

Abstract
The phosphatidylinositol 3-kinase (PI3K) pathway is frequently activated in human cancers. Class I PI3Ks are lipid kinases that phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) at the 3-OH of the inositol ring to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), which in turn activates Akt and the downstream effectors like mammalian target of rapamycin (mTOR) to play key roles in carcinogenesis. Therefore, PI3K has become an important anticancer drug target, and currently there is very high interest in the pharmaceutical development of PI3K inhibitors. Idelalisib has been approved in USA and Europe as the first-in-class PI3K inhibitor for cancer therapy. Dozens of other PI3K inhibitors including BKM120 and ZSTK474 are being evaluated in clinical trials. Multifaceted studies on these PI3K inhibitors are being performed, such as single and combinational efficacy, resistance, biomarkers, etc. This review provides an introduction to PI3K and summarizes key advances in the development of PI3K inhibitors.
KEY WORDS: Phosphatidylinositol 3-kinase, PI3K inhibitor, Drug candidate, Cancer therapy, PI3K/mTOR selectivity, Anticancer
Graphical abstract
PI3K catalyzes PIP2 to produce PIP3, therefore plays key roles in cell proliferation, survival, etc. Development of PI3K inhibitor as antitumor agent has become a hotspot area since 2006. A total of fifteen PI3K inhibitors approved or in clinical trials are summarized in this review.
1. Introduction
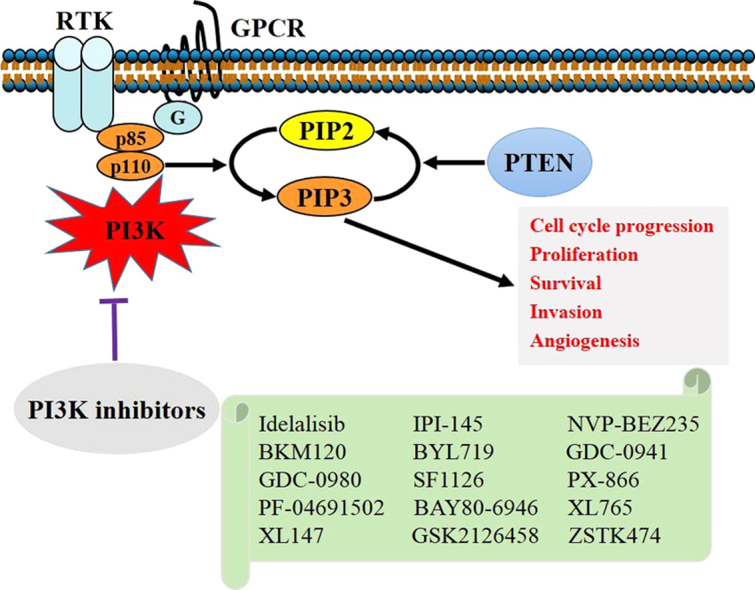
Phosphatidylinositol 3-kinases (PI3Ks) are a family of lipid kinases that phosphorylate the 3-OH of the inositol ring of phosphoinositides1, 2. Class I PI3Ks (generally called PI3Ks) are lipid kinases that phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 serves as a second messenger that plays important roles in fundamental cellular responses such as cell growth, survival, migration and metabolism3, 4. As a catalytic antagonist of PI3K, phosphatase and tensin homolog deleted on chromosome ten (PTEN) dephosphorylates PIP3 to PIP2 (Fig. 1). Since frequent gain-of-function mutations of PI3Ks and loss-of-function mutations of PTEN in human cancers suggest that PI3Ks are closely involved in tumorigenesis5, inhibitors targeting PI3Ks are expected to be promising anticancer drug candidates. In the past decade, dozens of PI3K inhibitors have been developed as potential chemotherapeutic drugs. Many of these have successfully entered clinical trials. In particular, idelalisib (CAL-101) has been approved in the USA and Europe as the first-in-class PI3K inhibitor for cancer therapy.
Figure 1.

Schematic structures of PI3K and PTEN, and the related lipid reactions they catalyze. PI3K phosphorylates PIP2 at 3-OH to generate PIP3. As a counterpart of PI3K, PTEN dephosphorylates PIP3 to produce PIP2. PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate.
In this review, we introduce PI3Ks and briefly describe the development of some representative PI3K inhibitors in clinical trials.
2. PI3K, critical element that is involved in carcinogenesis
PI3Ks belong to a family of lipid kinases that phosphorylate the 3-OH group of phosphoinositides1, 6, 7. Based on their primary structures and in vitro substrate specificity, PI3Ks are classified into three classes8, 9. Class I PI3Ks, which preferentially catalyze the phosphorylation of PIP2 to generate PIP3, are heterodimeric kinases as complexes of a catalytic subunit p110 with a regulatory subunit p85, p101, or p84. Members of this class are generally called PI3Ks because they have been investigated far more than the other two classes. PI3K-related kinases (PIKKs), which sometimes are termed Class IV PI3Ks, are protein kinases with a similar structure to the catalytic subunits of PI3Ks. Examples of PIKKs include mTOR and DNA-dependent protein kinase (DNA-PK), which are known to be involved in protein synthesis or DNA repair10. Class I PI3Ks are further divided into subclasses IA and IB based on their regulatory subunit and upstream activator7. Class IA PI3Ks are mainly activated by various receptor tyrosine kinases (RTKs) and RAS11. There are three isoforms in Class IA including PI3Kα, PI3Kβ, and PI3Kδ, with the respective p110 catalytic subunit bound to the p85 regulatory subunit. Class IB PI3Kγ, which consists of catalytic subunit p110γ and a regulatory subunit p101 or p84, is mainly activated by G-protein-coupled receptors (GPCRs) such as chemokine receptors12, 13, 14. While the PI3Kα and PI3Kβ are expressed ubiquitously, PI3Kδ and PI3Kγ are mainly in hemopoietic cells15. In particular, PI3Kα is known to play an important role in tumorigenesis because a high frequency of gain-of-function mutations and amplification of PIK3CA, which encodes p110α; this isoform has been found in human cancers16, 17, 18, 19, 20. Additionally, PI3Kα was found to be involved in insulin signaling and glucose metabolism21. PI3Kβ was reported to activate platelets, suggesting a role in the development of thrombotic diseases22. Recently, various reports showed that PI3Kβ predominantly contributed to PIP3 production in PTEN negative cancers, suggesting the key role of PI3Kβ in tumorigenesis with PTEN inactivation23, 24. PI3Kδ and/or γ inactivation leads to a severely impaired immune system25, 26, and blocks the recruitment of neutrophils to the sites of inflammation27, 28, suggesting that these two isoforms are involved in the immune system and inflammation. As the counterpart of PI3K, PTEN is also closely involved in cancer since frequent loss-of-function mutations were found in various human cancers29. In addition, PI3K mutation and PTEN inactivation were reported to cause resistance to cancer therapies targeting the RTKs30. Thus, PI3K is thought to be an attractive target for cancer chemotherapy.
PI3K pathway is closely involved in survival, growth, invasion of cancer cells and tumor angiogenesis. As shown in Fig. 2, after activation by RTK, GPCR or RAS, PI3K phosphorylates PIP2 to produce PIP3; this reaction is reversed by PTEN. PIP3 binds the pleckstrin homology (PH)-domain-containing protein kinases such as Akt and PDK1, to activate and recruit them to the plasma membrane. After recruitment by PIP3, Akt is activated by PDK1 and mTOR complex 2 (mTORC2)3. Activation of Akt promotes cell cycle progression by regulating glycogen synthesis kinase 3β (GSK3β) and the downstream cyclin D1. Akt also acts to maintain cell survival through inhibition of Bcl2-antagonist of cell death (BAD). Furthermore, Akt promotes cell growth by phosphorylation of the downstream mTOR complex 1 (mTORC1)31, which translates mRNAs to protein via the p70S6K-S6 and 4E-BP1-eIF4E pathways32. In addition, hypoxia-inducible factor 1α (HIF-1α) was reported to be up-regulated downstream of mTORC1, and therefore promotes tumor angiogenesis by transcribing vascular endothelial growth factor (VEGF)33. By activating NF-κB and inducing secretion of matrix metalloproteinase (MMP), Akt also promotes cell invasion34. However, phosphorylation of S6K negatively regulates insulin receptor substrate (IRS), leading to a negative feedback loop35, 36, 37. Therefore, inhibition of mTORC1 may activate upstream proteins such as PI3K and Akt38, and consequently reduces the inhibitory potency.
Figure 2.

PI3K/Akt/mTOR pathway involved in tumorigenesis and metastasis. After activation by RTKs, GPCR or RAS, PI3K catalyzes the phosphorylation of PIP2 to generate PIP3, which binds and recruits Akt and PDK1. Akt can be activated by PDK1 and mTORC2, after recruitment by PIP3. By increasing the level of cyclin D1, Akt promotes the cell cycle progression. Akt also acts to maintain cell survival by phosphorylation of BAD and release of the anti-apoptotic protein Bcl-2. Furthermore, Akt regulates cell growth by phosphorylation of the downstream mTORC1, which promotes translation of mRNAs to synthesize protein via p70S6K-S6 and 4E-BP1-eIF4E pathways. In addition, HIF-1α is up-regulated downstream of mTORC1, leading to angiogenesis. By activating NF-κB and inducing secretion of MMP, Akt promotes cell invasion. However, the mTORC1/S6K cascade negatively regulates IRS, which leads to a feedback loop. 4E-BP1, 4E-binding protein 1; GPCR, G protein-coupled receptor; GSK3β, glycogen synthesis kinase 3β; HIF-1, hypoxia-inducible factor 1; IRS, insulin receptor substrate; p70S6K, p70S6 kinase; PDK1, 3-phosphoinositide-dependent protein kinase 1; RTK, receptor tyrosine kinase.
Development of novel PI3K inhibitors attracted a great deal of attention from both academia and industry, while classic PI3K inhibitors LY294002 and wortmannin did not reach clinical trials due to the toxicity and poor druggability. Among the PI3K inhibitors under active development, some are PI3K isoform specific inhibitors like idelalisib (CAL-101) and IPI-145, but most are pan-PI3K inhibitors (Table 1). Besides, some exhibit selectivity over mTOR, such as BKM120, GDC-0941 and ZSTK4747, 39, whereas others show no obvious selectivity, such as NVP-BEZ235, GDC-0980 and GSK21264587, 40 (Table 1).
Table 1.
Selected PI3K inhibitors approved or in clinical trials.
| Inhibitor | Structure | IC50 (nmol/L) |
Isoform specificity | Selectivity over mTOR | Organization (development status) | |||
|---|---|---|---|---|---|---|---|---|
| p110α | p110β | p110δ | p110γ | |||||
| Idelalisib | 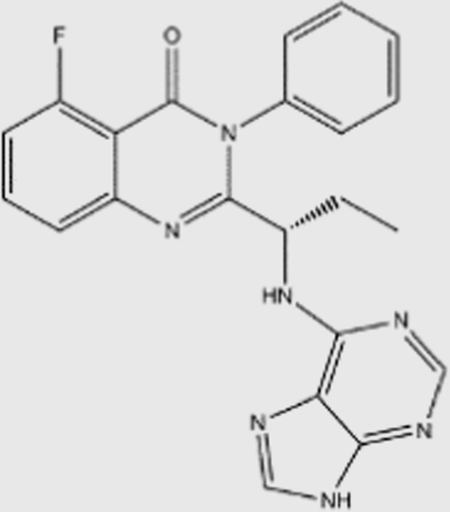 |
820 | 565 | 2.5 | 89 | p110δ specific | unknown | Gilead (launched) |
| IPI-145 | 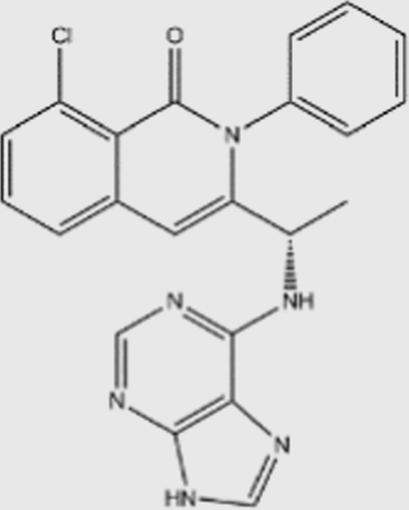 |
1602 | 85 | 3 | 27 | PI3Kδ, γ specific | unknown | Infinity Pharmaceuticals (phase III) |
| NVP-BEZ235 | 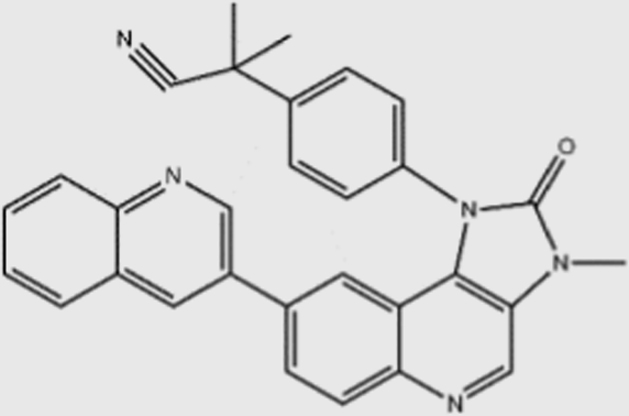 |
4 | 76 | 5 | 7 | pan | No | Novartis (phase I) |
| BKM-120 | 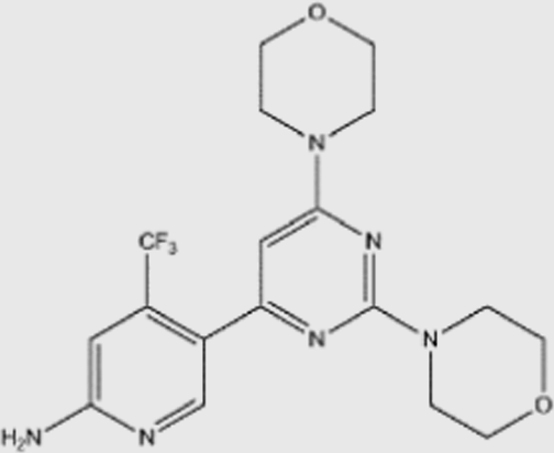 |
52 | 166 | 116 | 262 | pan | Yes | Novartis (phase III) |
| BYL-719 | 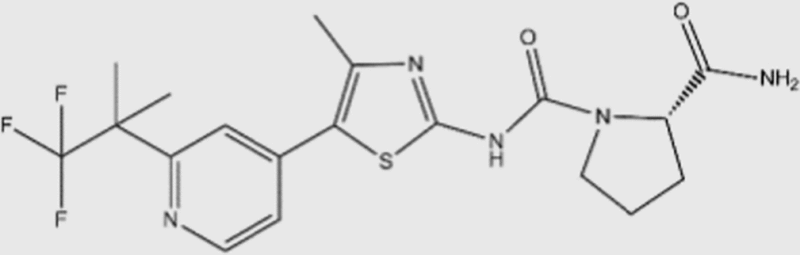 |
5 | 1200 | 290 | 250 | PI3Kα specific | Yes | Novartis (phase III) |
| GDC-0941 | 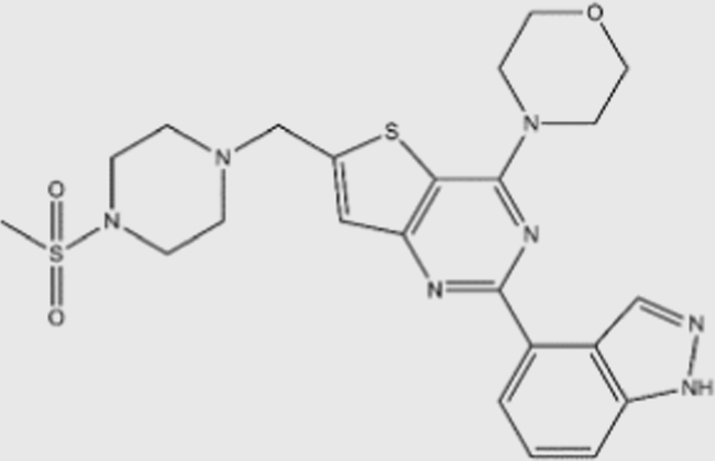 |
3 | 33 | 3 | 75 | pan | Yes | Genentech (phase I) |
| GDC-0980 | 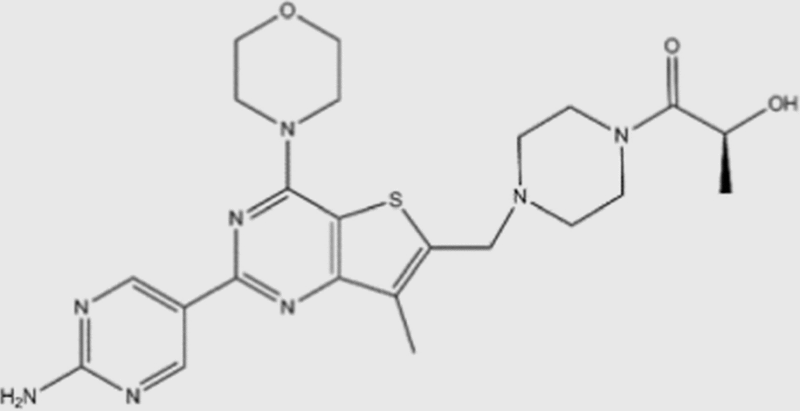 |
5 | 27 | 7 | 14 | pan | No | Genentech (phase II) |
| SF1126 | 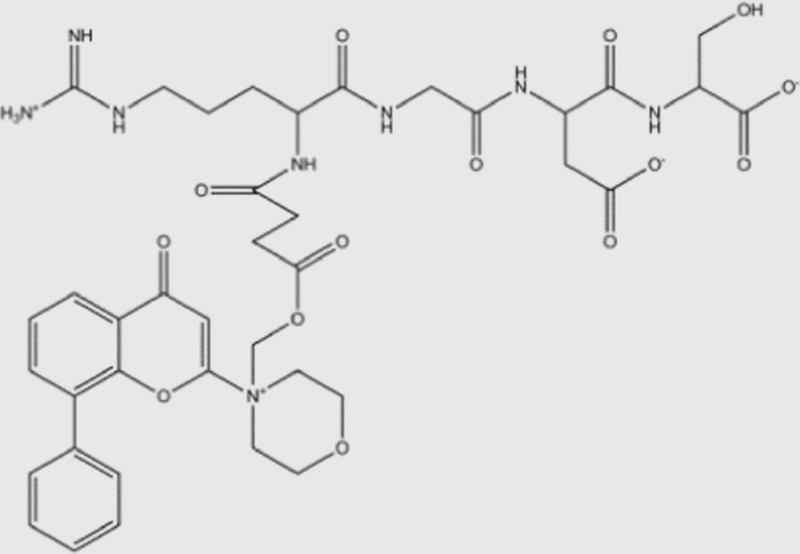 |
NA | NA | NA | NA | pan | No | SignalRx (phase I) |
| PX-866 | 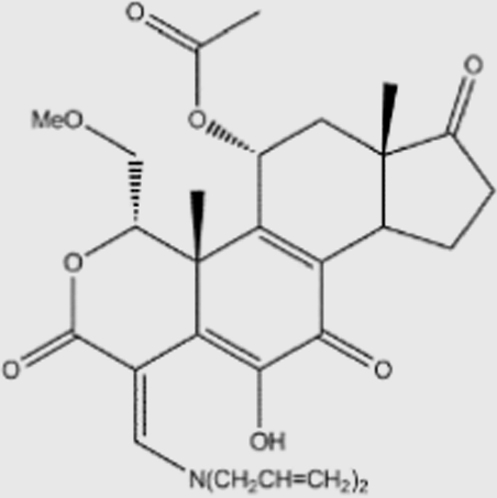 |
6 | >300 | 3 | 9 | pan | unknown | Oncothyreon (phase II) |
| PF-04691502 | 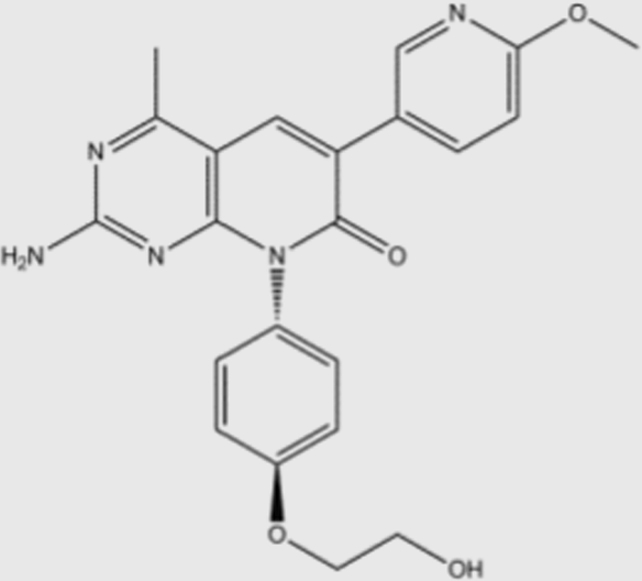 |
1.8a | 2.1a | 1.6a | 1.9a | pan | No | Pfizer (phase I) |
| BAY-80-6946 | 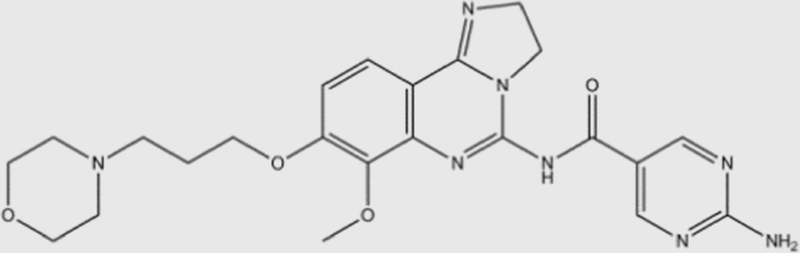 |
0.5 | 3.7 | 6.4 | 0.7 | pan | Yes | Bayer (phase III) |
| XL-765 | 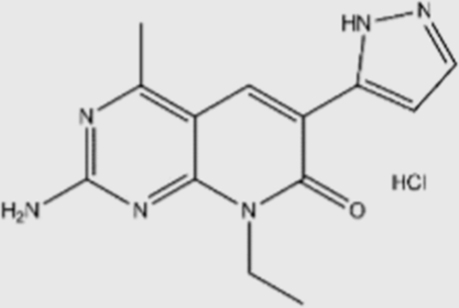 |
39 | 113 | 43 | 9 | pan | No | Sanofi (phase I/II) |
| XL-147 | 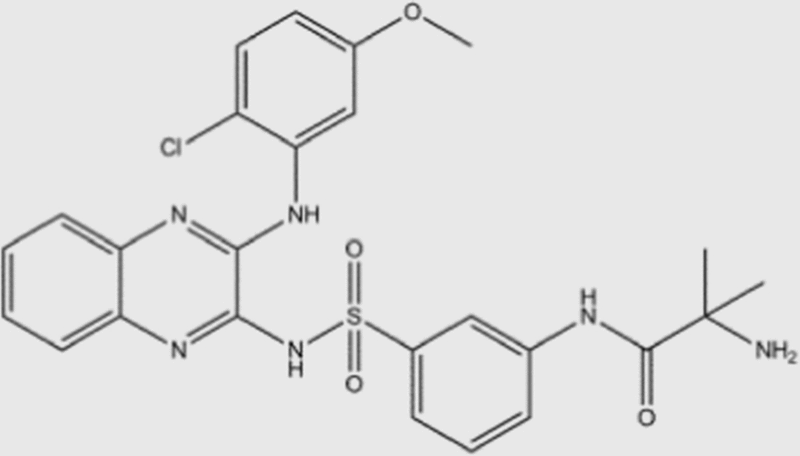 |
39 | 383 | 36 | 23 | pan | Yes | Sanofi (phase I) |
| GSK2126458 | 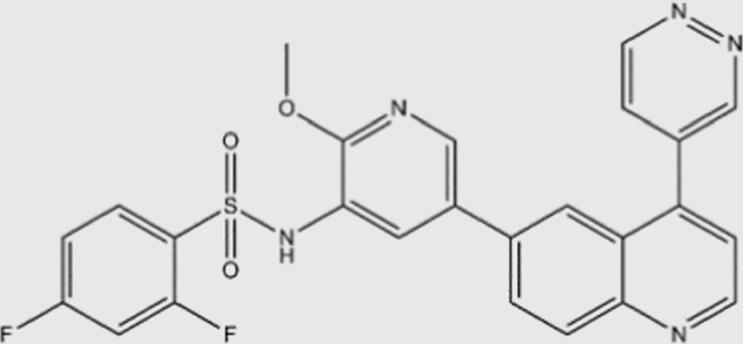 |
0.019 | 0.13 | 0.024 | 0.06 | pan | No | GlaxoSmithKline (phase I) |
| ZSTK474 | 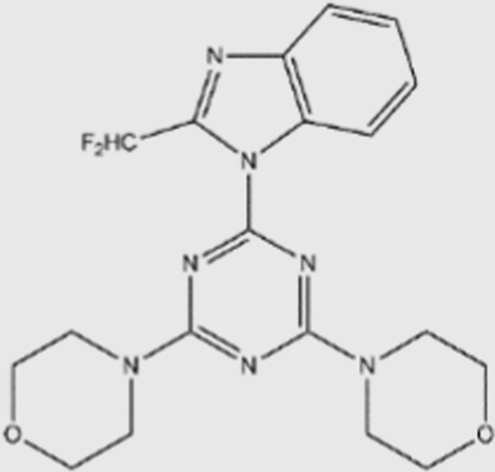 |
16 | 44 | 5 | 49 | pan | Yes | Zenyaku (phase I/II) |
NA, not available.
Ki.
3. PI3K inhibitors approved or in clinical trials
3.1. Idelalisib
Idelalisib was originally developed by Calistoga as CAL-101, but later transited to Gilead. Like the analog IC87114 which has been used as a chemical tool41, idelalisib also specifically targets PI3Kδ, with more than 30-fold selectivity over other PI3K isoforms42. In preclinical studies, idelalisib induces apoptosis in chronic lymphocytic leukemia (CLL), independently of patient genomic features43. In contrast, idelalisib does not exhibit cytotoxicity toward T cells or natural killer cells43. A phase I study in healthy volunteers demonstrated favorable pharmacokinetics with no lymphocyte count change and no notable toxicity during one week of oral administration. In CLL patients, a dose of 150 mg every 12 h was found to be optimal for the phase II study based on the tolerability and pharmacokinetics. In a phase II study, 84% of CLL patients achieved a reduction in lymph node and spleen size of no less than 50%. Median progression-free survival was 15.8 months, with median overall survival (OS) not reached44. Combination studies with other therapies have been investigated. For patients receiving idelalisib with rituximab or bendamustine, over 90% patients achieved a decrease in lymph node size of 50% or greater45. In a phase III trial of idelalisib (150 mg, twice a day) in combination with rituximab (n = 110) in heavily, pretreated patients (median of 3 prior therapies) with relapsed CLL, an overall response rate of 81% and overall survival of 91% at 12 months were obtained. The incidence of grade 3 or higher adverse events included neutropenia (34%), thrombocytopenia (10%), anemia (5%), elevation in transaminases (5%), and diarrhea (4%)46. Collectively, favorable efficacy and mild toxicity of idelalisib was observed in clinical studies. In addition, a population pharmacokinetic model has been established for idelalisib and its inactive metabolite from the data of phase I or II studies47. Idelalisib was approved in 2014 for therapy of relapsed CLL in combination with rituximab, and for monotherapy of relapsed follicular lymphoma (FL) or relapsed small lymphocytic lymphoma (SLL), as the first PI3K inhibitor licensed for cancer treatment.
3.2. IPI-145
IPI-145 was originally developed by Intellikine with the name of INK-1197. With a structure similar to idelalisib, IPI-145 also preferentially inhibits the PI3Kδ isoform with an IC50 of 3 nmol/L, but inhibits PI3Kγ potently as well with an IC50 of 27 nmol/L48. IPI-145 sensitizes B-cell receptor (BCR)–stimulated and/or stromal co-cultured primary CLL cells to apoptosis. This compound also potently inhibits cytokine-induced proliferation of CLL cells, without obvious cytotoxicity to normal B- and T-lymphocytes, suggesting the favorable efficacy on CLL49. Clinical results demonstrated the efficacy on indolent non-Hodgkin׳s lymphoma (NHL) and CLL49. The rate of nodal response exceeds 70% for treatment of CLL with oral IPI-14550. In addition, IPI-145 shows promise as an agent for the treatment of autoimmune and inflammatory diseases48. Presently, IPI-145 is in phase III clinical trials for therapy of NHL, CLL and follicular lymphoma.
3.3. NVP-BEZ235
NVP-BEZ235 was developed by Novartis as a PI3K inhibitor with IC50 values of 4, 76, 5 and 7 nmol/L for PI3Kα, β, δ and γ, respectively51. This agent is described as a dual PI3K/mTOR inhibitor since it also potently inhibits mTOR51, 52. We reported that DNA-PK was inhibited by NVP-BEZ235 with a similar potency to its PI3Kα inhibition53. NVP-BEZ235 potently inhibited growth of a panel of cancer cells, induced cell cycle arrest at G1 phase51, 54, and blocked tumor angiogenesis55. Interestingly, NVP-BEZ235 exhibited equal efficacy on HER2-positive breast cancer BT474 cells with p110α mutations compared to those cells with wild type p110α54, suggesting the potential application to patients with p110α mutations which are often resistant to Herceptin (a humanized monoclonal HER-2 antibody). NVP-BEZ235 is now undergoing evaluation in phase II clinical trials for the treatment of bladder cancer, pancreatic cancer as well as perivascular epithelioid cell tumors.
3.4. BKM120
Also developed by Novartis, BKM120 is a pan-PI3K inhibitor that targets all four isoforms of class I PI3K, but with more than 10-fold selectivity over mTOR39. Evaluation in a panel of 353 cancer cell lines showed a preferential inhibition of tumor cell lines with PIK3CA mutation for BKM120. Our multifaceted study on BKM120 and other PI3K inhibitors using various biochemical and cell-based assays suggest that BKM120 might have a unique mechanism, possibly independent of PI3K inhibition56. Potent antitumor effects in vivo were indicated in tumor bearing models. In addition, BKM120 blocks VEGF-induced neovascularization in vivo, suggesting antiangiogenic activity39. Combinational use of BKM120 with cisplatin exhibits enhanced efficacy57. A phase I clinical study in Japan showed that BKM120 had a manageable safety profile, and could be rapidly absorbed in a dose-proportional manner58. Clinical efficacy and tolerability of BKM120 were also evaluated in postmenopausal women with estrogen receptor–positive metastatic breast cancer, in combination with fulvestrant using daily or intermittent schedules (days 1–5 each week). Compared with daily dosing, intermittent use of BKM120 led to less frequent adverse effects. The maximum tolerated dose of BKM120 was defined as 100 mg daily. The clinical benefit rate was 58.6%, and the median progression-free survival was 12.4 months for all evaluable 29 patients59. While the response was not relevant to PIK3CA mutation, patients with PTEN negative, progesterone receptor (PgR) expression, or TP53 mutation exhibited resistance to BKM12059. BKM120 is now under study for breast cancer therapy in phase III clinical trials. It also holds promise for the treatment of head and neck squamous cell carcinoma (HNSCC), non-small cell lung carcinoma (NSCLC), lymphoma and glioblastoma multiforme60.
3.5. BYL719
BYL719 is another PI3K inhibitor developed by Novartis. Different from either NVP-BEZ235 or BKM-120, BYL719 is a PI3Kα specific inhibitor with an IC50 of 5 nmol/L61. In vivo results showed that BYL719 dose- and time-dependently inhibited the PI3K signaling, and exhibited robust antitumor efficacy with good tolerability. PIK3CA was indicated to be the foremost predictive biomarker for sensitivity to BYL71962. A combination study reported that BYL719 could enhance the effect of protein kinase C inhibitor in G-protein mutant uveal melanoma cells63. Presently, BYL719 is under evaluation for breast cancer therapy in phase III clinical trial, and phase II study for treatment of NSCLC and HNSCC are ongoing.
3.6. GDC-0941
GDC-0941 is developed by Genentech via structural modification of PI-10364, which was reported to have unfavorable pharmacokinetic property65. GDC-0941 inhibited PI3Kα, β, δ and γ with IC50 as 3, 33, 3 and 75 nmol/L, respectively. Weaker inhibition against mTOR with IC50 of 580 nmol/L suggested the PI3K inhibitory selectivity. GDC-0941 arrested cell cycle at G1 phase66, and induced apoptosis in a subset of cancer cells66. As an orally administered agent, GDC-0941 exhibited significant in vivo antitumor efficacy on human cancer xenografts64, 66, 67. Furthermore, combination of GDC-0941 with trastuzumab, petuzumab, or chemotherapeutic drug docetaxel enhanced their antitumor efficacy67. GDC0941 is presently under evaluation for treatment of solid tumors and NHL in phase I clinical trials.
3.7. GDC-0980
GDC-0980 is also a PI3K inhibitor developed by Genentech. In contrast to GDC-0941, GDC-0980 also potently inhibits mTOR and therefore is described as a dual PI3K/mTOR inhibitor40. In vitro and in vivo results indicated that GDC-0980 exhibited potent growth inhibition against breast, prostate, and lung cancer but relatively less activity on melanoma and pancreatic cancers with KRAS and BRAF mutations40. Combinational use of GDC-0980 enhanced the antitumor activity of docetaxel40. Salphati et al. investigated the absorption and disposition of GDC-0980, and suggested 55 mg once daily might be a clinically efficacious dose. In phase I trials, modest but durable activity was shown in patients with advanced solid tumors68. GDC-0980 is now under evaluation in phase II clinical trials for therapy of kidney, prostate, endometrial and breast cancers68.
3.8. SF1126
SF1126 is a prodrug of LY294002 by conjugation with Arg-Gly-Asp-Ser (RGDS), aiming to increase solubility and to target tumors via binding to specific integrins in the tumor microenvironment. Compared with the parent drug LY294002, SF1126 showed higher distribution in tumor tissues, and therefore more favorable in vivo antitumor efficacy without severe toxicity69. In addition, SF1126 exhibited anti-angiogenic activity, which might be attributed to the RGDS part since it targets the angiogenic integrins aνβ3 and a5β169. Moreover, combination of SF1126 with taxotere led to dramatic regression of PC3 tumors, superior to monotherapy using either taxotere or SF112669. SF1126 is under investigation in phase I clinical trials for therapy of solid cancer, and relapsed or refractory neuroblastoma.
3.9. PX-866
PX-866 is a structure-modified compound based on classic PI3K inhibitor wortmannin, which failed to enter clinical trial due to the bad stability and liver toxicity70. PX-866 overcomes such defects and exhibits remarkable in vivo antitumor efficacy via both oral and i.v. administrations70, 71. In contrast to most of the other novel PI3K inhibitors in clinical trials, PX-866 is an irreversible PI3K inhibitor. Interestingly, PX-866 was reported to show similar effect on tumors with RAS mutation which were regarded to be resistant to PI3K inhibitors, compared to those with wild type RAS72. PX-866 is presently evaluated for treatment of glioblastoma multiforme in phase II clinical trial.
3.10. PF-04691502
PF-04691502 was developed by Pfizer as an orally active, dual inhibitor of PI3K and mTOR73. In vitro, PF-04691502 inhibits proliferation of U87 (PTEN null) and SKOV3 (PIK3CA mutation) at sub-micromolar concentrations. Favorable antitumor efficacy was also confirmed in xenograft models of the above two cancer cells, as well as gefitinib- and erlotinib-resistant NSCLC cells73. Moreover, PF-04691502 can enhance TP53/p73 expression in human HNSCC xenograft with wild type TP53 and significantly inhibit tumor growth74. Phase I results indicated that oral administration of PF-04691502 was tolerable at 8 mg once daily75. PF-04691502 is now under evaluation in phase I clinical trials.
3.11. BAY 80-6946
BAY 80-6946 is a highly-selective pan-PI3K inhibitor developed by Bayer76. In vitro, BAY 80-6946 showed superior antitumor activity in breast cancer cell lines with PIK3CA and/or HER2 overexpression, compared to those with wild-type PIK3CA but without HER2. Distinct from most of the other novel PI3K inhibitors in clinical trials, BAY 80-6946 is administered intravenously. In vivo, BAY 80-6946 induced 100% complete tumor regression when dosed as a single agent every second day in rats bearing HER2-amplified and PIK3CA-mutant KPL4 breast tumors76. BAY 80-6946 is being evaluated in phase III clinical trial for therapy of B-cell lymphoma.
3.12. XL-765
Originally developed by Exelixis as a PI3K/mTOR inhibitor, XL-765 inhibits PI3Kα, β, δ and γ with IC50 values of 39, 383, 36 and 23 nmol/L, respectively, and also shows inhibition against mTOR with an IC50 of 157 nmol/L77. XL765 inhibits the phosphorylation of Akt, p70S6K, and S6 both in vitro and in vivo dose-dependently78. Besides, combination with autophagy inhibitor chloroquine showed efficacy on pancreatic adenocarcinoma79, and co-treatment with temozolomide enhanced the activity of the latter on invasive pituitary adenoma xenograft model80. Presently, XL765 is under evaluation in phase I/II clinical trials for therapy of patients with malignant neoplasms81.
3.13. XL147
Also first developed by Exelixis, XL-147 exhibits a comparable activity to XL-765 in terms of PI3K inhibition, but does not inhibit mTOR even at 10 μmol/L77, 82. Like XL765, XL147 also showed potent antitumor efficacies in xenograft models of various human cancer cells, via blockade of PI3K/Akt pathway78, 83. However, XL147 is less active than XL765 in hormone-insensitive prostate cancer models, which was attributed to its weak mTOR inhibition84. Preliminary result in phase I clinical trial already showed that both XL147 and XL765 are tolerable81, 85. XL147 is now under evaluation in phase I clinical trials for therapy of solid tumors as well as lymphoma.
3.14. GSK2126458
GSK2126458 was developed by GlaxoSmithKline as dual PI3K/mTOR inhibitor86. Durable responses were observed in its first-in-human phase I study in patients with solid tumors like sarcoma, kidney, breast, endometrial, oropharyngeal and bladder cancer87. Fasting insulin and glucose levels were indicated to act as pharmacodynamics biomarkers while PIK3CA mutation showed not predictive of response87. Additionally, combination with the MEK (MAPK/ERK kinase) inhibitor AZD6244 showed favorable efficacy on castration-resistant prostate cancer via blocking both the RAS/RAF/MEK/ERK and PI3K/Akt/mTOR pathways simultaneously88. Presently, GSK2126458 is undergoing phase I clinical trials for therapy of solid tumors, idiopathic pulmonary fibrosis and lymphoma.
3.15. ZSTK474
ZSTK474 is an S-triazine derivative synthesized by Zenyaku as an anticancer drug candidate together with more than 1500 other analogs89. It was identified as a PI3K inhibitor by Compare Analysis using the JFCR39 cancer cell line panel coupled with a drug-activity database90, 91. ZSTK474 competed with ATP (Fig. 3A) in inhibiting all four PI3K isoforms, with IC50 values of 16, 44, 5 and 49 nnol/L for PI3Kα, β, δ and γ, respectively, suggesting that it is a pan-PI3K inhibitor92. However, it showed far weaker inhibitory activity against mTOR and DNA-PK, compared to the PI3K inhibition52, 53, 92. Furthermore, it did not inhibit a panel of 139 protein kinases90. In vitro, ZSTK474 inhibited the growth of 39 human cancer cell lines with a mean 50% growth inhibition (GI50) value of 0.32 μmol/L90 (Fig. 3B), and blocked cell cycle progression at G1 phase in various human cancer cells (Fig. 3C)90, 93, 94. The G1 arrest effect might be attributed to downregulation of cyclin D1 and upregulation of p2793, 94. Moreover, ZSTK474 showed anti-angiogenic effect in vitro and in vivo95 (Fig. 3D). The in vivo anti-angiogenic effect was attributed to its dual inhibition mechanism: inhibition of VEGF secretion in cancer cells and direct inhibition of PI3K in endothelial cells96. In addition, ZSTK474 inhibited migration and invasion of prostate cancer cell PC3, and blocked the phosphorylation of Girdin and production of MMPs, suggesting the anti-metastatic activity34. Like other novel PI3K inhibitors such as NVP-BEZ235, BKM120 and GDC-0941, ZSTK474 inhibits both mutant PIK3CA and the wild type one56. While it does not induce obvious apoptosis generally, it indeed induces autophagy in MCF-7 breast cancer cells94. As an orally administered pan-PI3K inhibitor, ZSTK474 showed potent in vivo antitumor efficacy on cancer xenografts at both early and advanced stages (Fig. 3E), without obvious toxicity being observed90, 93, 95. However, acquired resistance after long term treatment of ZSTK474 has been observed, which might be attributed to over-expression of insulin-like growth factor 1 receptor (IGF1R)95. In addition, combination with imatinib exhibited synergic antileukemia activity on chronic myeloid leukemia K562 cells as well as adriamycin-resistant K562/A02 cells96. ZSTK474 is now under evaluation in phase I/II clinical trials for treatment of solid tumors.
Figure 3.

Antitumor activities of ZSTK474. (A) ZSTK474 inhibits PI3K by occupying the ATP binding pocket89. (B) Fingerprint of ZSTK474 for the JFCR39 panel. Fingerprint indicates the differential growth inhibition pattern of ZSTK474 for the cell lines in JFCR39 panel. The X-axis shows difference in logarithmic scale between the mean of logGI50 values for all 39 cell lines (MG-MID, expressed as 0 in the fingerprint) and the logGI50 for each cell line in JFCR39 panel. Columns to the right of 0 indicate the sensitivity of the cell lines to a given compound and columns to the left indicate the resistance. MG-MID is mean of logGI50 values for all 39 cell lines89. (C) ZSTK474 induces G1 arrest in breast cancer MCF-7 cells94. (D) In vitro antiangiogenic effect of ZSTK474. ZSTK474 potently blocks the in vitro tube formation by HUVECs95. (E) In vivo antitumor efficacy of ZSTK474. Oral administration of ZSTK474 at 400 mg/kg to WiDr xenograft daily from day 0 to 26, except for days 6, 13 and 20, leads to obvious tumor growth inhibition90.
4. Discussion
An exciting race for development of PI3K inhibitor as anticancer drug candidate between pharmaceutical companies has lasted since 2006 when ZSTK474 was first reported90. Currently, over a dozen of PI3K inhibitors are still in clinical trials for cancer therapy. Among them, there are PI3K isoform specific inhibitors, pan-PI3K inhibitors, dual PI3K/mTOR inhibitors, as well as a prodrug of PI3K inhibitor. Interestingly, the main companies involved in the development of PI3K inhibitors, including Novartis, Genentech, as well as Sanofi (Table 1), have various types of PI3K inhibitors evaluated in clinical trials. The reason could be to maximize chances of clinical success because a key question still remains to be answered: what is the most effective and tolerable inhibition profile of an inhibitor targeting PI3K superfamily? That is, whether an isoform-specific PI3K inhibitor is superior to that targeting all PI3K isoforms, and whether dual inhibition of PI3K and mTOR is superior to specifically targeting PI3K. The PI3Kδ-specific inhibitor idelalisib has been approved for treatment of acute lymphocytic leukemia (ALL), suggesting that at least targeting PI3Kδ is enough for lymphoma and leukemia therapy. Considering that the existence of PI3Kδ is mainly restricted to hemopoietic cells, inhibitors specifically targeting this isoform might be preferred to avoid possible metabolic side effects due to inhibition of PI3Kα and β. On the other hand, since the PI3Kα-specific inhibitor BYL719, the pan-PI3K inhibitor BKM120 and the dual PI3K/mTOR inhibitor GDC-0980 have passed phase I clinical trials, the tolerability might not be a problem for PI3K inhibitors with or without PI3Kα isoform specificity as well as PI3K/mTOR selectivity. Regarding the selectivity over mTOR, it was reported that additional inhibition of mTOR is required for the sensitivity to PI3Kα specific inhibitor BYL719 in PIK3CA mutant breast cancer97, although clinical evidence is needed. To answer the question of what is the most desirable type of PI3K inhibitor, the tumor lineage and the genetic status of cancer should also be considered. As an example, while many reports suggest targeting PI3Kβ could treat PTEN negative cancers, one study showed that PI3Kβ-specific inhibitors had no effect on endometrial cancer (EEC) cell lines98. In any case, the more results of clinical trials are reported, the more we will know about which type of PI3K inhibitor is most favorable for specific types of cancer.
Biomarker development has become tremendously important for precision therapy of cancer. Predicative biomarker can help to identify patients who are most likely to benefit from specific treatments, thereby optimizing personalized medical treatment. While some preclinical reports indicated that PIK3CA mutation could serve as predictive biomarkers for treatment with various types of PI3K inhibitors39, 40, 62, 70, contrary conclusions have also been obtained in other reports54, 56. Moreover, early clinical trial results do not support the conclusion as well99.
Since PI3K inhibitors are generally cytostatic but not cytotoxic agents, their antitumor efficacy should be tumor stabilization rather than tumor regression in patients with advanced solid tumors. On the other hand, besides PI3K, Akt can be activated by other kinases like Ack1, TBK1 and DNA-PK100. In addition, it was reported that PI3Kα inhibition led to rebound of PIP3, which was produced by the PI3Kβ isoform101. Therefore, combination of PI3K inhibitors with standard chemotherapeutic drugs, radiation therapy, as well as other molecular-targeted drugs should increase efficacy. This conclusion is supported by growing evidence. For example, when combined with paclitaxel, a lower dose of BAY 80-6946 was sufficient to induce complete tumor regression in mice76. In contrast to a single use of PF-04691502 which failed to stop tumor progression, combination with MEK inhibitor PD-0325901 led to obvious tumor regression in a model with KRAS mutation102.
A large body of scientific work has demonstrated that targeting PI3K is an important cancer therapy strategy. The ongoing clinical evaluation on PI3K inhibitors is expected to provide a new class of molecular targeted anticancer drugs.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Toker A., Cantley L.C. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 2.Fruman D.A., Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke T.F., Kaplan D.R., Cantley L.C. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd P.R., Withers D.J., Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J. 1998;333:471–490. doi: 10.1042/bj3330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong D.X., Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99:1734–1740. doi: 10.1111/j.1349-7006.2008.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong D., Yamori T. Advances in development of phosphatidylinositol 3-kinase inhibitors. Curr Med Chem. 2009;16:2839–2854. doi: 10.2174/092986709788803222. [DOI] [PubMed] [Google Scholar]
- 8.Wymann M.P., Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck B., Leevers S.J., Ahmadi K., Timms J., Katso R., Driscoll P.C. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 10.Marone R., Cmiljanovic V., Giese B., Wymann M.P. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Viciana P., Warne P.H., Dhand R., Vanhaesebroeck B., Gout I., Fry M.J. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 12.Hennessy B.T., Smith D.L., Ram P.T., Lu Y.L., Mills G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 13.Vanhaesebroeck B., Ali K., Bilancio A., Geering B., Foukas L.C. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Rommel C., Camps M., Ji H. PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 15.Vanhaesebroeck B., Welham M.J., Kotani K., Stein R., Warne P.H., Zvelebil M.J. P110δ, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94:4330–4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samuels Y., Wang Z.H., Bardelli A., Silliman N., Ptak J., Szabo S. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 17.Levine D.A., Bogomolniy F., Yee C.J., Lash A., Barakat R.R., Borgen P.I. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 18.Whyte D.B., Holbeck S.L. Correlation of PIK3CA mutations with gene expression and drug sensitivity in NCI-60 cell lines. Biochem Biophys Res Commun. 2006;340:469–475. doi: 10.1016/j.bbrc.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 19.Shayesteh L., Lu Y., Kuo W.L., Baldocchi R., Godfrey T., Collins C. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 20.Campbell I.G., Russell S.E., Choong D.Y., Montgomery K.G., Ciavarella M.L., Hooi C.S. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 21.Knight Z.A., Gonzalez B., Feldman M.E., Zunder E.R., Goldenberg D.D., Williams O. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson S.P., Schoenwaelder S.M., Goncalves I., Nesbitt W.S., Yap C.L., Wright C.E. PI 3-kinase p110β: a new target for antithrombotic therapy. Nat Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 23.Jia S.D., Liu Z.N., Zhang S., Liu P.X., Zhang L., Lee S.H. Essential roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wee S., Wiederschain D., Maira S.M., Loo A., Miller C., deBeaumont R. PIK3CB-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okkenhaug K., Bilancio A., Farjot G., Priddle H., Sancho S., Peskett E. Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 26.Del Prete A., Vermi W., Dander E., Otero K., Barberis L., Luini W. Defective dendritic cell migration and activation of adaptive immunity in PI3Kγ-deficient mice. EMBO J. 2004;23:3505–3515. doi: 10.1038/sj.emboj.7600361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirsch E., Katanaev V.L., Garlanda C., Azzolino O., Pirola L., Silengo L. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 28.Puri K.D., Doggett T.A., Douangpanya J., Hou Y.H., Tino W.T., Wilson T. Mechanisms and implications of phosphoinositide 3-kinase δ in promoting neutrophil trafficking into inflamed tissue. Blood. 2004;103:3448–3456. doi: 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- 29.Yuan T.L., Cantley L.C. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LoPiccolo J., Blumenthal G.M., Bernstein W.B., Dennis P.A. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wendel H.G., de Stanchina E., Fridman J.S., Malina A., Ray S., Kogan S. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 32.Giles F.J., Albitar M. Mammalian target of rapamycin as a therapeutic target in leukemia. Curr Mol Med. 2005;5:653–661. doi: 10.2174/156652405774641034. [DOI] [PubMed] [Google Scholar]
- 33.Skinner H.D., Zheng J.Z., Fang J., Agani F., Jiang B.H. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1α, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279:45643–45651. doi: 10.1074/jbc.M404097200. [DOI] [PubMed] [Google Scholar]
- 34.Zhao W.N., Guo W.Z., Zhou Q.X., Ma S.N., Wang R., Qiu Y.L. In vitro antimetastatic effect of phosphatidylinositol 3-kinase inhibitor ZSTK474 on prostate cancer PC3 cells. Int J Mol Sci. 2013;14:13577–13591. doi: 10.3390/ijms140713577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrington L.S., Findlay G.M., Gray A., Tolkacheva T., Wigfield S., Rebholz H. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Vogt P.K., Kang S. Kinase inhibitors: vice becomes virtue. Cancer Cell. 2006;9:327–328. doi: 10.1016/j.ccr.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 38.Sun S.Y., Rosenberg L.M., Wang X.R., Zhou Z.M., Yue P., Fu H.A. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 39.Maira S.M., Pecchi S., Huang A., Burger M., Knapp M., Sterker D. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 40.Wallin J.J., Edgar K.A., Guan J., Berry M., Prior W.W., Lee L. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10:2426–2436. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 41.Sadhu C., Masinovsky B., Dick K., Sowell C.G., Staunton D.E. Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J Immunol. 2003;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 42.Lannutti B.J., Meadows S.A., Herman S.E., Kashishian A., Steiner B., Johnson A.J. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herman S.E., Gordon A.L., Wagner A.J., Heerema N.A., Zhao W.Q., Flynn J.M. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown J.R., Byrd J.C., Coutre S.E., Benson D.M., Flinn I.W., Wagner-Johnston N.D. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–3397. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrd J.C., Woyach J.A., Johnson A.J. Translating PI3K-δ inhibitors to the clinic in chronic lymphocytic leukemia: the story of CAL-101 (GS1101) Am Soc Clin Oncol Educ Book. 2012;2012:691–694. doi: 10.14694/EdBook_AM.2012.32.75. [DOI] [PubMed] [Google Scholar]
- 46.Shah A., Mangaonkar A. Idelalisib: a novel PI3Kδ inhibitor for chronic lymphocytic leukemia. Ann Pharmacother. 2015;49:1162–1170. doi: 10.1177/1060028015594813. [DOI] [PubMed] [Google Scholar]
- 47.Jin F., Gao Y.Y., Zhou H.F., Fang L., Li X.M., Ramanathan S. Population pharmacokinetic modeling of idelalisib, a novel PI3Kδ inhibitor, in healthy subjects and patients with hematologic malignancies. Cancer Chem Pharm. 2016;77:89–98. doi: 10.1007/s00280-015-2891-8. [DOI] [PubMed] [Google Scholar]
- 48.Winkler D.G., Faia K.L., DiNitto J.P., Ali J.A., White K.F., Brophy E.E. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol. 2013;20:1364–1374. doi: 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Balakrishnan K., Peluso M., Fu M., Rosin N.Y., Burger J.A., Wierda W.G. The phosphoinositide-3-kinase (PI3K)-δ and γ inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29:1811–1822. doi: 10.1038/leu.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang J.E., Kahl B.S. PI3-kinase inhibitors in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2014;9:33–43. doi: 10.1007/s11899-013-0189-7. [DOI] [PubMed] [Google Scholar]
- 51.Maira S.M., Stauffer F., Brueggen J., Furet P., Schnell C., Fritsch C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 52.Kong D.X., Dan S., Yamazaki K., Yamori T. Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR39. Eur J Cancer. 2010;46:1111–1121. doi: 10.1016/j.ejca.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Kong D.X., Yaguchi S.I., Yamori T. Effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor, on DNA-dependent protein kinase. Biol Pharm Bull. 2009;32:297–300. doi: 10.1248/bpb.32.297. [DOI] [PubMed] [Google Scholar]
- 54.Serra V., Markman B., Scaltriti M., Eichhorn P.J., Valero V., Guzman M. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 55.Schnell C.R., Stauffer F., Allegrini P.R., O׳Reilly T., McSheehy P.M., Dartois C. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68:6598–6607. doi: 10.1158/0008-5472.CAN-08-1044. [DOI] [PubMed] [Google Scholar]
- 56.Kong D.X., Yamori T., Yamazaki K., Dan S. In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA. Invest New Drugs. 2014;32:1134–1143. doi: 10.1007/s10637-014-0152-z. [DOI] [PubMed] [Google Scholar]
- 57.Bassi C., Ho J., Srikumar T., Dowling R.J., Gorrini C., Miller S.J. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–399. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ando Y., Inada-Inoue M., Mitsuma A., Yoshino T., Ohtsu A., Suenaga N. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105:347–353. doi: 10.1111/cas.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma C.X., Luo J., Naughton M., Ademuyiwa F., Suresh R., Griffith M. A phase I trial of BKM120 (Buparlisib) in combination with fulvestrant in postmenopausal women with estrogen receptor–positive metastatic breast cancer. Clin Cancer Res. 2016;22:1583–1591. doi: 10.1158/1078-0432.CCR-15-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massacesi C., Di Tomaso E., Urban P., Germa C., Quadt C., Trandafir L. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–210. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Furet P., Guagnano V., Fairhurst R.A., Imbach-Weese P., Bruce I., Knapp M. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase α inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 62.Fritsch C., Huang A., Chatenay-Rivauday C., Schnell C., Reddy A., Liu M. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 63.Musi E., Ambrosini G., de Stanchina E., Schwartz G.K. The phosphoinositide 3-kinase α selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol Cancer Ther. 2014;13:1044–1053. doi: 10.1158/1535-7163.MCT-13-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Folkes A.J., Ahmadi K., Alderton W.K., Alix S., Baker S.J., Box G. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 65.Raynaud F.I., Eccles S., Clarke P.A., Hayes A., Nutley B., Alix S. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 66.O׳Brien C., Wallin J.J., Sampath D., GuhaThakurta D., Savage H., Punnoose E.A. Predictive biomarkers of sensitivity to the phosphatidylinositol 3ʹ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16:3670–3683. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 67.Yao E., Zhou W., Lee-Hoeflich S.T., Truong T., Haverty P.M., Eastham-Anderson J. Suppression of HER2/HER3-mediated growth of breast cancer cells with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and pertuzumab. Clin Cancer Res. 2009;15:4147–4156. doi: 10.1158/1078-0432.CCR-08-2814. [DOI] [PubMed] [Google Scholar]
- 68.Salphati L., Pang J., Plise E.G., Lee L.B., Olivero A.G., Prior W.W. Preclinical assessment of the absorption and disposition of the phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor GDC-0980 and prediction of its pharmacokinetics and efficacy in human. Drug Metab Dispos. 2012;40:1785–1796. doi: 10.1124/dmd.112.046052. [DOI] [PubMed] [Google Scholar]
- 69.Garlich J.R., De P., Dey N., Su J.D., Peng X.D., Miller A. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 70.Ihle N.T., Williams R., Chow S., Chew W., Berggren M.I., Paine-Murrieta G. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:763–772. [PubMed] [Google Scholar]
- 71.Ihle N.T., Paine-Murrieta G., Berggren M.I., Baker A., Tate W.R., Wipf P. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther. 2005;4:1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ihle N.T., Lemos R., Jr, Wipf P., Yacoub A., Mitchell C., Siwak D. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–150. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan J., Mehta P.P., Yin M.J., Sun S.X., Zou A.H., Chen J. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10:2189–2199. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]
- 74.Herzog A., Bian Y.S., Vander Broek R., Hall B., Coupar J., Cheng H. PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res. 2013;19:3808–3819. doi: 10.1158/1078-0432.CCR-12-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Britten C.D., Adjei A.A., Millham R., Houk B.E., Borzillo G., Pierce K. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs. 2014;32:510–517. doi: 10.1007/s10637-013-0062-5. [DOI] [PubMed] [Google Scholar]
- 76.Liu N.S., Rowley B.R., Bull C.O., Schneider C., Haegebarth A., Schatz C.A. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 77.Patnaik A., LoRusso P.M., Tabernero J., Laird A.D., Aggarwal S.K., Papadopoulos K.P. Biomarker development for XL765, a potent and selective oral dual inhibitor of PI3K and mTOR currently being administered to patients in a Phase I clinical trial. Mol Cancer Ther. 2007;6:3516S. [Google Scholar]
- 78.Yu P.W., Laird A.D., Du X.N., Wu J.M., Won K.A., Yamaguchi K. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014;13:1078–1091. doi: 10.1158/1535-7163.MCT-13-0709. [DOI] [PubMed] [Google Scholar]
- 79.Mirzoeva O.K., Hann B., Hom Y.K., Debnath J., Aftab D., Shokat K. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med. 2011;89:877–889. doi: 10.1007/s00109-011-0774-y. [DOI] [PubMed] [Google Scholar]
- 80.Dai C.X., Zhang B., Liu X.H., Ma S.H., Yang Y.K., Yao Y. Inhibition of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity in pituitary adenoma cell lines in vitro and xenografted pituitary adenoma in female nude mice. Endocrinology. 2013;154:1247–1259. doi: 10.1210/en.2012-1908. [DOI] [PubMed] [Google Scholar]
- 81.Papadopoulos K.P., Tabernero J., Markman B., Patnaik A., Tolcher A.W., Baselga J. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res. 2014;20:2445–2456. doi: 10.1158/1078-0432.CCR-13-2403. [DOI] [PubMed] [Google Scholar]
- 82.Shapiro G.I., Edelman G., Calvo E., Aggarwal S.K., Laird A.D. Targeting aberrant PI3K pathway signaling with XL147, a potent, selective and orally bioavailable PI3K inhibitor. Mol Cancer Ther. 2007;6:3594S. [Google Scholar]
- 83.Foster P., Yamaguchi K., Hsu P.P., Qian F., Du X.N., Wu J.M. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther. 2015;14:931–940. doi: 10.1158/1535-7163.MCT-14-0833. [DOI] [PubMed] [Google Scholar]
- 84.Gravina G.L., Mancini A., Scarsella L., Colapietro A., Jitariuc A., Vitale F. Dual PI3K/mTOR inhibitor, XL765 (SAR245409), shows superior effects to sole PI3K [XL147 (SAR245408)] or mTOR [rapamycin] inhibition in prostate cancer cell models. Tumour Biol. 2016;37:341–351. doi: 10.1007/s13277-015-3725-3. [DOI] [PubMed] [Google Scholar]
- 85.Shapiro G.I., Rodon J., Bedell C., Kwak E.L., Baselga J., Braña I. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:233–245. doi: 10.1158/1078-0432.CCR-13-1777. [DOI] [PubMed] [Google Scholar]
- 86.Knight S.D., Adams N.D., Burgess J.L., Chaudhari A.M., Darcy M.G., Donatelli C.A. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munster P., Aggarwal R., Hong D., Schellens J.H., van der Noll R., Specht J. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin Cancer Res. 2016;22:1932–1939. doi: 10.1158/1078-0432.CCR-15-1665. [DOI] [PubMed] [Google Scholar]
- 88.Park H., Kim Y., Sul J.W., Jeong I.G., Yi H.J., Ahn J.B. Synergistic anticancer efficacy of MEK inhibition and dual PI3K/mTOR inhibition in castration-resistant prostate cancer. Prostate. 2015;75:1747–1759. doi: 10.1002/pros.23057. [DOI] [PubMed] [Google Scholar]
- 89.Kong D.X., Yamori T. ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor identified using the JFCR39 drug discovery system. Acta Pharmacol Sin. 2010;31:1189–1197. doi: 10.1038/aps.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yaguchi S.I., Fukui Y., Koshimizu I., Yoshimi H., Matsuno T., Gouda H. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst. 2006;98:545–556. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- 91.Kong D.X., Yamori T. JFCR39, a panel of 39 human cancer cell lines, and its application in the discovery and development of anticancer drugs. Bioorg Med Chem. 2014;20:1947–1951. doi: 10.1016/j.bmc.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 92.Kong D.X., Yamori T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007;98:1638–1642. doi: 10.1111/j.1349-7006.2007.00580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dan S., Yoshimi H., Okamura M., Mukai Y., Yamori T. Inhibition of PI3K by ZSTK474 suppressed tumor growth not via apoptosis but G0/G1 arrest. Biochem Biophys Res Commun. 2009;379:104–109. doi: 10.1016/j.bbrc.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y., Liu J., Qiu Y., Jin M., Chen X., Fan G. ZSTK474, a specific class I phosphatidylinositol 3-kinase inhibitor, induces G1 arrest and autophagy in human breast cancer MCF-7 cells. Oncotarget. 2016 doi: 10.18632/oncotarget.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kong D.X., Okamura M., Yoshimi H., Yamori T. Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur J Cancer. 2009;45:857–865. doi: 10.1016/j.ejca.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 96.Zhou Q.X., Chen Y.L., Chen X., Zhao W.N., Zhong Y.X., Wang R. In vitro antileukemia activity of ZSTK474 on K562 and multidrug resistant K562/A02 cells. Int J Biol Sci. 2016;12:631–638. doi: 10.7150/ijbs.14878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elkabets M., Vora S., Juric D., Morse N., Mino-Kenudson M., Muranen T. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weigelt B., Warne P.H., Lambros M.B., Reis-Filho J.S., Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013;19:3533–3544. doi: 10.1158/1078-0432.CCR-12-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodon J., Dienstmann R., Serra V., Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 100.Mahajan K., Mahajan N.P., PI3K-independent A.K.T. activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol. 2012;227:3178–3184. doi: 10.1002/jcp.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Costa C., Ebi H., Martini M., Beausoleil S.A., Faber A.C., Jakubik C.T. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108. doi: 10.1016/j.ccell.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kinross K.M., Brown D.V., Kleinschmidt M., Jackson S., Christensen J., Cullinane C. In vivo activity of combined PI3K/mTOR and MEK inhibition in a KrasG12D;Pten deletion mouse model of ovarian cancer. Mol Cancer Ther. 2011;10:1440–1449. doi: 10.1158/1535-7163.MCT-11-0240. [DOI] [PubMed] [Google Scholar]