

Abstract

Herein we report the discovery and hit-to-lead optimization of a series of spirocyclic piperidine aldosterone synthase (CYP11B2) inhibitors. Compounds from this series display potent CYP11B2 inhibition, good selectivity versus related CYP enzymes, and lead-like physical and pharmacokinetic properties.
Keywords: aldosterone synthase, CYP11B2, hypertension
Hypertension is the most prevalent chronic disease state, afflicting an estimated 80 million people in the United States and >1 billion people worldwide.1 Roughly half of that patient population exhibits resistant hypertension, blood pressure that remains high even after treatment with a combination of three or more antihypertensive agents.2 Such uncontrolled hypertension can contribute to organ damage, cardiovascular disease, stroke, heart attack, and heart failure.3 Current treatment options notwithstanding, there remains a significant need for additional, mechanistically novel, antihypertensive therapies.
One novel approach involves the reduction of plasma aldosterone levels.4 Elevated plasma aldosterone levels promote sodium and water retention, vasoconstriction, and increased sympathetic drive, ultimately leading to increased blood pressure. Aldosterone synthase, also known as CYP11B2, catalyzes the final three rate-limiting steps of aldosterone’s biosynthesis. Based on this rationale, inhibitors of CYP11B2 should prevent aldosterone biosynthesis, thereby leading to a reduction in plasma aldosterone and consequently blood pressure.
The role of aldosterone synthase inhibitors (ASIs) has received much attention in recent years.5−15 Of note, the inhibitor LCI-699 (Figure 1) has been shown to lower plasma aldosterone levels and blood pressure in the clinic, thus providing proof of concept for the use of ASIs to treat hypertension.16,17 LCI-699 is a modestly selective CYP11B2 inhibitor, displaying ∼4-fold selectivity for inhibition of CYP11B2 versus a close structural homologue, CYP11B1, an enzyme that catalyzes the biosynthesis of cortisol.12 In the clinic, LCI-699 produced an undesired impairment of the stress response, an adverse effect presumed to be caused by inhibition of CYP11B1. Given the physiological importance of cortisol in glucose metabolism and stress response, selectivity for inhibition of CYP11B2 over CYP11B1 is required in a clinically viable candidate.
Figure 1.

Aldosterone synthase inhibitors.
With this in mind the goal was to discover a structurally novel class of potent and selective inhibitors of aldosterone synthase and to assess their potential therapeutic value in models of resistant hypertension. In this regard spiro-fused scaffolds were explored as replacements for the aromatic, planar-fused bicyclic and tricyclic scaffolds that are present in previously described inhibitors.5 Recent literature reports have highlighted the benefits that such spirocyclic scaffolds can impart, including increased sp3 character, reduced planarity, improved solubility, and structural novelty.18
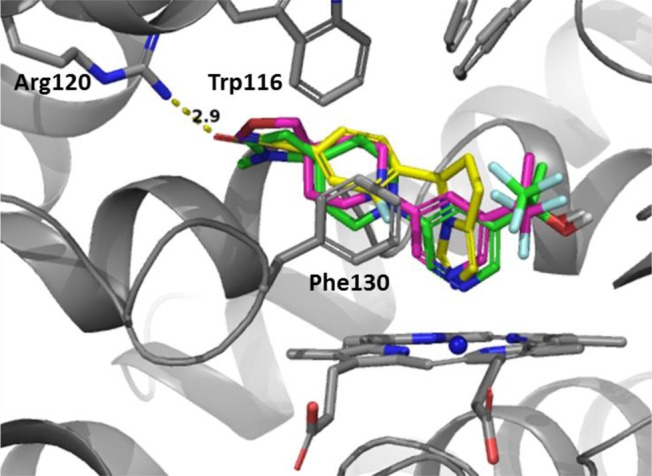
Modeling studies were performed to evaluate the ability of spirocyclic analogues such as compound 1 to bind to the CYP11B2 active site. Cocrystal structures of CYP11B2 in complex with fadrozole, deoxycorticosterone, and a tetrahydroisoquinoline inhibitor have been reported.19,20 These structures were used to support our spirocycle docking experiments. Figure 2 depicts an overlay of LCI-699, a lactam-based inhibitor,21 and spirocycle 1 (Figure 1) in the CYP11B2 binding pocket. As illustrated, there was good overlap between the proposed spirocycle and the aforementioned aldosterone synthase inhibitors. The positioning of the isoquinoline appeared to be in an optimal position to interact with the heme cofactor. These results motivated us to pursue this novel class of spirocyclic inhibitors.
Figure 2.

Overlay of aldosterone synthase inhibitors with a proposed spirocyclic piperidine analogue: LCI-699 (yellow), a lactam inhibitor (cyan), and spirocyclic compound 1 (green) are shown as docked to CYP11B2 (PDB code 4ZGX, shown as gray ribbons with the heme cofactor as gray tubes).
Encouraged by the modeling studies, a variety of spiro-fused analogues were synthesized and evaluated for CYP11B2 activity.22 The initial series of compounds incorporated an isoquinoline moiety, which has been used in previous work and provided a critical interaction with the heme binding domain of CYP11B2.5,8 Spirocyclic isoquinoline 1, our proposed spirocycle, displayed potent inhibition of human CYP11B2 and modest selectivity against CYP11B1. In contrast, related analogue 2, which contains a larger spiro-fused cyclohexane, proved less potent and less selective than 1. These initial results suggested the spirocycles would be tolerated in the CYP11B2 binding pocket and provided a starting point for optimization of this series.
Once the effectiveness of the spirocyclic skeleton was demonstrated with compound 1, a series of compounds were synthesized to test whether the addition of a hydrogen bond acceptor might improve the potency. The docking studies revealed an arginine residue, Arg120, was proximal to our spirocyclic core. It was hypothesized that adding appropriate functionality in the optimal position might increase the likelihood of reaching this residue. As a first attempt, an ether oxygen was installed at the 2-position in 3, which appeared to be in a similar trajectory as the nitrile group in LCI-699 and positioned to interact with the Arg120. Improved CYP11B2 inhibition was observed relative to 1. When the oxygen was repositioned to the interior 1-position in 4, a significant loss of CYP11B2 potency was observed with a modest decrease in selectivity. The related spirooxetane derivative 5 displayed potency and selectivity similar to that of 3, with the added benefits of reduced molecular weight and lipophilicity.
The modeling overlay of the spirocyclic compound with the lactam suggested that an amide might be tolerated in the spirocyclic ring. It was also thought that the lactam carbonyl could be positioned to form a hydrogen bond with the Arg120. With this in mind, lactam 6, which placed the carbonyl oxygen in a similar trajectory as the nitrile in LCI-699 and oxygen ether in 3, was synthesized and displayed good potency and CYP11B1 selectivity. In contrast, lactam 7, which moves the carbonyl to an interior position away from the arginine, proved significantly less potent than 6, a trend that is in concordance with our modeling studies. In addition to an improvement in CYP11B2 potency and selectivity, addition of a hydrogen bond acceptor as in the amide and ether moieties favorably impacted the Log D, solubility, and in vitro intrinsic clearance in rat microsomes compared to compound 1 (Table 2).
Table 2. Physiochemical Properties of Selected Compounds.
| Cpd | Log Da | MWb | Soly pH2/7c | Rat Mic Clintd |
|---|---|---|---|---|
| 1 | 5.6 | 266 | 176/3 | 1291 |
| 3 | 2.6 | 268 | 196/165 | <20 |
| 6 | 1.6 | 295 | 149/167 | 76 |
Determined experimentally by HPLC.
Molecular weight.
Solubility concentration (μM) in PBS buffer at pH 2 and 7.
Rat microsomal intrinsic clearance (mL/min/kg).
Further attempts to reach the Arg120 led to extending the HBA closer to the residue. In this regard, acetamide derivative 9, which extends the amide functionality off the spirocyclic ring, showed reduced CYP11B2 inhibition with respect to lactam 6 and ether 3 and was nonselective versus CYP11B1. Additional amide derivatives with larger amide R groups were also prepared, but they were consistently less potent than acetamide 9 (data not shown). Carbamates such as compound 10 displayed similarly modest potency and selectivity. This data suggested the carbonyl was not optimally positioned to form a hydrogen bond with the arginine in these analogues.
Given the promising CYP11B2 selectivity of the initial spirocyclic compounds coupled with the improved Log D and low intrinsic clearance of 3 and 6, this was a good starting place to begin further optimization of the series. Taking this into account we next examined the impact of varying the isoquinoline moiety with the better spirocyclic cores identified in Table 1. Previous work had shown that the isoquinoline typically imparted high potency, but also a liability in the form of high plasma clearance.8 Surrogate heme binding moieties with improved potency, selectivity, and pharmacokinetic properties have been reported.7 In this regard, we focused on combining optimized spiro-fused cores with heme binding moieties identified by our group that have shown improved pharmacokinetic properties.
Table 1. Effect of Hydrogen Bond Acceptor Positioning on CYP11B2 and CYP11B1 Inhibition.
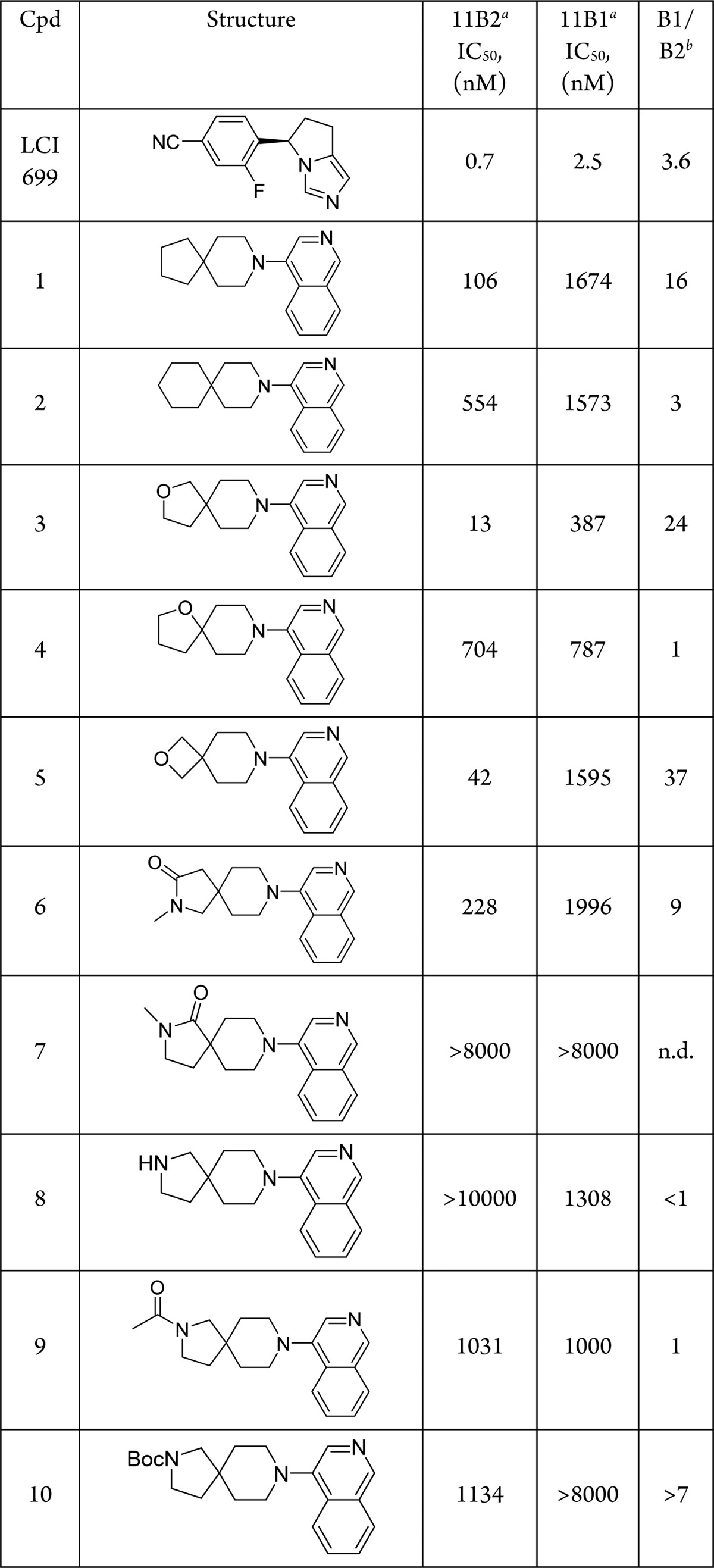
IC50s calculated from n ≥ 1, see Supporting Information for details. For compounds n > 1 the geometric mean is reported.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
As shown in Table 3, the isoquinoline moiety in N-methyl lactam 6 was replaced with an azetidine-substituted pyrazine to afford compound 11. This analogue displayed improved potency and B1/B2 selectivity when compared with isoquinoline 6. Overall, the azetidine proved generally beneficial as demonstrated by compounds 12 and 13. The N-methyl lactam spirocycle present in 6 and 11 was also combined with a tertiary alcohol-substituted pyridine to afford enantiomeric products 14 and 15. As shown in Table 3, the (S)-enantiomer 14 exhibited notably better potency than the (R)-enantiomer 15. There did not appear to be any differences in binding with specific residues between the two enantiomers in our docking model; however, the preference for the (S)-enantiomer has been reported in previous work.8 Finally, the spirocyclic ether core embodied in 3 was combined with the (S)-tertiary alcohol-substituted pyridine to yield compound 16.
Table 3. Effect of Putative Heme Binding Moiety on CYP11B2 and CYP11B1 Inhibition.
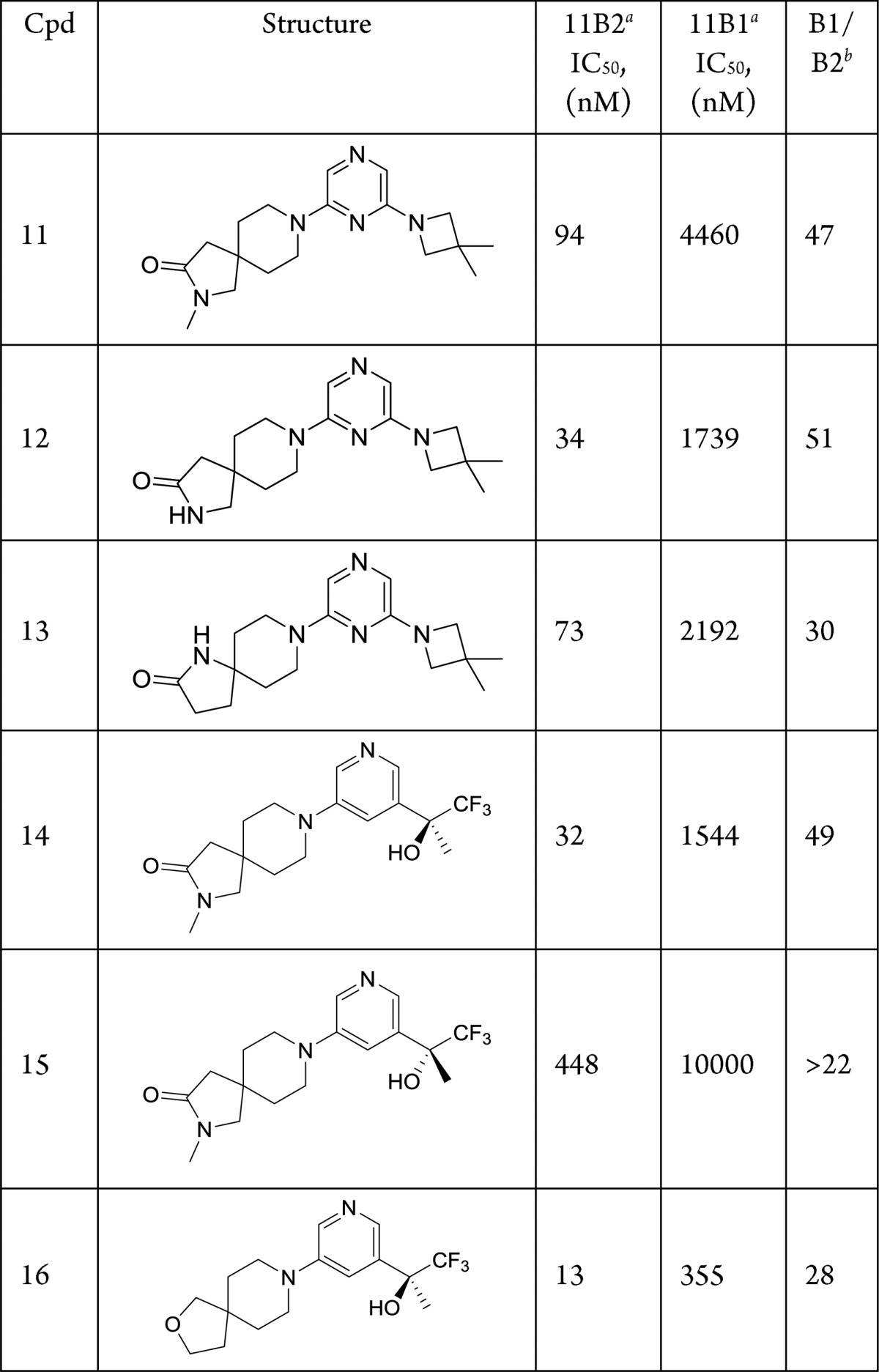
IC50s calculated from n ≥ 1, see Supporting Information for details. For compounds n > 1 the geometric mean is reported.
Ratio of hCYP11B1 IC50/hCYP11B2 IC50.
Motivated by the CYP11B2 potency and selectivity of compounds 14 and 16, additional studies to assess the lead-like properties were conducted. As shown in Table 4, both compounds were initially profiled for stability in rat liver microsomes where they demonstrated low rates of intrinsic clearance. Encouraged by this result, compounds 14 and 16 were also profiled in a rat pharmacokinetic experiment. Both compounds displayed high oral bioavailability, good oral exposure, and modest rates of plasma clearance. The comparatively short half-lives of compounds 14 and 16 reflect both their low volumes of distribution and moderate clearance rates. In addition to the ADME properties, the off-target profile of compounds 14 and 16 were also investigated. In this regard the compounds showed good selectivity versus hepatic CYPs and ion channels. Most notably compound 14 was >200-fold selective for the related steroidal CYPs 17, 19A1, and 21.23
Table 4. Rat Pharmacokinetic Profilesa of Compounds 14 and 16.
| Cpd | Rat Mic Clintb | Dose (iv/po)c | F (%)d | AUCN (po)e | Clpf | Vdg | t1/2h |
|---|---|---|---|---|---|---|---|
| 14 | <20 | 1.0/2.0 | 91 | 2.15 | 24 | 1.9 | 1.2 |
| 16 | <20 | 0.5/1.0 | 100 | 0.85 | 36 | 1.2 | 0.5 |
n = 2 (iv), n = 3 (po), Sprague–Dawley rats.
Intrinsic clearance in rat liver microsomes (mL/min/kg).
Dose (mg/kg).
Bioavailability.
Area under the plasma concentration vs time curve, normalized for dose (μM·h·kg/mg).
Plasma clearance (mL/min/kg).
Volume of distribution.
Half-life (h).
In summary, we have reported the discovery and hit-to-lead optimization of a novel class of spirocyclic piperidine aldosterone synthase inhibitors. The work demonstrated the value of using structure-based drug design to guide the discovery of the spirocyclic series. Lead compounds from this series, 14 and 16, showed good overlap with LCI699 as shown in Figure 3. These compounds displayed potent inhibition of CYP11B2 as well as selectivity over CYP11B1, other related CYPs, and pharmacologically relevant off-targets. In addition, they displayed lead-like physical and pharmacokinetic properties. On the basis of its promising overall profile, the spirocyclic piperidine series was progressed into lead optimization.
Figure 3.
Overlay of LCI-699 with lead spirocyclic piperidine compounds: LCI-699 (yellow), compound 14 (green), and compound 16 (pink) are shown as docked to CYP11B2 (PDB code 4ZGX, shown as gray ribbons with the heme cofactor as gray tubes).
Glossary
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, excretion
- Arg
arginine
- ASI
aldosterone synthase inhibitor
- AUC
area under the plasma concentration vs time curve
- Boc
tert-butyloxycarbonyl
- CYP
cytochrome P450
- HBA
hydrogen bond acceptor
- HPLC
high-performance liquid chromatography
- PBS
phosphate buffered saline
- PK
pharmacokinetic
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00455.
Assay protocols, synthesis schemes, representative procedures for the synthesis of compounds 1, 11, and 14 and related intermediates, and X-ray crystallographic data for intermediate used in compounds 14 and 16 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nwankwo T.; Yoon S. S.; Burt V.; Gu Q. Hypertension among adults in the US: national health and nutrition examination survey, 2011–2012. NCHS Data Brief, No. 133; National Center for Health Statistics, Centers for Disease Control and Prevention, US Dept of Health and Human Services, Hyattsville, MD, 2013. [PubMed] [Google Scholar]
- Calhoun D.; Jones D.; Textor S.; Goff D.; Murphey T.; Toto R.; White A.; Cushman W.; White W.; Sica D.; Ferdinand K.; Giles T.; Falkner B.; Carey R. Resistant hypertension: diagnosis, evaluation, and treatment. Hypertension 2008, 51, 1403–1419. 10.1161/HYPERTENSIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D.; Benjamin E.; Go A.; Arnett D.; Blaha M.; Cushman M.; de Ferranti S.; Després J.-P.; Fullerton H.; Howard V.; Huffman M.; Judd S.; Kissela B.; Lackland D.; Lichtman J.; Lisabeth L.; Liu S.; Mackey R.; Matchar D.; McGuire D.; Mohler E. III; Moy C.; Muntner P.; Mussolino M.; Nasir K.; Neumar R.; Nichol G.; Palaniappan L.; Pandey D.; Reves M.; Rodriguez C.; Sorlie P.; Stein J.; Towfighi A.; Turan T.; Virani S.; Willey J.; Woo D.; Yeh W.; Turner M. Executive summary: heart disease and stroke statistics – 2015 update. Circulation 2015, 131, e29–e322. 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- Tomaschitz A.; Pilz S.; Ritz E.; Obermayer-Pietsch B.; Pieber T. Aldosterone and arterial hypertension. Nat. Rev. Endocrinol. 2010, 6, 83–93. 10.1038/nrendo.2009.263. [DOI] [PubMed] [Google Scholar]
- For a recent review of aldosterone synthase inhibitors, see:Hu Q.; Yin L.; Hartmann R. W. Aldosterone synthase inhibitors as promising treatments for mineralocorticoid dependent cardiovascular and renal diseases. J. Med. Chem. 2014, 57, 5011–5022. 10.1021/jm401430e. [DOI] [PubMed] [Google Scholar]
- Cerny M. A. Progress toward clinically useful aldosterone synthase inhibitors. Curr. Top. Med. Chem. 2013, 13, 1385–1401. 10.2174/1568026611313120003. [DOI] [PubMed] [Google Scholar]
- Hoyt S. B.; Park M.; London C.; Xiong Y.; Tata J.; Bennett D. J.; Cooke J.; Cai J.; Carswell E.; Robinson J.; MacLean J.; Brown L.; Belshaw S.; Clarkson T.; Liu K.; Liang G.-B.; Struthers M.; Cully D.; Wisniewski T.; Ren N.; Bopp C.; Sok A.; Cai T.-Q.; Stribling S.; Pai L.-Y.; Ma X.; Metzger J.; Verras A.; McMasters D.; Chen Q.; Tung E.; Tang W.; Salituro G.; Buist N.; Kuethe J.; Rivera N.; Clemas J.; Zhou G.; Gibson J.; Maxwell C. A.; Lassman M.; McLaughlin T.; Castro-Perez J.; Szeto D.; Forrest G.; Hajdu R.; Rosenbach M.; Ali A. Discovery of benzimidazole CYP11B2 inhibitors with in Vivo activity in rhesus monkeys. ACS Med. Chem. Lett. 2015, 6, 573–578. 10.1021/acsmedchemlett.5b00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt S. B.; Petrilli W.; London C.; Liang G.-B.; Tata J.; Hu Q.; Yin L.; van Koppen C. J.; Hartmann R. W.; Struthers M.; Cully D.; Wisniewski T.; Ren N.; Bopp C.; Sok A.; Cai T.-Q.; Stribling S.; Pai L.-Y.; Ma X.; Metzger J.; Verras A.; McMasters D.; Chen Q.; Tung E.; Tang W.; Salituro G.; Buist N.; Clemas J.; Zhou G.; Gibson J.; Maxwell C. A.; Lassman M.; McLaughlin T.; Castro-Perez J.; Szeto D.; Forrest G.; Hajdu R.; Rosenbach M.; Xiong Y. Discovery of triazole CYP11B2 inhibitors with in vivo activity in rhesus monkeys. ACS Med. Chem. Lett. 2015, 6, 861–865. 10.1021/acsmedchemlett.5b00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.; Yin L.; Ali A.; Cooke A. J.; Bennett J.; Ratcliffe P.; Lo M. M.-C.; Metzger E.; Hoyt S.; Hartmann R. W. Novel pyridyl substituted 4,5-dihydro-[1,2,4]-triazolo[4,3-a]quinolines as potent and selective aldosterone synthase inhibitors with improved in vitro metabolic stability. J. Med. Chem. 2015, 58, 2530–2537. 10.1021/acs.jmedchem.5b00079. [DOI] [PubMed] [Google Scholar]
- Grombein C. M.; Hu Q.; Rau S.; Zimmer C.; Hartmann R. W. Heteroatom insertion into 3,4-dihydro-1H-quinolin-2-ones leads to potent and selective inhibitors of human and rat aldosterone synthase. Eur. J. Med. Chem. 2015, 90, 788–796. 10.1016/j.ejmech.2014.12.022. [DOI] [PubMed] [Google Scholar]
- Yin L.; Hu Q.; Emmerich J.; Lo M. M.-C.; Metzger E.; Ali A.; Hartmann R. W. J. Med. Chem. 2014, 57, 5179–5189. 10.1021/jm500140c. [DOI] [PubMed] [Google Scholar]
- Meredith E. L.; Ksander G.; Monovich L. G.; Papillon J. P. N.; Liu Q.; Miranda K.; Morris P.; Rao C.; Burgis R.; Capparelli M.; Hu Q.-Y.; Singh A.; Rigel D. F.; Jeng A. Y.; Beil M.; Fu F.; Hu C.-W.; LaSala D. Discovery and in vivo evaluation of potent dual CYP11B2 (Aldosterone Synthase) and CYP11B1 inhibitors. ACS Med. Chem. Lett. 2013, 4, 1203–1207. 10.1021/ml400324c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon J. P. N.; Adams C. M.; Hu Q.-Y.; Lou C.; Singh A. K.; Zhang C.; Carvahlo J.; Rajan S.; Amaral A.; Beil M. E.; Fu F.; Gangl E.; Hu C.-W.; Jeng A. Y.; LaSala D.; Liang G.; Logman M.; Maniara W. M.; Rigel D. F.; Smith S. A.; Ksander G. M. Structure-activity relationships, pharmacokinetics, and in vivo activity of CYP11B2 and CYP11B1 inhibitors. J. Med. Chem. 2015, 58, 4749–4770. 10.1021/acs.jmedchem.5b00407. [DOI] [PubMed] [Google Scholar]
- Papillon J. P. N.; Lou C.; Singh A. K.; Adams C. M.; Ksander G. M.; Beil M. E.; Chen W.; Leung-Chu J.; Fu F.; Gan L.; Hu C.-W.; Jeng A. Y.; LaSala D.; Liang G.; Rigel D. F.; Russell K. S.; Vest J. A.; Watson C. Discovery of N-[5-(6-chloro-3-cyano-1-methyl-1H-indol-2-yl)-pyridin-3-ylmethyl]-ethanesulfonamide, a cortisol-sparing CYP11B2 inhibitor that lowers aldosterone in human subjects. J. Med. Chem. 2015, 58, 9382–9384. 10.1021/acs.jmedchem.5b01545. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Aebi J. D.; Hornsperger B.; Krebs H.-J.; Kuhn B.; Kuglstatter A.; Alker A. M.; Marki H. P.; Muller S.; Burger D.; Ottaviani G.; Riboulet W.; Verry P.; Tan X.; Amrein K.; Mayweg A. V. Discovery of 4-Aryl-5,6,7,8-tetrahydroisoquinolines as potent, selective, and orally active aldosterone synthase (CYP11B2) inhibitors: in vivo evaluation in rodents and cynomologous monkeys. J. Med. Chem. 2015, 58, 8054–8065. 10.1021/acs.jmedchem.5b00851. [DOI] [PubMed] [Google Scholar]
- Calhoun D. C.; White W. B.; Krum H.; Guo W.; Bermann G.; Trapani A.; Lefkowitz M. P.; Menard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension. Circulation 2011, 124, 1945–1955. 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]
- Azizi M.; Amar L.; Menard J. Aldosterone synthase inhibition in humans. Nephrol., Dial., Transplant. 2013, 28, 36–43. 10.1093/ndt/gfs388. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Tice C. M.; Singh S. B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. 10.1016/j.bmcl.2014.06.081. [DOI] [PubMed] [Google Scholar]
- Strushkevich N.; Gilep A.; Shen L.; Arrowsmith C.; Edwards A.; Usanov S.; Park H.-W. Structural Insights into Aldosterone Synthase Substrate Specificity and Targeted Inhibition. Mol. Endocrinol. 2013, 27, 315–324. 10.1210/me.2012-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. E.; Aebi J. D.; Hornsperger B.; Krebs H. J.; Kuhn B.; Kuglstatter A.; Alker A. M.; Marki H. P.; Muller S.; Burger D.; Ottaviani G.; Riboulet W.; Verry P.; Tan X.; Amrein K.; Mayweg A. V. Discovery of 4-Aryl-5,6,7,8-tetrahydroisoquinolines as Potent, Selective, and Orally Active Aldosterone Synthase (CYP11B2) Inhibitors: In Vivo Evaluation in Rodents and Cynomolgus Monkeys. J. Med. Chem. 2015, 58, 8054–8065. 10.1021/acs.jmedchem.5b00851. [DOI] [PubMed] [Google Scholar]
- Lucas S.; Heim R.; Ries C.; Schewe K. E.; Birk B.; Hartmann R. W. In vivo active aldosterone synthase inhibitors with improved selectivity: lead optimization providing a series of pyridine substituted 3,4-dihydro-1H-quinolin-2-one derivatives. J. Med. Chem. 2008, 51, 8077–8087. 10.1021/jm800888q. [DOI] [PubMed] [Google Scholar]
- Compounds were synthesized as described in the Supporting Information and were initially assayed for their ability to inhibit human CYP11B2 and CYP11B1. Assays were run in V79 cells stably expressing either CYP11B2 or CYP11B1, and employed HTRF detection to measure the conversion of 11-deoxycorticosterone to aldosterone (in the case of CYP11B2) or 11-deoxycortisol to cortisol (in the case of CYP11B1).
- Selectivity data including IC50 is reported in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.