

Abstract
Guided by antiproliferative activity in MIA PaCa-2 cells, we have performed preliminary structure–activity relationship studies on N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides. Two selected compounds showed submicromolar antiproliferative activity and good metabolic stability. Both compounds reduced mTORC1 activity and increased autophagy at the basal level. In addition, they disrupted autophagic flux by interfering with mTORC1 reactivation and clearance of LC3-II under starvation/refeed conditions, as evidenced by accumulation of LC3-II and abnormal LC3 labeled punctae. Therefore, N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides may represent a new class of autophagy modulators that possesses potent anticancer activity and potentially a novel mechanism of action.
Keywords: Autophagy, autophagy modulator, mTOR, pancreatic cancer, anticancer agents
Macroautophagy (hereafter referred to as autophagy) is a conserved cellular process through which cytosolic components, such as proteins and organelles, are sequestered, degraded, and recycled.1 The autophagy machinery requires a number of autophagy-related (Atg) proteins and functional complexes, including the UNC51-like kinase 1 (ULK1) complex, the class III phosphoinositide 3-kinase (PI3K) complex, the Atg12-Atg5-Atg16 ubiquitin-like conjugation system, and the microtubule-associated protein light chain 3 (LC3)-phosphatidylethanolamine (PE) conjugation system. Among these autophagy components, LC3-II, which is the PE lipidated form of LC3-I, plays a key role. LC3-II is generated after a series of processing by the Atg4-Atg7-Atg3 conjugation machinery. Since it is closely involved in autophagy, the level of LC3-II is commonly used as a marker to monitor autophagy.2
Autophagy is regulated by a network of complex signaling pathways, where the class I phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin complex 1 (PI3K-AKT-mTORC1) pathway is a well-established negative regulator. While a basal level of autophagy is required to maintain cellular homeostasis, it is upregulated in response to lack of nutrients, unfavorable energy status, attenuated growth signaling, virus/bacteria invasion, and a number of cellular stresses including endoplasmic reticulum stress, oxidative stress, and hypoxia.3 Activation of autophagy is generally accomplished by inhibiting mTORC1, which in turn increases autophagy by unblocking ULK1. In addition to its role in autophagy, mTORC1 also phosphorylates and regulates substrates including ribosomal protein S6 kinase beta-1 (P70S6K1, hereafter referred to as P70) and 4E-binding protein 1 (4EBP1), and levels of phosphorylated substrates are routinely used to gauge mTORC1 activity.
The precise role of autophagy in tumorigenesis and cancer therapy still remains to be defined due to the heterogeneous nature of cancers and the complexity of the autophagy machinery.4 Studies have shown that autophagy plays different or even opposite roles depending on the cancer type and the stage of tumor progression. Currently, it is generally accepted that autophagy behaves as a tumor suppressor in normal cells in part because it removes damaged cellular components. Nevertheless, increasing evidence shows that autophagy is a cytoprotective mechanism often exploited by established cancer cells to cope with their harsh microenvironment and cellular stresses induced by chemotherapies. Inhibition of autophagy has been proposed as a promising strategy to combat pancreatic ductal adenocarcinoma (PDAC), the most lethal cancer among major malignancies. It has been shown that autophagy is elevated and required for de novo tumor growth and that inhibition of autophagy leads to robust tumor regression;5,6 however, the status of p53 may determine the outcome of autophagy inhibition.7,8 Despite the complexity of autophagy as a therapeutic target, these studies suggest that disabling the pro-survival autophagy machinery might represent a viable approach to promote cell death and reduce drug resistance in refractory cancers such as pancreatic cancer.3,9
Because of their potential application as anticancer agents used alone or in combination with other cancer therapeutics, autophagy inhibitors have been actively pursued even though early inhibitors generally lack selectivity or target the lysosomal function.10 Chloroquine and hydrochloroquine, two lysosomotropic agents, have been commonly used as autophagy inhibitors. However, it has been proposed that chloroquine’s anticancer property is independent of its effect on autophagy.11,12 These findings underscore the importance of discovering new and specific autophagy inhibitors, preferably those with a novel mechanism of action. In recent years, in addition to the continued search for improved lysosomotropic autophagy inhibitors,13−16 there has been a growing list of selective autophagy inhibitors, especially those targeting Beclin 1,17 ULK1 kinase,18−20 Vps34 (Class III PI3K),21−23 or Atg4B.24 Herein we report our discovery of N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides that possess antiproliferative activity and elicit differential effects on autophagy under different nutrient conditions. Structure–activity relationship (SAR) studies of this class of compounds, assessment of in vitro absorption, distribution, metabolism, and excretion (ADME) properties, and evaluation of effects on mTORC1 and autophagy are presented.
During our search for active compounds in pancreatic cancer cells, we identified compound 1, which possessed an EC50 value of 10 μM in MIA PaCa-2, a pancreatic cancer line (Table 1). When compared with known autophagy inhibitors, compound 1 was more active than 3-methyladenine (3 mA) and spautin-117 while possessing an antiproliferative capacity similar to that of chloroquine. Furthermore, it was more potent than erlotinib, a tyrosine kinase inhibitor that has been approved for use in combination with gemcitabine. Moreover, our initial examination of compound 1’s effect on autophagy suggested that it disrupted the autophagy pathway. Taken together, these results suggested that compound 1 could serve as a starting point for SAR studies and the subsequent exploration of improved compounds for their effects on autophagy.
Table 1. SAR Studies: Substituents on Ring Aa.
| compd | R1 | R2 | R3 | EC50 (μM) |
|---|---|---|---|---|
| 1 | H | H | H | 10 |
| 2 | C(O)NH2 | H | H | >100 |
| 3 | H | C(O)NH2 | H | 9.2 |
| 4 | H | H | C(O)NH2 | 42 |
| 5 | H | C(O)NHMe | H | 6.2 |
| 6 | H | C(O)N(Me)2 | H | 6.4 |
| 7 | H | C(O)NHEt | H | 11 |
| 8 | H | C(O)NHiPr | H | 8.9 |
| 9 | H | C(O)NHPh | H | 6.4 |
| gemcitabine | 0.0015 | |||
| erlotinib | 27 | |||
| 3 mA | >100 | |||
| chloroquine | 14 | |||
| spautin-1 | 20 |
EC50 values were determined in triplicate in MIA PaCa-2 cells.
First, we investigated ring A by introducing a functional group, as represented by R1, R2, and R3, at the para, meta, and ortho positions, respectively. Because an amide provided an opportunity to serve as both a hydrogen bond donor and a hydrogen bond acceptor, we chose to first explore amide functionalities (Table 1). When a primary amide was introduced, a negative effect was observed for a substitution at the para (compound 2) or ortho (compound 4) position. However, compound 3, which contained a primary amide at the meta position, retained activity nearly identical to that of parent compound 1, suggesting that the meta position might be more amenable to structural modifications. Indeed, incorporation of a methyl amide as seen in compound 5 slightly improved anticancer activity in comparison with its primary amide counterpart. To further explore amide substitutions, we prepared compounds 6–9, which contained a dimethyl, ethyl, isopropyl, and phenyl amide, respectively, at the meta position. Comparable activities exhibited by these compounds indicated that further structural elaboration beyond a methyl amide was not productive.
With a methyl amide fixed at the meta position of ring A, we proceeded to investigate the phenyl ring D (Table 2). Compound 10, in which the phenyl ring was replaced with a saturated cyclohexyl group, showed reduced activity, highlighting the importance of an aromatic ring. Subsequently, we studied the substituents on ring D by modifying parent compound 5. To that end, we performed a substitution scan at the ortho, meta, and para positions, as represented by R1, R2, and R3, respectively. To gauge the impact of electron-donating or -withdrawing groups, we used groups with different electrosteric properties, including Me, OMe, Cl, and CF3. First, we conducted a methyl scan on ring D (compounds 11–13), which revealed that a methyl group at the ortho or para position enhanced activity, while one at the meta position had no effect. When a similar scan was performed using a methoxy (14–16) or chloro (17–19) group, the resulting compounds failed to significantly improve anticancer activity relative to compound 5. Remarkably, introducing a trifluoromethyl group at the para position led to compound 22, which possessed a submicromolar EC50 value. Because a methyl group at the ortho position resulted in an increase in activity as seen for compound 11, we chose to investigate a potential additive effect by preparing compound 23, which featured a trifluoromethyl and methyl group at the para and ortho position, respectively. Compound 23 exhibited an EC50 value of 0.62 μM without significant improvement over 22. In summary, a trifluoromethyl group was the preferred substituent at the para position of ring D, while a methyl at the ortho position could be beneficial.
Table 2. SAR Studies: Substituents on Ring Da.
| compd | R1 | R2 | R3 | EC50 (μM) |
|---|---|---|---|---|
| 10 | 14 | |||
| 11 | Me | H | H | 2.1 |
| 12 | H | Me | H | 6.2 |
| 13 | H | H | Me | 2.6 |
| 14 | OMe | H | H | 8.5 |
| 15 | H | OMe | H | 6.2 |
| 16 | H | H | OMe | 8.0 |
| 17 | Cl | H | H | 6.3 |
| 18 | H | Cl | H | 5.9 |
| 19 | H | H | Cl | 4.2 |
| 20 | CF3 | H | H | 10 |
| 21 | H | CF3 | H | 2.3 |
| 22 | H | H | CF3 | 0.80 |
| 23 | Me | H | CF3 | 0.62 |
EC50 values were determined in triplicate in MIA PaCa-2 cells.
To evaluate in vitro ADME properties of selected compounds, we tested compounds 22 and 23 for their metabolic stabilities (Table S1). Both compounds possessed excellent stability in mouse and human plasma as well as very good stability in mouse and human liver microsomes. Furthermore, both compounds showed good aqueous solubility with 23 being 2-fold more soluble than 22. This enhanced solubility could be explained by reduced coplanarity of ring D (in relation to the oxymethyl moiety within the phenoxymethyl linker) induced by the extra methyl group on that ring in compound 23. Taken together, compounds 23 and 22 showed both relatively good in vitro ADME properties and antiproliferative activity; therefore, their effects on autophagy were examined.
Effects on autophagy were studied in ARPE-19, a human retinal pigment epithelial cell line. Markedly higher EC50 values of compounds 23 (7.1 μM) and 22 (47.9 μM) in ARPE-19 cells indicated that both compounds were less cytotoxic in noncancer ARPE-19 cells than in MIA PaCa-2 pancreatic cancer cells. This finding is not unexpected since cancer cells often rely on autophagy and are hence more vulnerable to disruption of the autophagy machinery.
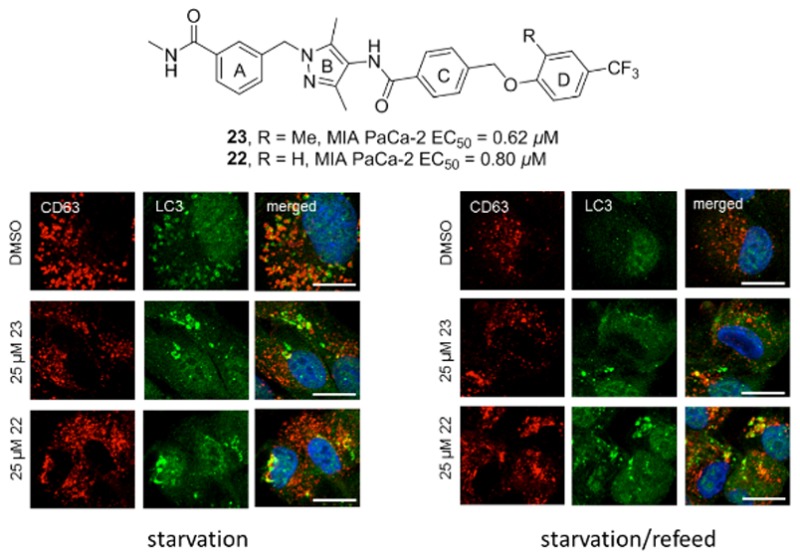
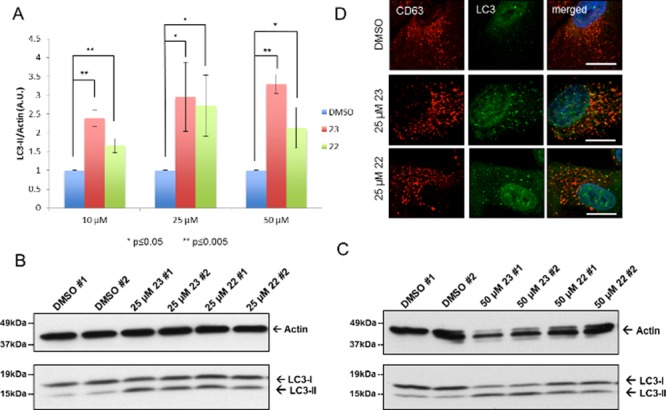
Basal autophagy was measured by treating ARPE-19 cells with increasing concentrations of compound 23 or 22 (or DMSO control) for 4 h in complete medium. Autophagy induction was quantified by measuring the fold increase in LC3-II/actin ratio compared to control cells.2 A low dose (10 μM) of both compounds resulted in significant increase of LC3-II conversion compared to control (Figure 1A and Figure S1). This response was dose dependent for compound 23 as judged by enhanced effects at the higher concentrations of 25 μM (Figure 1A,B) and 50 μM (Figure 1A,C). Compound 23 was slightly more effective at LC3 turnover (3.3-fold increase compared to control) at 50 μM, while compound 22 peaked at 25 μM (2.7-fold increase compared to control). This observation was consistent with compound 23’s slightly higher antiproliferative ability and improved aqueous solubility. Furthermore, in line with the biochemical data, an increase in the number of LC3 labeled punctae was detected in cells treated with 25 μM of either compound 23 or 22 (Figure 1D). In our immunofluorescence experiment, CD63 was used as a late endosomal/lysosomal marker. Limited colocalization between LC3 and CD63 in normal (Figure 1D) and starvation conditions (Figure 3C,D) indicated that the LC3-positive punctae corresponded to autophagosomes instead of degradation-deficient autolysosomes. Enhanced colocalization between LC3 and CD63 was observed in bafilomycin-treated ARPE-19 cells and LC3/CD63-positive structures corresponded to degradation-deficient autolysosomes (Figure S2).
Figure 1.
Induction of basal autophagy. ARPE-19 cells were treated for 4 h with DMSO, 23, or 22 without starvation. (A) Fold increase of LC3-II/actin ratios normalized to the DMSO control (n = 3). (B) Representative Western blot of LC-3 after treatment with 23 or 22 (25 μM). (C) Representative Western blot of LC-3 after treatment with 23 or 22 (50 μM). (D) Representative immunofluorescence detection of LC3 punctae after treatment with 23 or 22 (25 μM). Scale bar is 10 μm. CD63 was used as a late endosomal/lysosomal marker.
Figure 3.
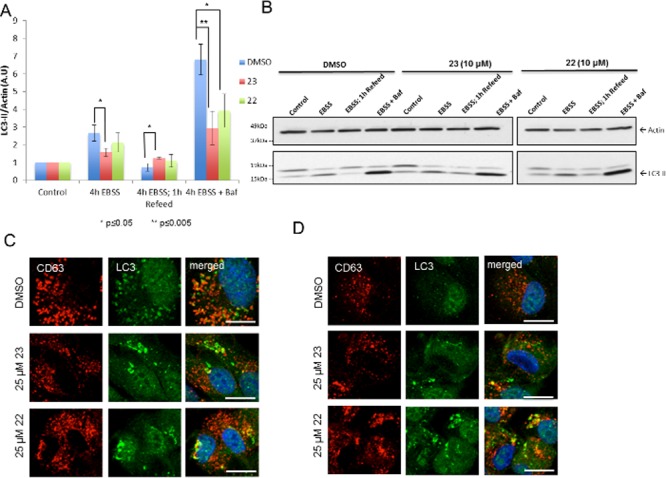
Impaired autophagy flux. ARPE-19 cells were treated with DMSO, 23, or 22 under starvation (4 h EBSS), starvation/refeed (4 h EBSS followed by 1 h complete medium recovery), and starvation/bafilomycin (4 h EBSS containing 100 nM bafilomycin) conditions (n = 3). (A) Fold increase LC3-II/actin normalized to DMSO control after treatment with 23 or 22 (10 μM) under starvation/refeed conditions. All the three controls were fixed at value = 1. (B) Representative Western blot of LC3 after treatment with 23 or 22 (10 μM). (C) Representative immunofluorescence detection of LC3 punctae after treatment with 23 or 22 (25 μM) under starvation conditions. (D) Representative immunofluorescence detections of LC3 punctae after treatment with 23 or 22 (25 μM) under starvation/refeed conditions. Scale bar is 10 μm. CD63 was used as a late endosomal/lysosomal marker.
Because mTORC1 is a negative regulator of autophagy, we tested for mTORC1 activity in cells treated with compound 23 or 22. Basal mTORC1 activity was measured by the phosphorylation level of the mTORC1 substrate P70 (Figure 2). Levels of phosphorylated P70/total P70 were decreased after treatment with 10 μM compound 23 or 22 (Figure 2A). Similar to the induction of basal autophagy, the decrease in mTORC1 activity was also dose dependent. Compound 23 was more responsive than compound 22, decreasing mTORC1 activity as much as 78% compared to control treated cells, whereas compound 22 peaked at a decrease of only 46% (Figure 2A,B and Figure S3). Taken together, these results suggested that compounds 23 and 22 reduced basal mTORC1 activity, thereby inducing an increase in autophagy.
Figure 2.
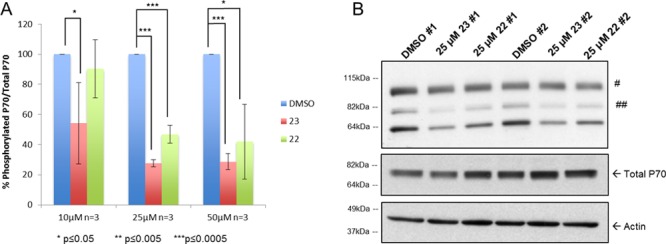
Decreased basal mTORC1 activity. ARPE-19 cells were treated for 4 h with DMSO, 23, or 22 without starvation. (A) Level of phosphorylated P70/total P70 after treatment with increasing concentrations of 23 or 22 vs DMSO (100%). (B) Representative Western blot of phosphorylated P70 after treatment with 23 or 22 (25 μM). #, nonspecific; ##, phosphorylated P85 S6 kinase (an isoform of P70 S6 kinase).
To better understand the effects of compounds 23 and 22 on autophagy, we performed an autophagic flux assay2 and used bafilomycin to block the late fusion/degradation stage of autophagy. ARPE-19 cells were pretreated for 30 min with a low dose (10 μM) of compound 23 or 22 (or DMSO). Cells were then incubated for 4 h with compound 23 or 22 in Earle’s balanced salt solution (EBSS, starvation medium) or EBSS with 100 nM bafilomycin and the fold increase in LC3-II/actin was quantified (Figure 3A,B and Figure S4). Under starvation conditions, cells treated with compounds had a decrease in LC3 turnover compared to DMSO treated cells (1.7-fold decrease for 23 and 1.2-fold decrease for 22; Figure 3A). Treatment with bafilomycin induced a strong increase in LC3-II/actin both in control and compound-treated cells, indicating that compounds 23 and 22 did not inhibit fusion between autophagosomes and lysosomes. However, the LC3-II/actin ratios were decreased in compound-treated cells, suggesting that autophagosome degradation might be affected at some extent by the compounds. Moreover, upon treatment with 25 μM of either compound under starvation conditions, LC3 labeled structures were enlarged and nonuniform in shape, whereas DMSO treated cells had uniform LC3 labeled punctae that were evenly distributed throughout the cell periphery (Figure 3C).
To measure autophagy under starvation/refeed conditions, cells starved for 4 h in EBSS were then incubated in complete medium with either compound or DMSO for 1 h to recover from autophagy induction (refeed conditions). DMSO treated cells refed for 1 h had a near complete recovery in LC3 turnover to basal levels, whereas cells treated with 10 μM of compound 23 were not able to recover LC3 turnover (1.7-fold increase compared to DMSO treatment; Figure 3A). LC3-II accumulation after refeed was more pronounced when treated with increasing concentrations (25 and 50 μM) of either compound (Figures S5 and S6). Lingering LC3-II was also observed in cells treated with 25 μM compound 23 or 22 under starvation/refeed conditions, where abnormal LC3 punctae were still detected even after 1 h recovery with complete medium (Figure 3D), suggesting that there was a problem with the clearance of LC3 punctae in compound treated cells.
Autophagic flux was also measured in the presence of 100 nM bafilomycin in full medium. However, no statistically significant reduction in autophagic flux was observed (Figure S7). While further studies are needed, this finding could be partly due to the fact that the basal autophagy level in ARPE-19 cells is not particularly high and suffers from consequent experimental variability.
The impact of these two compounds on mTORC1 activity was assessed under the same starvation/refeed conditions as described above. Treatment of cells with 10 μM compound 23 resulted in reduced mTORC1 reactivation following starvation and subsequent refeed, as judged by the phosphorylation levels of P70 and 4EBP1, two mTORC1 specific substrates (Figure 4A,B). No significant effects were observed for compound 22 at 10 μM. However, treatment with higher concentrations of either compound led to reduced mTORC1 reactivation (Figures S5 and S6). Taken together, these results suggested that both compounds 23 and 22 disrupted autophagic flux by inhibiting mTORC1 under basal conditions and interfering with its reactivation under refeed conditions.
Figure 4.
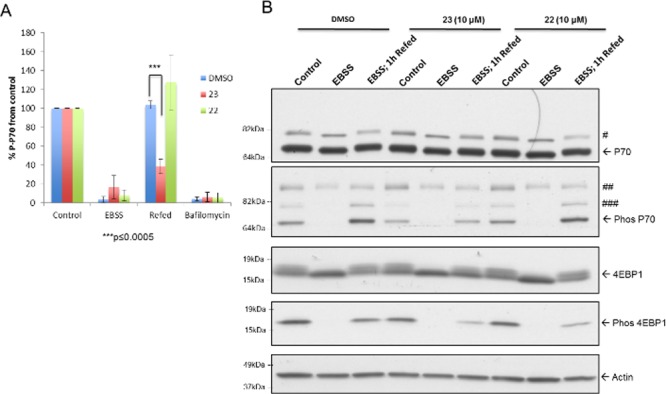
Reduced mTORC1 reactivation under refeed conditions. ARPE-19 cells were treated with DMSO, 23, or 22 (10 μM) under starvation (4 h EBSS) and starvation/refed (4 h EBSS followed by 1 h complete medium recovery) conditions. (A) Level of phosphorylated P70 normalized to control after treatment with 23 or 22 (10 μM) under starvation/refeed conditions (n = 3). (B) Representative Western blot of P70, phosphorylated P70, 4E-BP1, and phosphorylated 4E-BP1. #, P85 S6 kinase (an isoform of P70 S6 kinase); ##, nonspecific; ###, phosphorylated P85 S6 kinase.
The synthesis of N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides is straightforward (Scheme 1). Alkylation of pyrazole 24 with benzyl bromide followed by reduction of the nitro group with NaBH4 in the presence of NiCl2 gave amine 25a, which in turn underwent an amide coupling reaction to afford compound 1. An identical sequence also produced methyl esters 26b–d, which were then converted into the corresponding primary amides 2–4 after treatment with methanolic ammonia. Upon treatment with methylamine, methyl ester 26c was transformed into methyl amide 5. Hydrolysis of 26c gave carboxylic acid 27, which was used to prepare compounds 6–9 under standard HBTU-mediated amide formation conditions.
Scheme 1.

Reagents and conditions: (a) ArCH2Br, Cs2CO3, DMF; (b) NaBH4, NiCl2·6H2O, MeOH; (c) 4-(phenoxymethyl)benzoic acid, EDC, DMF/CH2Cl2; (d) 7 N methanolic NH3, 70 °C; (e) MeNH2, EtOH, 70 °C; (f) NaOH, MeOH, H2O; (g) amine, HBTU, Et3N, DMF/CH2Cl2; (h) methyl 3-(bromomethyl)benzoate, Cs2CO3, DMF; (i) for 10, 4-((cyclohexyloxy)methyl)benzoic acid, HBTU, Et3N; for 30, 4-(chloromethyl)benzoyl chloride, Et3N, CH2Cl2; (j) ArOH, Cs2CO3, DMF, 50 °C.
To prepare compounds 10–23, pyrazole 24 was alkylated with methyl 3-(bromomethyl)benzoate followed by treatment with methylamine to give 28, which was then reduced to amine 29. Routine amide coupling between amine 29 and 4-((cyclohexyloxy)methyl)benzoic acid gave 10. However, amine 29 was acylated with 4-(chloromethyl)benzoyl chloride to give intermediate 30, whose benzyl chloride moiety underwent nucleophilic substitutions with a set of substituted phenol to afford compounds 11–23 in a range of yields. Preparation of phenol 33, which was needed for compound 23, was depicted in Scheme S1. Commercially available bromide 31 was converted into boronic pinacolate ester 32, which was subsequently oxidized with mCPBA to give 33 in good yields.
In summary, guided by antiproliferative activity in MIA PaCa-2 cells, our preliminary SAR studies on the N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamide core structure have led to compounds 23 and 22, which show submicromolar antiproliferative activity and good metabolic stability. We have also demonstrated that both compounds increase basal autophagy but impair autophagic flux under starvation and starvation/refeed conditions. To our best knowledge, no compound with such differential activities on autophagy has been reported previously. Therefore, compound 23 and its analogues might represent a new class of autophagy modulators that possess remarkable anticancer activity. Furthermore, it is tempting to speculate that these compounds might be able to selectively target solid tumor cells that are under metabolic stress and hypoxia due to poor vasculature and impeded delivery of nutrients and oxygen. The harsh tumor microenvironment has been shown to induce autophagy as a survival mechanism, which might prove to be a weakness that can be selectively disrupted by compound 23 under nutrient-deficient conditions. At the same time, compound 23 might spare normal cells by allowing or even prompting basal autophagy that is expected to reduce tumorigenesis. Therefore, at least in principle, a compound like 23 is expected to be advantageous over a conventional autophagy inhibitor that unselectively suppresses autophagy in both normal and cancerous cells. Taken together, N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides may hold promise as a new class of anticancer agents against PDAC. Further SAR studies and elucidation of their mechanism of action are therefore warranted.
Acknowledgments
This work was supported by the Center for Drug Design in the Academic Health Center of the University of Minnesota (to L. C.). R.W. and R.P. were supported by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI). D.-H.K. was supported by the National Institute of General Medical Sciences (NIGMS) (R01GM097057). We thank Dr. Huaqing Cui for the initial synthesis of compound 1.
Glossary
ABBREVIATIONS
- PDAC
pancreatic ductal adenocarcinoma
- Atg
autophagy-related
- ULK1
UNC51-like kinase 1
- PI3K
phosphoinositide 3-kinase
- LC3
microtubule-associated protein light chain 3
- PE
phosphatidylethanolamine
- mTORC1
mammalian target of rapamycin complex 1
- P70S6K1 (P70)
ribosomal protein S6 kinase beta-1
- 4EBP1
4E-binding protein 1
- SAR
structure–activity relationship
- ADME
absorption, distribution, metabolism, and excretion
- 3 mA
3-methyladenine
- EBSS
Earle’s balanced salt solution
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00392.
In vitro ADME properties of compounds 22 and 23; Western blots of ARPE-19 cells treated with compounds 23 or 22 under basal, starvation, and starvation/refeed conditions; synthesis of phenol 33; and experimental methods (PDF)
Author Contributions
∥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- He C.; Klionsky D. J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X.; Chen R.; Wang Z.; Huang Z.; Kong N.; Zhang M.; Han W.; Lou F.; Yang J.; Zhang Q.; Wang X.; He C.; Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Pietrocola F.; Bravo-San Pedro J. M.; Amaravadi R. K.; Baehrecke E. H.; Cecconi F.; Codogno P.; Debnath J.; Gewirtz D. A.; Karantza V.; Kimmelman A.; Kumar S.; Levine B.; Maiuri M. C.; Martin S. J.; Penninger J.; Piacentini M.; Rubinsztein D. C.; Simon H. U.; Simonsen A.; Thorburn A. M.; Velasco G.; Ryan K. M.; Kroemer G. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S.; Mitsunaga S.; Yamazaki M.; Hasebe T.; Ishii G.; Kojima M.; Kinoshita T.; Ueno T.; Esumi H.; Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819. 10.1111/j.1349-7006.2008.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Wang X.; Contino G.; Liesa M.; Sahin E.; Ying H.; Bause A.; Li Y.; Stommel J. M.; Dell’antonio G.; Mautner J.; Tonon G.; Haigis M.; Shirihai O. S.; Doglioni C.; Bardeesy N.; Kimmelman A. C. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt M. T.; O’Prey J.; Morton J. P.; Nixon C.; MacKay G.; Mrowinska A.; Au A.; Rai T. S.; Zheng L.; Ridgway R.; Adams P. D.; Anderson K. I.; Gottlieb E.; Sansom O. J.; Ryan K. M. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- Yang A.; Rajeshkumar N. V.; Wang X.; Yabuuchi S.; Alexander B. M.; Chu G. C.; Von Hoff D. D.; Maitra A.; Kimmelman A. C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery 2014, 4, 905–913. 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez V. E.; Giovannetti E.; Peters G. J. Unraveling the complexity of autophagy: Potential therapeutic applications in Pancreatic Ductal Adenocarcinoma. Semin. Cancer Biol. 2015, 35, 11–19. 10.1016/j.semcancer.2015.09.011. [DOI] [PubMed] [Google Scholar]
- Wang C.; Hu Q.; Shen H. M. Pharmacological inhibitors of autophagy as novel cancer therapeutic agents. Pharmacol. Res. 2016, 105, 164–175. 10.1016/j.phrs.2016.01.028. [DOI] [PubMed] [Google Scholar]
- Maycotte P.; Aryal S.; Cummings C. T.; Thorburn J.; Morgan M. J.; Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C. H.; Wang Z.; Tkach D.; Toral-Barza L.; Ugwonali S.; Liu S.; Fitzgerald S. L.; George E.; Frias E.; Cochran N.; De Jesus R.; McAllister G.; Hoffman G. R.; Bray K.; Lemon L.; Lucas J.; Fantin V. R.; Abraham R. T.; Murphy L. O.; Nyfeler B. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 182–187. 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee Q.; Zhang Z.; Samanta A.; Levi S. M.; Ma X. H.; Piao S.; Lynch J. P.; Uehara T.; Sepulveda A. R.; Davis L. E.; Winkler J. D.; Amaravadi R. K. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8253–8258. 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom L. U.; Sironi J.; Aranda E.; Maisonet J.; Perez-Soler R.; Wu P.; Schwartz E. L. Discovery of autophagy inhibitors with antiproliferative activity in lung and pancreatic cancer cells. ACS Med. Chem. Lett. 2015, 6, 134–139. 10.1021/ml500348p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente E.; Parodi C.; Ercolani L.; De Mei C.; Ferrari A.; Scarpelli R.; Grimaldi B. Synthesis and in vitro anticancer activity of the first class of dual inhibitors of REV-ERBbeta and autophagy. J. Med. Chem. 2015, 58, 5900–5915. 10.1021/acs.jmedchem.5b00511. [DOI] [PubMed] [Google Scholar]
- Wang T.; Goodall M. L.; Gonzales P.; Sepulveda M.; Martin K. R.; Gately S.; MacKeigan J. P. Synthesis of improved lysomotropic autophagy inhibitors. J. Med. Chem. 2015, 58, 3025–3035. 10.1021/jm501586m. [DOI] [PubMed] [Google Scholar]
- Liu J.; Xia H.; Kim M.; Xu L.; Li Y.; Zhang L.; Cai Y.; Norberg H. V.; Zhang T.; Furuya T.; Jin M.; Zhu Z.; Wang H.; Yu J.; Li Y.; Hao Y.; Choi A.; Ke H.; Ma D.; Yuan J. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M. B.; Novotny C. J.; Shokat K. M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M. B.; Shokat K. M. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 2015, 23, 5483–5488. 10.1016/j.bmc.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D. F.; Chun M. G.; Vamos M.; Zou H.; Rong J.; Miller C. J.; Lou H. J.; Raveendra-Panickar D.; Yang C. C.; Sheffler D. J.; Teriete P.; Asara J. M.; Turk B. E.; Cosford N. D.; Shaw R. J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 2015, 59, 285–297. 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier B.; El-Ahmad Y.; Filoche-Romme B.; Dureuil C.; Fassy F.; Abecassis P. Y.; Mathieu M.; Bertrand T.; Benard T.; Barriere C.; El Batti S.; Letallec J. P.; Sonnefraud V.; Brollo M.; Delbarre L.; Loyau V.; Pilorge F.; Bertin L.; Richepin P.; Arigon J.; Labrosse J. R.; Clement J.; Durand F.; Combet R.; Perraut P.; Leroy V.; Gay F.; Lefrancois D.; Bretin F.; Marquette J. P.; Michot N.; Caron A.; Castell C.; Schio L.; McCort G.; Goulaouic H.; Garcia-Echeverria C.; Ronan B. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)- 3,4-dihydro-2H-pyrimido[1,2-a]pyrimidin-6-one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 2015, 58, 376–400. 10.1021/jm5013352. [DOI] [PubMed] [Google Scholar]
- Dowdle W. E.; Nyfeler B.; Nagel J.; Elling R. A.; Liu S.; Triantafellow E.; Menon S.; Wang Z.; Honda A.; Pardee G.; Cantwell J.; Luu C.; Cornella-Taracido I.; Harrington E.; Fekkes P.; Lei H.; Fang Q.; Digan M. E.; Burdick D.; Powers A. F.; Helliwell S. B.; D’Aquin S.; Bastien J.; Wang H.; Wiederschain D.; Kuerth J.; Bergman P.; Schwalb D.; Thomas J.; Ugwonali S.; Harbinski F.; Tallarico J.; Wilson C. J.; Myer V. E.; Porter J. A.; Bussiere D. E.; Finan P. M.; Labow M. A.; Mao X.; Hamann L. G.; Manning B. D.; Valdez R. A.; Nicholson T.; Schirle M.; Knapp M. S.; Keaney E. P.; Murphy L. O. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- Honda A.; Harrington E.; Cornella-Taracido I.; Furet P.; Knapp M. S.; Glick M.; Triantafellow E.; Dowdle W. E.; Wiedershain D.; Maniara W.; Moore C.; Finan P. M.; Hamann L. G.; Firestone B.; Murphy L. O.; Keaney E. P. Potent, selective, and orally bioavailable inhibitors of VPS34 provide chemical tools to modulate autophagy in vivo. ACS Med. Chem. Lett. 2016, 7, 72–76. 10.1021/acsmedchemlett.5b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z.; Kuhn B.; Aebi J.; Lin X.; Ding H.; Zhou Z.; Xu Z.; Xu D.; Han L.; Liu C.; Qiu H.; Zhang Y.; Haap W.; Riemer C.; Stahl M.; Qin N.; Shen H. C.; Tang G. Discovery of fluoromethylketone-based peptidomimetics as covalent ATG4B (autophagin-1) inhibitors. ACS Med. Chem. Lett. 2016, 7, 802–806. 10.1021/acsmedchemlett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.