCL-2, BCL-xL or MCL-1. The effectiveness of these types of inhibitors depends on the functional intact of death executioners BAX/BAK or BOK, and depends on the overall priming of cells, which can be fluctuated by co-expression levels of BCL-2 family proteins. Resistant or refractory to killing can occur if anti-apoptotic factors can׳t be overcome by the inhibitors. A new type of peptide, amphipathic tail-anchoring peptide (ATAP) derived from BFL-1, a bifunctional BCL-2 family member, can bear pore forming property, trigger MOMP and release cytochrome c independent of BAX/BAK and other BCL-2 family proteins, even other cellular factors100. This unique feature relies on its intrinsic mitochondrial targeting sequence101. Once linked to an internalized RGD peptide, selective targeting for ATAP to tumor cells can be achieved and their ability to kill cancer cells has been demonstrated in prostate cancer cell lines and their xenograft models102. While these data suggest modified ATAP peptides can potentially be therapeutic agents, the mechanism of action of this type of peptides, especially their connection with BCL-2 family or other mitochondrial proteins, and the determinants of future patient response, remain to be identified.
5. Combination therapies and predicting patient response with BH3 profiling
Despite the high response rates reported in recent clinical trials in CLL for the second generation BTK inhibitor acalabrutinib (95%)68 and BCL-2 inhibitor venetoclax (79%)6, the complete response (CR) rate is low. All responses for acalabrutinib are partial, and only 20% CR is achieved by venetoclax. Furthermore, resistance to mono-therapy will inevitably occur103. This raises the question of what should be combined with these novel agents in order to achieve better efficacy in treating patients. Answering this question is even more important in liquid cancers with less optimal response, or in solid tumors, which have been in general very refractory to the BCL-2 inhibitors described here (with the exception for some SCLCs that are dependent on BCL-2 for survival)87.
For liquid cancers, another example supporting the need for combination therapy comes from the clinical trial with elderly acute myelogenous leukemia (AML) patients. Used as a single agent in elderly AML patients, venetoclax had only achieved a modest response rate (19% objective response rate including 6% CR104). Indeed, AML is a much different disease than CLL, and AML in the elderly is known to have a poor response to the standard induction therapies like cytarabine and daunorubicin due to its more aggressive disease biology and toxicity from these therapies. However, combining venetoclax with hypo-methylating agents (decitabine or azacitidine) has led to overall response rates of 75% (combining with decitabine) and 70% (with azacitidine) in treatment-naïve elderly patients (≥ 65 years), who were not eligible for standard therapies105. Thus the combination therapies have greatly improved the drug׳s efficacy.
So, what should be combined? Since BCL-2 family members play such a central role in regulating intrinsic mitochondrial cell death, they may have the potential to serve as biomarkers - predicting treatment responses or guiding the choice of combination therapies. A functional assay developed by the Letai group known as BH3 profiling measures mitochondrial priming resulting from the interaction of BCL-2 family proteins29, 90. Using synthetic BH3 peptides based on the BH3 domains of the BH3-only proteins, cells can be profiled to measure their distance from the apoptotic threshold106, 107 (Fig. 2). Highly primed cells, containing mitochondria that can be permeabilized with relatively low doses of BH3 peptides, are those that are very close to the threshold of apoptosis. BH3 profiling, or dynamic BH3 profiling108 (DBP, a variant of BH3 profiling focusing on priming changes after drug treatments), have been tested in various preclinical models109, 110, 111, 112. Using pretreatment samples from patients on the venetoclax trial (M12-175), we performed BH3 profiling and found that the extent of mitochondrial depolarization induced by a BIM BH3 peptide in vitro was correlated with percentage reduction of CLL in the blood and bone marrow in vivo, while the lethal concentration (LC50) derived from standard cytotoxicity assays was not; and the mitochondrial responses to peptides were independent of 17p status113. These data support further assessment of BH3 profiling as a predictive biomarker for this novel agent and other therapies.
In addition to baseline BH3 profiling of patient samples, DBP can be even more useful in assessing mitochondrial priming or apoptotic threshold changes after therapeutic intervention. Theoretically, two drugs from distant pathways would be less likely to develop overlapping resistance or feedback inhibition. Using DBP, we explored the possible combination of ibrutinib with venetoclax, and the mechanism underlying the efficacy of this combination. We have discovered that even though ibrutinib does not elicit frank cell death, it can increase mitochondrial BCL-2 dependence, and sensitize CLL cells to the BCL-2 inhibitor venetoclax114. Thus, by increasing our understanding of the mechanism of action of novel agents on apoptosis, BH3 profiling can provide guiding information on what compounds to combine to achieve enhanced drug efficacy.
6. In conclusion
Cancer therapy has entered a new era, with huge data sets generated over the last decade from cancer genome sequencing and other big data molecular approaches using cancer patient samples. This provides a greater opportunity to understand the cause of tumorigenesis, and uncover novel therapeutic targets. Nonetheless, the overwhelming information could also hinder the process of turning these discoveries into clinical benefits. The ultimate goal for any therapy is to kill cancer cells, and to cure patients115. With apoptosis converging on the BCL-2 family to execute the killing decision, understanding BCL-2 family proteins, their interactions and their context within other singling networks, will shed light on the effectiveness of chemotherapies and optimal combinations. The continued development of our BH3 profiling system and other biomarkers116 (for instance, DR_ MOMP)117, 118, perhaps can help to provide guiding information in patient and drug selection in the future.
Acknowledgments
The author would like to thank Dr. Anthony Letai and the Letai group (Dana-Farber Cancer Institute, Harvard Medical School, USA) for their support, helpful discussion and the research cited in this review; and Dr. Patrick Bhola and Ms. Alexandra Pourzia for their critical review of this manuscript.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 2.National Human Genome Research Institute. DNA sequencing costs. Available from: 〈https://www.genome.gov/27541954/dna-sequencing-costs/〉.
- 3.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Eng J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Food and Drug Administration. Novel drug approvals for 2015. Available from: 〈http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm430302.htm〉.
- 6.Roberts A.W., Davids M.S., Pagel J.M., Kahl B.S., Puvvada S.D., Gerecitano J.F. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Eng J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stilgenbauer S., Eichhorst B., Schetelig J., Coutre S., Seymour J.F., Munir T. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–778. doi: 10.1016/S1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- 8.U.S. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality; [updated 2016 April 11]. Available from: 〈http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm〉.
- 9.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 10.Bhola P.D., Letai A. Mitochondria-judges and executioners of cell death sentences. Mol Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsujimoto Y., Cossman J., Jaffe E., Croce C.M. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 12.Callagy G.M., Pharoah P.D., Pinder S.E., Hsu F.D., Nielsen T.O., Ragaz J. Bcl-2 is a prognostic marker in breast cancer independently of the Nottingham Prognostic Index. Clin Cancer Res. 2006;12:2468–2475. doi: 10.1158/1078-0432.CCR-05-2719. [DOI] [PubMed] [Google Scholar]
- 13.Catz S.D., Johnson J.L. BCL-2 in prostate cancer: a minireview. Apoptosis. 2003;8:29–37. doi: 10.1023/a:1021692801278. [DOI] [PubMed] [Google Scholar]
- 14.Nunez G., Seto M., Seremetis S., Ferrero D., Grignani F., Korsmeyer S.J. Growth- and tumor-promoting effects of deregulated BCL2 in human B-lymphoblastoid cells. Proc Natl Acad Sci U S A. 1989;86:4589–4593. doi: 10.1073/pnas.86.12.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H., Xue J., Hessler P., Tahir S.K., Chen J., Jin S. Genomic analysis and selective small molecule inhibition identifies BCL-XL as a critical survival factor in a subset of colorectal cancer. Mol Cancer. 2015;14:126. doi: 10.1186/s12943-015-0397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho-Vega J.H., Rassidakis G.Z., Admirand J.H., Oyarzo M., Ramalingam P., Paraguya A. MCL-1 expression in B-cell non-Hodgkin׳s lymphomas. Hum Pathol. 2004;35:1095–1100. doi: 10.1016/j.humpath.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Song L., Coppola D., Livingston S., Cress W.D., Haura E.B. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H., Guttikonda S., Roberts L., Uziel T., Semizarov D., Elmore S.W. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene. 2011;30:1963–1968. doi: 10.1038/onc.2010.559. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B., Gojo I., Fenton R.G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- 20.Rassidakis G.Z., Lai R., McDonnell T.J., Cabanillas F., Sarris A.H., Medeiros L.J. Overexpression of Mcl-1 in anaplastic large cell lymphoma cell lines and tumors. Am J Pathol. 2002;160:2309–2310. doi: 10.1016/S0002-9440(10)61178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Placzek W.J., Wei J., Kitada S., Zhai D., Reed J.C., Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40. doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly P.N., Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyamoto Y., Hosotani R., Wada M., Lee J.U., Koshiba T., Fujimoto K. Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology. 1999;56:73–82. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 25.Strasser A., Harris A.W., Bath M.L., Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 26.Llambi F., Wang Y.M., Victor B., Yang M., Schneider D.M., Gingras S. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell. 2016;165:421–433. doi: 10.1016/j.cell.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuwana T., Mackey M.R., Perkins G., Ellisman M.H., Latterich M., Schneiter R. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 28.Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S.A., Mannella C.A. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 29.Deng J., Carlson N., Takeyama K., Dal Cin P., Shipp M., Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Luciano F., Jacquel A., Colosetti P., Herrant M., Cagnol S., Pages G. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 31.Essafi A., Fernández de Mattos S., Hassen Y.A., Soeiro I., Mufti G.J., Thomas N.S. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene. 2005;24:2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- 32.Möller C., Alfredsson J., Engström M., Wootz H., Xiang Z., Lennartsson J. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106:1330–1336. doi: 10.1182/blood-2004-12-4792. [DOI] [PubMed] [Google Scholar]
- 33.del Peso L., González-Garcia M., Page C., Herrera R., Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 34.Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi H., Wang H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20:7779–7786. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- 36.Gardai S.J., Hildeman D.A., Frankel S.K., Whitlock B.B., Frasch S.C., Borregaard N. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 37.Sax J.K., Fei P., Murphy M.E., Bernhard E., Korsmeyer S.J., El-Deiry W.S. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 38.Nakano K., Vousden K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 39.Villunger A., Michalak E.M., Coultas L., Müllauer F., Böck G., Ausserlechner M.J. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 40.Toshiyuki M., Reed J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 41.You H., Pellegrini M., Tsuchihara K., Yamamoto K., Hacker G., Erlacher M. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Catz S.D., Johnson J.L. Transcriptional regulation of bcl-2 by nuclear factor κB and its significance in prostate cancer. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 43.Chen C., Edelstein L.C., Gélinas C. The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-xL. Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zong W.X., Edelstein L.C., Chen C., Bash J., Gélinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-κB that blocks TNFα-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng X., Gao F., Flagg T., May W.S., Jr. Mono- and multisite phosphorylation enhances Bcl2׳s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A. 2004;101:153–158. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng X., Gao F., Flagg T., Anderson J., May W.S. Bcl2׳s flexible loop domain regulates p53 binding and survival. Mol Cell Biol. 2006;26:4421–4434. doi: 10.1128/MCB.01647-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basu A., Haldar S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003;538:41–47. doi: 10.1016/s0014-5793(03)00131-5. [DOI] [PubMed] [Google Scholar]
- 48.Ding Q., He X., Hsu J.M., Xia W., Chen C.T., Li L.Y. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y., Altman B.J., Coloff J.L., Herman C.E., Jacobs S.R., Wieman H.L. Glycogen synthase kinase 3α and 3β mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inuzuka H., Shaik S., Onoyama I., Gao D., Tseng A., Maser R.S. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wertz I.E., Kusam S., Lam C., Okamoto T., Sandoval W., Anderson D.J. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 52.Tsujimoto Y., Finger L.R., Yunis J., Nowell P.C., Croce C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 53.Wong H.K., Fricker M., Wyttenbach A., Villunger A., Michalak E.M., Strasser A. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol Cell Biol. 2005;25:8732–8747. doi: 10.1128/MCB.25.19.8732-8747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrans S.L., Evans P.A., O׳Connor S.J., Kendall S.J., Owen R.G., Haynes A.P. The t(14;18) is associated with germinal center-derived diffuse large B-cell lymphoma and is a strong predictor of outcome. Clin Cancer Res. 2003;9:2133–2139. [PubMed] [Google Scholar]
- 55.Cimmino A., Calin G.A., Fabbri M., Iorio M.V., Ferracin M., Shimizu M. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimizu S., Takehara T., Hikita H., Kodama T., Miyagi T., Hosui A. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52:698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 57.Nakano H., Miyazawa T., Kinoshita K., Yamada Y., Yoshida T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-XL in colorectal cancer cells. Int J Cancer. 2010;127:1072–1080. doi: 10.1002/ijc.25143. [DOI] [PubMed] [Google Scholar]
- 58.Ji F., Zhang H., Wang Y., Li M., Xu W., Kang Y. MicroRNA-133a, downregulated in osteosarcoma, suppresses proliferation and promotes apoptosis by targeting Bcl-xL and Mcl-1. Bone. 2013;56:220–226. doi: 10.1016/j.bone.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 59.Yang X., Yin J., Yu J., Xiang Q., Liu Y., Tang S. miRNA-195 sensitizes human hepatocellular carcinoma cells to 5-FU by targeting BCL-w. Oncol Rep. 2012;27:250–257. doi: 10.3892/or.2011.1472. [DOI] [PubMed] [Google Scholar]
- 60.Cragg M.S., Kuroda J., Puthalakath H., Huang D.C., Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4:e316. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng J., Shimamura T., Perera S., Carlson N.E., Cai D., Shapiro G.I. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 62.Gong Y., Somwar R., Politi K., Balak M., Chmielecki J., Jiang X. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cragg M.S., Jansen E.S., Cook M., Harris C., Strasser A., Scott C.L. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Investig. 2008;118:3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuroda J., Puthalakath H., Cragg M.S., Kelly P.N., Bouillet P., Huang D.C. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Faber A.C., Li D., Song Y., Liang M.C., Yeap B.Y., Bronson R.T. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davids M.S., Brown J.R. Ibrutinib: a first in class covalent inhibitor of Bruton׳s tyrosine kinase. Future Oncol. 2014;10:957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byrd J.C., Furman R.R., Coutre S.E., Flinn I.W., Burger J.A., Blum K.A. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Eng J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Byrd J.C., Harrington B., O׳Brien S., Jones J.A., Schuh A., Devereux S. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Eng J Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Portell C.A., Axelrod M., Brett L.K., Gordon V.L., Capaldo B., Xing J.C. Synergistic cytotoxicity of ibrutinib and the BCL2 antagonist, ABT-199(GDC-0199) in mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL): molecular analysis reveals mechanisms of target interactions. Blood. 2014;124:509. [Google Scholar]
- 70.LaBelle J.L., Katz S.G., Bird G.H., Gavathiotis E., Stewart M.L., Lawrence C. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walensky L.D., Kung A.L., Escher I., Malia T.J., Barbuto S., Wright R.D. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oliver C.L., Miranda M.B., Shangary S., Land S., Wang S., Johnson D.E. (–)-Gossypol acts directly on the mitochondria to overcome Bcl-2– and Bcl-XL–mediated apoptosis resistance. Mol Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- 73.Oblimersen: augmerosen, BCL-2 antisense oligonucleotide-Genta, G 3139, GC 3139, oblimersen sodium. Drugs R D. 2007;8:321–334. doi: 10.2165/00126839-200708050-00006. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen M., Marcellus R.C., Roulston A., Watson M., Serfass L., Murthy Madiraju S.R. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O׳Brien S.M., Claxton D.F., Crump M., Faderl S., Kipps T., Keating M.J. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parikh S.A., Kantarjian H., Schimmer A., Walsh W., Asatiani E., El-Shami K. Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin Lymphoma Myeloma Leuk. 2010;10:285–289. doi: 10.3816/CLML.2010.n.059. [DOI] [PubMed] [Google Scholar]
- 77.Paik P.K., Rudin C.M., Pietanza M.C., Brown A., Rizvi N.A., Takebe N. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer. 2011;74:481–485. doi: 10.1016/j.lungcan.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 79.Tse C., Shoemaker A.R., Adickes J., Anderson M.G., Chen J., Jin S. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 80.Anderson M.A., Huang D., Roberts A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin Hematol. 2014;51:219–227. doi: 10.1053/j.seminhematol.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 81.Wilson W.H., O׳Connor O.A., Czuczman M.S., LaCasce A.S., Gerecitano J.F., Leonard J.P. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lessene G., Czabotar P.E., Sleebs B.E., Zobel K., Lowes K.N., Adams J.M. Structure-guided design of a selective BCL-XL inhibitor. Nat Chem Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 83.Tao Z.F., Hasvold L., Wang L., Wang X., Petros A.M., Park C.H. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5:1088–1093. doi: 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leverson J.D., Phillips D.C., Mitten M.J., Boghaert E.R., Diaz D., Tahir S.K. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa4642. 279ra40. [DOI] [PubMed] [Google Scholar]
- 85.Rudin C.M., Hann C.L., Garon E.B., Ribeiro de Oliveira M., Bonomi P.D., Camidge D.R. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gandhi L., Camidge D.R., Ribeiro de Oliveira M., Bonomi P., Gandara D., Khaira D. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vogler M. Targeting BCL2-proteins for the treatment of solid tumours. Adv Med. 2014;2014:943648. doi: 10.1155/2014/943648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 89.Gibson C.J., Davids M.S. BCL-2 antagonism to target the intrinsic mitochondrial pathway of apoptosis. Clin Cancer Res. 2015;21:5021–5029. doi: 10.1158/1078-0432.CCR-15-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Certo M., Del Gaizo Moore V., Nishino M., Wei G., Korsmeyer S., Armstrong S.A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 91.Le Gouill S., Podar K., Harousseau J.L., Anderson K.C. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 2004;3:1259–1262. doi: 10.4161/cc.3.10.1196. [DOI] [PubMed] [Google Scholar]
- 92.Shigemasa K., Katoh O., Shiroyama Y., Mihara S., Mukai K., Nagai N. Increased MCL-1 expression is associated with poor prognosis in ovarian carcinomas. Jpn J Cancer Res. 2002;93:542–550. doi: 10.1111/j.1349-7006.2002.tb01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mazumder S., Choudhary G.S., Al-Harbi S., Almasan A. Mcl-1 phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012;72:3069–3079. doi: 10.1158/0008-5472.CAN-11-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simonin K., N׳Diaye M., Lheureux S., Loussouarn C., Dutoit S., Briand M. Platinum compounds sensitize ovarian carcinoma cells to ABT-737 by modulation of the Mcl-1/Noxa axis. Apoptosis. 2013;18:492–508. doi: 10.1007/s10495-012-0799-x. [DOI] [PubMed] [Google Scholar]
- 95.Ma Y., Cress W.D., Haura E.B. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther. 2003;2:73–81. [PubMed] [Google Scholar]
- 96.Raje N., Kumar S., Hideshima T., Roccaro A., Ishitsuka K., Yasui H. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl−1 in multiple myeloma. Blood. 2005;106:1042–1047. doi: 10.1182/blood-2005-01-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Varadarajan S., Butterworth M., Wei J., Pellecchia M., Dinsdale D., Cohen G.M. Sabutoclax (BI97C1) and BI112D1, putative inhibitors of MCL-1, induce mitochondrial fragmentation either upstream of or independent of apoptosis. Neoplasia. 2013;15:568–578. doi: 10.1593/neo.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pan R., Ruvolo V.R., Wei J., Konopleva M., Reed J.C., Pellecchia M. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (–)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126:363–372. doi: 10.1182/blood-2014-10-604975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Belmar J., Fesik S.W. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84. doi: 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ko J.K., Choi K.H., Peng J., He F., Zhang Z., Weisleder N. Amphipathic tail-anchoring peptide and Bcl-2 homology domain-3 (BH3) peptides from Bcl-2 family proteins induce apoptosis through different mechanisms. J Biol Chem. 2011;286:9038–9048. doi: 10.1074/jbc.M110.198457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ko J.K., Choi K.H., Pan Z., Lin P., Weisleder N., Kim C.W. The tail-anchoring domain of Bfl1 and HCCS1 targets mitochondrial membrane permeability to induce apoptosis. J Cell Sci. 2007;120:2912–2923. doi: 10.1242/jcs.006197. [DOI] [PubMed] [Google Scholar]
- 102.De G., Ko J.K., Tan T., Zhu H., Li H., Ma J. Amphipathic tail-anchoring peptide is a promising therapeutic agent for prostate cancer treatment. Oncotarget. 2014;5:7734–7747. doi: 10.18632/oncotarget.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woyach J.A., Furman R.R., Liu T.M., Ozer H.G., Zapatka M., Ruppert A.S. Resistance mechanisms for the Bruton׳s tyrosine kinase inhibitor ibrutinib. N Eng J Med. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Konopleva M., Pollyea D.A., Potluri J., Chyla B.J., Busman T., McKeegan E. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukemia (AML) Blood. 2014;124:118. [Google Scholar]
- 105.DiNardo C., Pollyea D., Pratz K., Thirman M.J., Letai A., Frattini M. A phase 1b study of venetoclax (ABT-199/GDC-0199) in combination with decitabine or azacitidine in treatment-naive patients with acute myelogenous leukemia who are ≥ to 65 years and not eligible for standard induction therapy. Blood. 2015;126:327. [Google Scholar]
- 106.Ryan J., Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013;61:156–164. doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ryan J., Montero J., Rocco J., Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem. 2016;397:671–678. doi: 10.1515/hsz-2016-0107. [DOI] [PubMed] [Google Scholar]
- 108.Montero J., Sarosiek K.A., DeAngelo J.D., Maertens O., Ryan J., Ercan D. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160:977–989. doi: 10.1016/j.cell.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Davids M.S., Deng J., Wiestner A., Lannutti B.J., Wang L., Wu C.J. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–3509. doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ni Chonghaile T., Sarosiek K.A., Vo T.T., Ryan J.A., Tammareddi A., Moore V.D. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vo T.T., Ryan J., Carrasco R., Neuberg D., Rossi D.J., Stone R.M. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Touzeau C., Ryan J., Guerriero J., Moreau P., Chonghaile T.N., Le Gouill S. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30:761–764. doi: 10.1038/leu.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Anderson M.A., Deng J., Seymour J.F., Tam C., Kim S.Y., Fein J. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127:3215–3224. doi: 10.1182/blood-2016-01-688796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Deng J., Isik E., Fernandes S.M., Brown J.R., Letai A., Davids M.S. Ibrutinib therapy increases BCL-2 dependence and enhances sensitivity to venetoclax in CLL. Blood. 2015;126:490. doi: 10.1038/leu.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Letai A. Cell death and cancer therapy: don׳t forget to kill the cancer cell. Clin Cancer Res. 2015;21:5015–5020. doi: 10.1158/1078-0432.CCR-15-1204. [DOI] [PubMed] [Google Scholar]
- 116.Friedman A.A., Letai A., Fisher D.E., Flaherty K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747–756. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lindner A.U., Concannon C.G., Boukes G.J., Cannon M.D., Llambi F., Ryan D. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013;73:519–528. doi: 10.1158/0008-5472.CAN-12-2269. [DOI] [PubMed] [Google Scholar]
- 118.Flanagan L., Lindner A.U., de Chaumont C., Kehoe J., Fay J., Bacon O. BCL2 protein signalling determines acute responses to neoadjuvant chemoradiotherapy in rectal cancer. J Mol Med. 2015;93:315–326. doi: 10.1007/s00109-014-1221-7. [DOI] [PubMed] [Google Scholar]