

Abstract
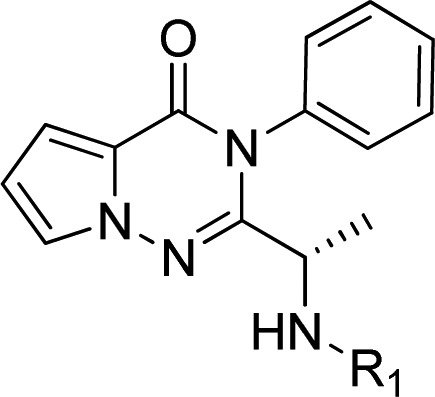
The delta isoform of the phosphatidylinositol 3-kinase (PI3Kδ) has been shown to have an essential role in specific immune cell functions and thus represents a potential therapeutic target for autoimmune and inflammatory diseases. Herein, the optimization of a series of pyrrolotriazinones as potent and selective PI3Kδ inhibitors is described. The main challenge of the optimization process was to identify an orally available compound with a good pharmacokinetic profile in preclinical species that predicted a suitable dosing regimen in humans. Structure–activity relationships and structure–property relationships are discussed. This medicinal chemistry exercise led to the identification of LAS191954 as a candidate for clinical development.
Keywords: Phosphoinositide-3-kinase delta inhibitor, PI3Kδ inhibitor, structure−activity relationship, autoimmune diseases, inflammatory diseases, lead optimization
The phosphatidylinositol-3-kinase (PI3K) signal transduction pathway is central to a plethora of different cellular processes involving metabolism, proliferation, differentiation, or activation. Hence, manipulation of the PI3K pathway represents an interesting approach for treatment of a number of different pathological conditions such as cancer, where inhibitors of class IA PI3K with varying isoform selectivity profiles have already been established as treatment options for different indications.1
In the immune system, the PI3K delta isoform plays a central role in both innate and adaptive immune cell functions. Taking advantage of the strong dependency of B cells on functional PI3Kδ,2 the oral PI3Kδ inhibitor Idelalisib has been successfully developed as a novel treatment for different types of B-cell malignancies.3 In addition, the involvement of PI3Kδ in central immune functions suggests a therapeutic potential of PI3Kδ inhibitors as novel broad-acting anti-inflammatory agents for autoimmune diseases4 and pathologies with an allergic or inflammatory component such as allergic rhinitis,5 asthma,6 or COPD.7 Apart from inhibiting B cell activation, pharmacological inhibition of PI3K delta has been demonstrated to attenuate T cell receptor induced cytokine production,8 to inhibit degranulation of basophils and mast cells,5,9 and to suppress the oxidative burst response by neutrophils.10 In addition, PI3Kδ seems to be involved in corticosteroid resistance induced by oxidative stress in macrophages,11 thus indicating an activity profile complementary to other current treatments such as corticosteroids.
The discovery of the propeller-shaped inhibitor 1 (IC87114, ICOS) and the mechanisms by which δ isoform selectivity can be accomplished focused our attention.12 Following this strategy, our efforts toward the identification of new PI3Kδ inhibitors were based on a pyrrolotriazinone scaffold as shown in Scheme 1. Our initial lead 2 showed similar PI3Kδ inhibitor potency to IC87114 (110 nM versus 130 nM) and also high selectivity against the other class I PI3K isoforms. Moving the methyl group from the phenyl ring in compound 2 to the linker (compound 3), slightly improved the PI3Kδ inhibitor potency to 75 nM and removed the potential for atropisomerism13 present in ortho-substituted compounds 1 and 2. Taking into account the stereochemistry of known PI3Kδ selective inhibitors in the clinic such as Idelalisib14 and Duvelisib,15 the S-enantiomers were initially targeted.
Scheme 1. Pyrrolotriazinone Scaffold.

Thus, an extensive structure–activity relationship (SAR) study was then performed around compound 3 to improve the potency and modulate ADME properties.
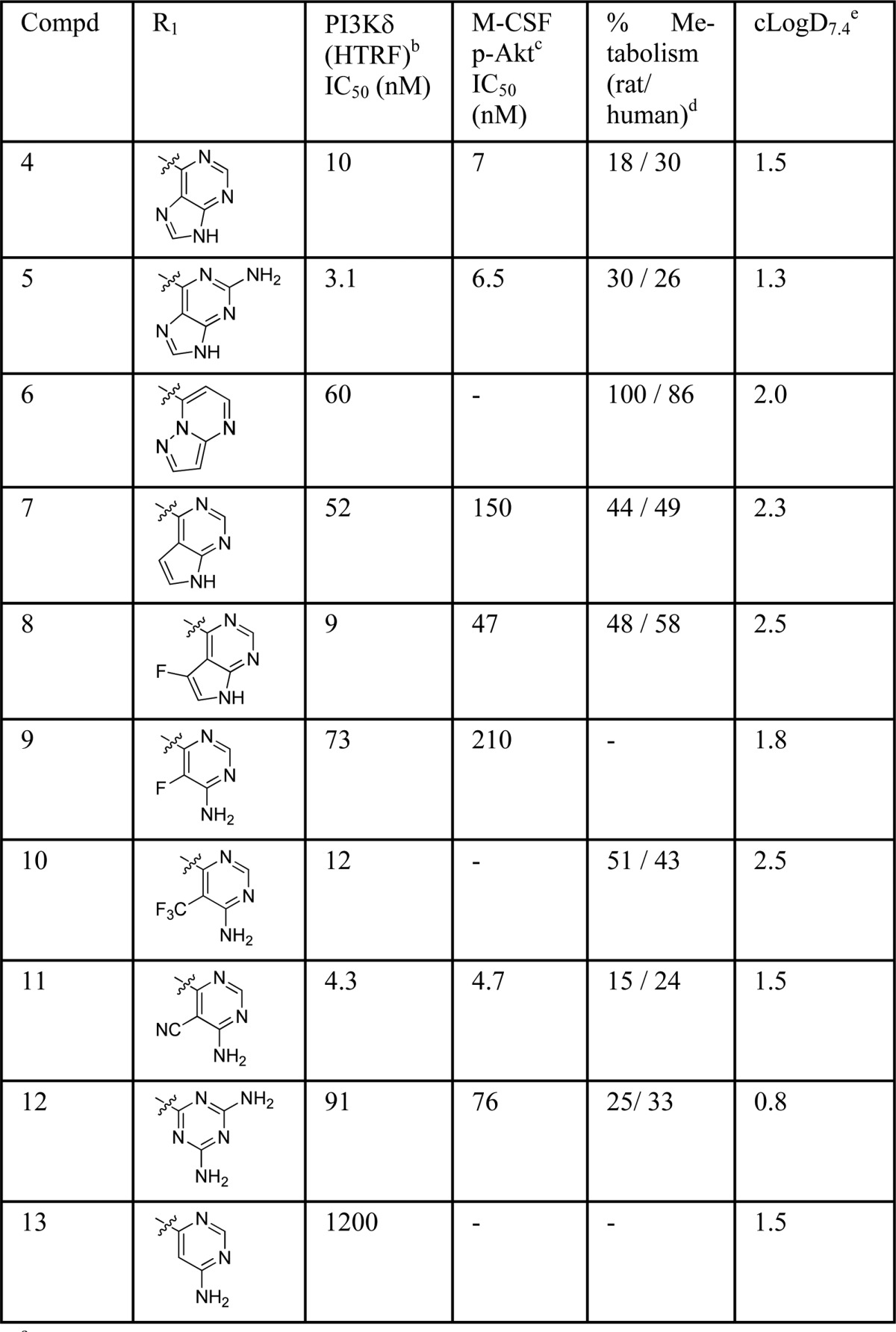
Most kinase inhibitors are known to bind to a region in the kinase termed the hinge binder region, usually with an H-bond acceptor–donor pair. Initially, an exploration of this region was undertaken, and PI3Kδ inhibitory potency in both enzymatic and cellular assays was assessed together with in vitro metabolism16 for each compound (Table 1). Introduction of other purine-like hinge binders (4, 5) increased the in vitro potency for PI3Kδ considerably, and the compounds showed low in vitro metabolism. Other bicyclic rings (6–8) demonstrated lower potency and decreased metabolic stability when compared to compounds 4 and 5. Monocyclic rings such as those in compounds 9–13 (Table 1) were then studied and cyanopyrimidine 11 showed the best overall balance between potency and in vitro metabolism. A clear correlation between metabolism rate and lipophilicity could not be established; however, a tendency was observed for those compounds with a cLogD7.4 around 2.0 or above all showing low metabolic stability.
Table 1. SAR Exploration of the Hinge Bindera.
All assay results are reported as the geometric mean of at least two separate runs.
PI3Kδ activities were measured with an ATP concentration fixed at the Km of PI3Kδ by HTRF, where the PIP3 product is detected by displacement of biotin-PIP3 from an energy transfer complex.
THP-1 cells were treated with compounds for 30 min, stimulated with M-CSF at EC80 for 3 min, and then lysed to measure (by ELISA) pAkt (Thr308) produced through PI3Kδ.
Percent metabolism expressed as disappearance of parent compound after microsomal incubation for 30 min (1 mg/mL protein and 5 μM compound at 37 °C).
Calculated LogD value at pH = 7.4.
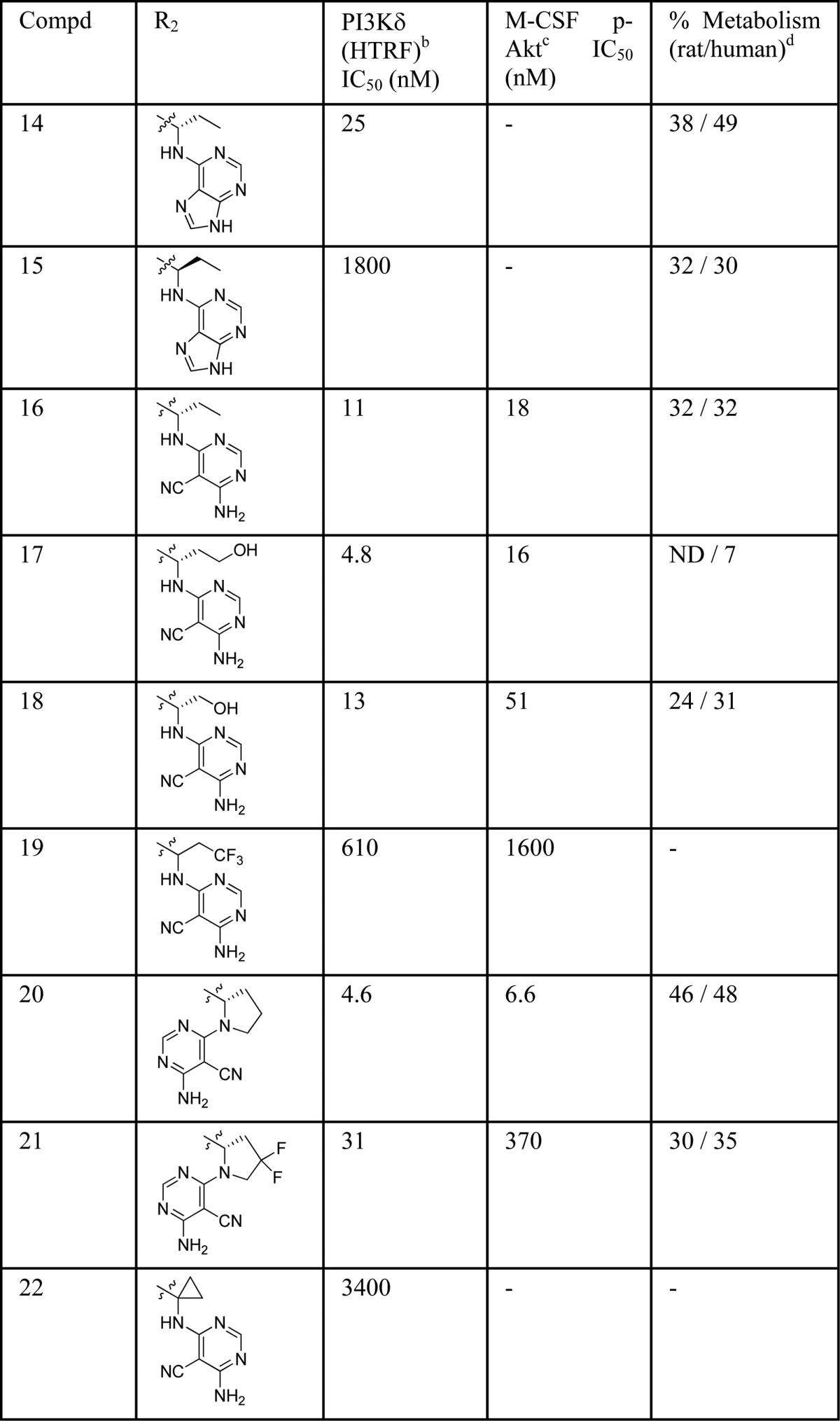
Optimization of the linker was then explored as shown in Table 2. An ethyl group was well tolerated giving a compound (14) of similar potency to the methyl derivative 4 but poorer microsomal stability. The corresponding (R)-enantiomer of 14 was synthesized (compound 15) and the resultant drop off in potency confirmed the preference of PI3Kδ for the (S)-enantiomer. Replacing the hinge binder of compound 14 for the cyanopyrimide (16) slightly improved potency and in vitro metabolism. Appending a hydroxyl group onto compounds 11 and 16 resulted in derivative 18 and 17, respectively, but only compound 17 showed an improved microsomal stability while maintaining good PI3Kδ potency. In contrast, introduction of a trifluoroethyl group (compound 19) was less well tolerated. Cyclic analogue 20 kept in vitro potency but was metabolically less stable. Introduction of fluorine atoms in the cyclopentyl ring of 20 with the aim of increasing metabolic stability led to a less potent compound 21. Spirocyclic ring variation (compound 22) resulted in a large drop off in potency.
Table 2. Linker SAR Explorationa.
All assay results are reported as the geometric mean of at least two separate runs.
PI3Kδ activities were measured with an ATP concentration fixed at the Km of PI3Kδ by HTRF, where the PIP3 product is detected by displacement of biotin-PIP3 from an energy transfer complex.
THP-1 cells were treated with compounds for 30 min, stimulated with M-CSF at EC80 for 3 min, and then lysed to measure (by ELISA) pAkt (Thr308) produced through PI3Kδ.
Percent metabolism expressed as disappearance of parent compound after microsomal incubation for 30 min (1 mg/mL protein and 5 μM compound at 37 °C).
Compounds 11 and 17 were then profiled in a rat in vivo pharmacokinetic (PK) experiment. However, for both compounds, in vivo clearance in rat was higher than expected from the microsomal stability data, resulting in moderate systemic exposure and short half-lives (Table 3).
Table 3. Rat PK Profiles of Compounds 11 and 17.
Mean values (n = 2) in Wistar rat after an administration of 1 mg/kg i.v. Parameters calculated from plasma samples: t1/2 = half-life, Cl = clearance, AUC = area under the curve, Vss = volume of distribution at steady state.
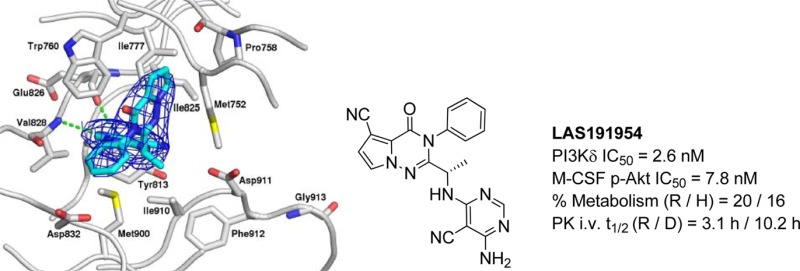
To better understand the binding mode of the ligands an X-ray cocrystal of human PI3Kδ in complex with compound 11 was performed. The structure was solved at a resolution of 2.85 Å, revealing the detailed binding mode of the ligand. Like IC87114, compound 11 adopted a propeller-shaped conformation where the pyrrolotriazinone moiety was sandwiched into the induced hydrophobic specificity pocket between Trp760 and Met752. The cyanopyrimidine ring of 11 served as the hinge binder and formed two specific hydrogen bonds to the main chain atoms of Val828 and Glu826. The following residues were found in the vicinity of the ligand with a maximum distance of 3.9 Å: Met752, Pro758, Trp 760, Ile777, Tyr813, Ile825, Glu826, Val827, Val828, Ser831, Asp832, Met900, Ile910, and Asp911. A closer look at the binding pocket surface (Figure 1) suggested that the pyrrolotriazinone core ring could be substituted at the 5-, 6- or 7-position to further modulate potency.
Figure 1.
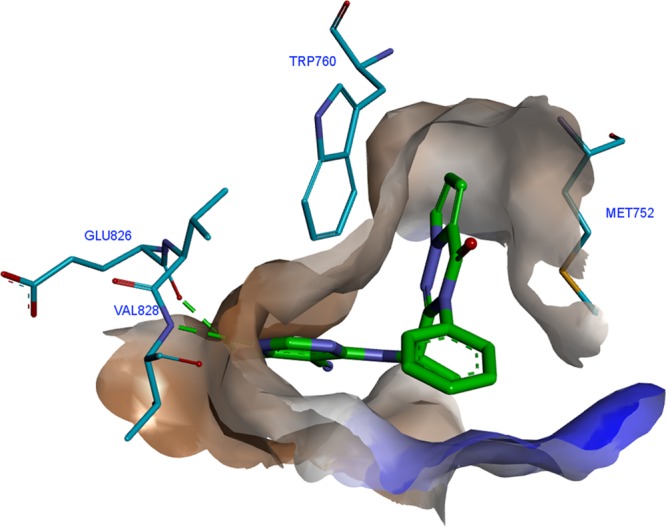
X-ray cocrystal structure of the binding pocket surface of human PI3Kδ containing compound 11.
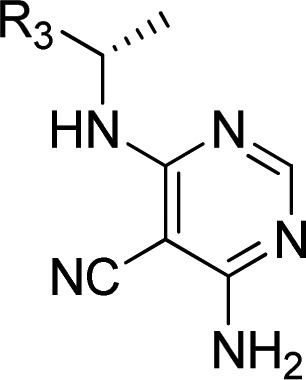
Thus, several analogues with different substitutions on the pyrrolo ring were synthesized and tested. A methyl group was well tolerated in different positions (compounds 23 and 29), but the resultant derivatives were metabolically unstable (Table 4). Appending a hydroxyl group onto the methyl group of 23 resulted in a compound 24 with reasonable PI3Kδ inhibitor potency and improved metabolic stability. After further study it was found that placement of other substituents at position 5 (25, 26, and 27) gave rise to compounds with excellent potencies and good metabolic stability. Further characterization of compound 25 showed the formation of GSH adducts following incubation of 25 with microsomes in the presence of glutathione (GSH). As the formation of reactive metabolites is potentially involved in severe adverse drug reactions, this compound was not further profiled. Moving this fluorine atom to position 6 (compound 28) slightly diminished potency. At position 7, compounds bearing small groups such as methyl (29) or cyano (30) were better tolerated than a bulkier trifluoromethyl group (31). Finally, the imidazotriazinone 32 showed a substantially diminished PI3Kδ inhibitor potency in comparison to the corresponding pyrrolotriazinone 11.
Table 4. Distal Core SAR Explorationa.
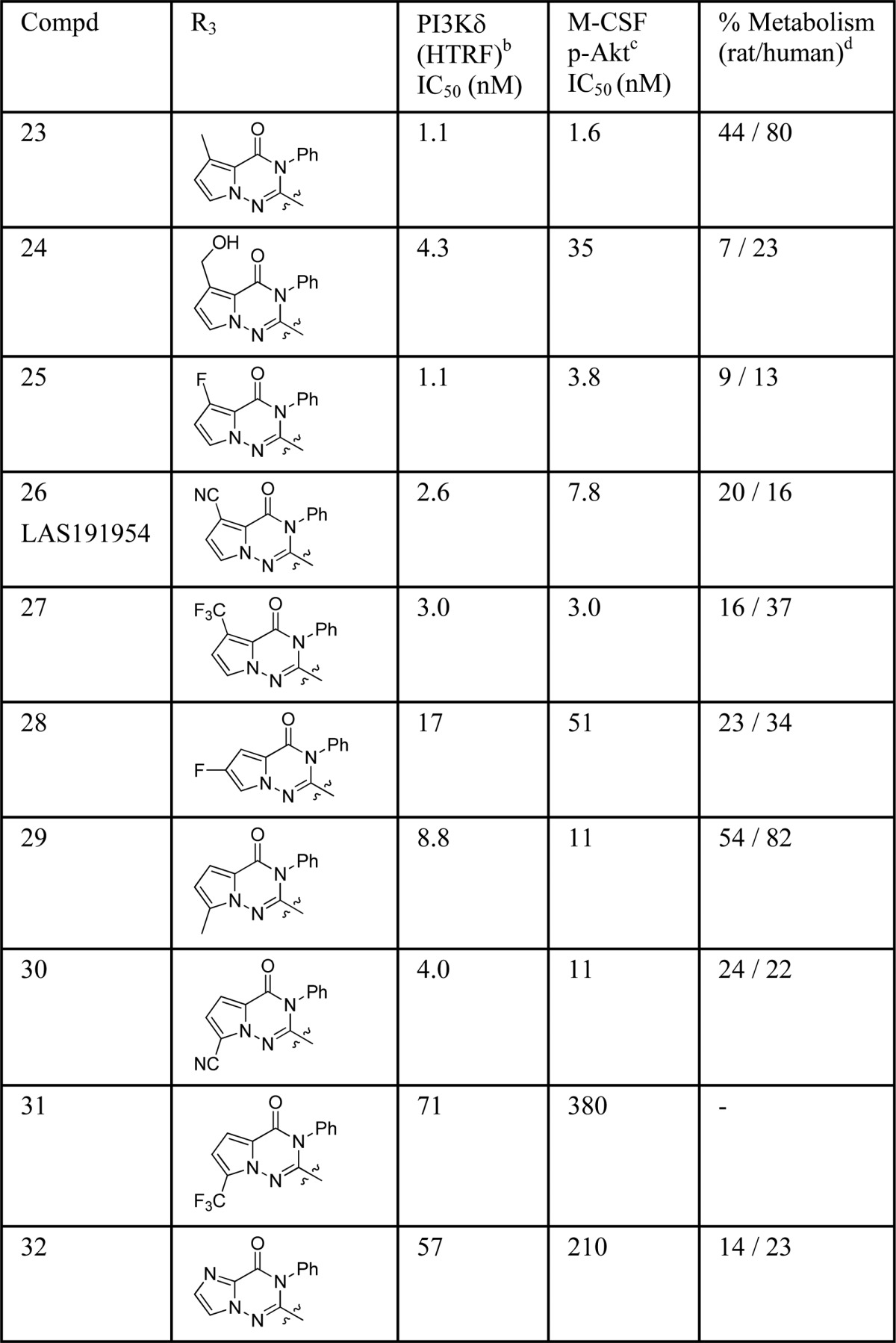
All assay results are reported as the geometric mean of at least two separate runs.
PI3Kδ activities were measured with an ATP concentration fixed at the Km of PI3Kδ by HTRF, where the PIP3 product is detected by displacement of biotin-PIP3 from an energy transfer complex.
THP-1 cells were treated with compounds for 30 min, stimulated with M-CSF at EC80 for 3 min and then lysed to measure (by ELISA) pAkt (Thr308) produced through PI3Kδ.
Percent metabolism expressed as disappearance of parent compound after microsomal incubation for 30 min (1 mg/mL protein and 5 μM compound at 37 °C).
From this set of compounds, 26 and 27 were further profiled in a rat in vivo PK experiment (Table 5) to determine if the in vitro metabolism results were, in these cases, predictive of the in vivo clearance values. These results were compared with Idelalisib, the most advanced compound in clinical development at that moment.
Table 5. Rat PK Profiles of Compounds 26 and 27 in Comparison with Idelalisib.
| compd | t1/2 (h)a | AUC (ng·h/mL)a | Cl (mL/min/kg)a | Vz (L/kg)a | F (%)b |
|---|---|---|---|---|---|
| Idelalisib | 0.8 | 512 | 32.8 | 2.2 | 68 |
| 26 | 3.1 | 1729 | 9.6 | 2.6 | 101 |
| 27 | 3.3 | 2040 | 8.6 | 2.4 | 113 |
Mean values (n = 2) in Wistar rat after an administration of 1 mg/kg i.v.
Mean values (n = 2) in Wistar rat after an administration of 1 mg/kg p.o.
As seen in Table 5, compounds 26 and 27, with substituents at position 5 of the pyrrolo ring, showed increased plasma exposure compared to unsubstituted compounds 11 and 17, excellent oral bioavailability and reduced clearance in rat, which was more consistent with the observed microsomal stability values. One plausible hypothesis could be that, although moderate metabolism was observed for unsubstituted pyrrolo derivatives such as 11 and 17, many metabolites (>5) were identified after microsomal incubation for both compounds and those metabolites might be magnified in vivo. The same behavior was observed for Idelalisib, a compound also showing moderate in vitro metabolism (20% and 21% in rat and human liver microsomes, respectively) with seven metabolites identified. In contrast, compounds with substitution on the pyrrolo ring formed fewer metabolites in microsomes (except for compounds bearing a methyl group), and this could explain the lower clearance observed in vivo.
Additionally, the PK properties of both compounds 26 and 27 were studied in dog as a second preclinical species to allow for estimation of the human half-life based on allometric scaling. The dog in vivo PK data are shown in Table 6. Both compounds 26 and 27 displayed a superior PK profile to Idelalisib in the two preclinical species. Compound 26 (LAS191954), with excellent oral bioavailability and low clearance in the two species, gave a superior predicted half-life in humans and was further profiled.
Table 6. Dog PK Profiles of Compounds 26 and 27 in Comparison with Idelalisib.
| compd | t1/2 (h)a | AUC (ng·h/mL)a | Cl (mL/min/kg)a | Vz (L/kg)a | F (%)b |
|---|---|---|---|---|---|
| Idelalisib | 1.6 | 924 | 21.5 | 2.5 | |
| 26 | 10.2 | 9441 | 1.4 | 1.2 | 98 |
| 27 | 4.2 | 3937 | 4.2 | 1.5 |
Mean values (n = 2) in Beagle dog after an administration of 1 mg/kg i.v.
Mean values (n = 3) in Beagle dog after an administration of 1 mg/kg p.o.
For the assessment of selectivity, the enzymatic potency of LAS191954 on the four class I PI3K recombinant human isoforms was determined by HTRF with a compound preincubation time of 30 min. Assays were run at ATP concentration equal to the respective Km and using a subnanomolar concentration of the enzyme. In these conditions, LAS191954 showed a potency on the target of 2.6 nM, with the highest selectivity versus PI3Kα (8.2 μM) and the lowest versus PI3Kγ and PI3Kβ (72 and 94 nM, respectively). The compound was then tested in a primary PI3Kδ-dependent cellular assay based on M-CSF-induced AKT phosphorylation, a downstream effector of PI3Kδ, in the human monocytic cell line THP-1. An IC50 of 7.8 nM was obtained indicating that the compound had excellent permeability and cellular activity. To evaluate the cellular inhibition of PI3Kβ, an assay based on stimulation of HUVEC cells with sphingosine-1-P was employed.17 The results (IC50 = 295 nM) indicated that the enzymatic selectivity between δ and β isoforms (36-fold) was maintained at the cell-based level (38-fold). The compound was also selective against an extensive panel of protein and lipid kinases and GPCRs.
PI3Kδ kinase is involved in the activation of B cells upon antigen binding to the B cell receptor (BCR),18 and thus, inhibitors of PI3Kδ are expected to inhibit BCR activation. The effect of LAS191954 on the function of human B cells was assessed in vitro by cross-linking the B-cell receptor with anti-IgD antibodies and assessing the early activation marker CD69 in the CD19+ B cell subset by flow cytometry. On isolated PBMC, LAS191954 showed an IC50 of 4.6 nM. Similar assay performed in human whole blood yielded an IC50 of 47 nM.
From an ADME perspective, further in vitro characterization of LAS191954 was performed as summarized in Table 7. Plasma protein binding19 was low in preclinical species (60–65% in mouse/rat/dog) in contrast to human (95%), which might be the major factor accounting for the difference in potency between the isolated human PBMC and whole blood assays previously described. The compound showed a good permeability20 and did not show any signs of plasma instability. Drug–drug interactions are not expected in a clinical setting since neither inhibition nor induction of the major CYP450 isoforms was observed in vitro in humans. Opposite to compound 25, no formation of GSH-adducts was observed following incubation of LAS191954 with rat and human liver microsomes in the presence of glutathione (GSH).
Table 7. In Vitro ADME Profile of LAS191954.
| human PPB (% bound)a | PAMPA Papp (× 10–6 cm/s)b | plasma stability (24 h)c | CYP450 inhibitiond | CYP450 inductione |
|---|---|---|---|---|
| 95.4 | 2.8 | 100% | >25 μM | No issues |
Mean PPB values determined by equilibrium dialysis at a plasma concentration of 1 μM at 37 °C.
Permeability values at RT in PBS (containing 2% DMSO),; compound concentration in donor compartment: 20 μM.
LAS191954 stability at 37 °C at 2 μg/mL in plasma (anticoagulant: sodium heparin).
CYP450 in HLM with and without preincubation for CYP1A2, 3A4, 2D6, 2C9, and 2C19 isoforms.
Induction of CYP1A and 3A4 isoforms was assessed in cultured human cryopreserved hepatocytes at concentrations up to 50 μM.
In terms of cardiac safety, LAS191954 did not show any activity on the hERG channel up to 10 μM.
As a consequence of these encouraging results, the compound was further characterized in different in vivo models.
PI3Kδ has been implicated in T-cell proliferation in vitro and in vivo. Cytokine production induced by concanavalin A (ConA) or anti-CD3 antibody activation of naïve CD4+ T cells in mice is blocked by PI3Kδ inhibition.21 To assess the effect of LAS191954 on T cell cytokine production in vivo, a rat model of ConA induced IL2 production was employed in which the compound was administered orally 1 h prior to an intravenous ConA challenge (10 mg/kg) and IL2 levels were measured 90 min later. LAS191954 inhibited IL2 production with an ID50 of 0.13 mg/kg as compared to an ID50 of 1.6 mg/kg of Idelalisib as shown in Figure 2.
Figure 2.
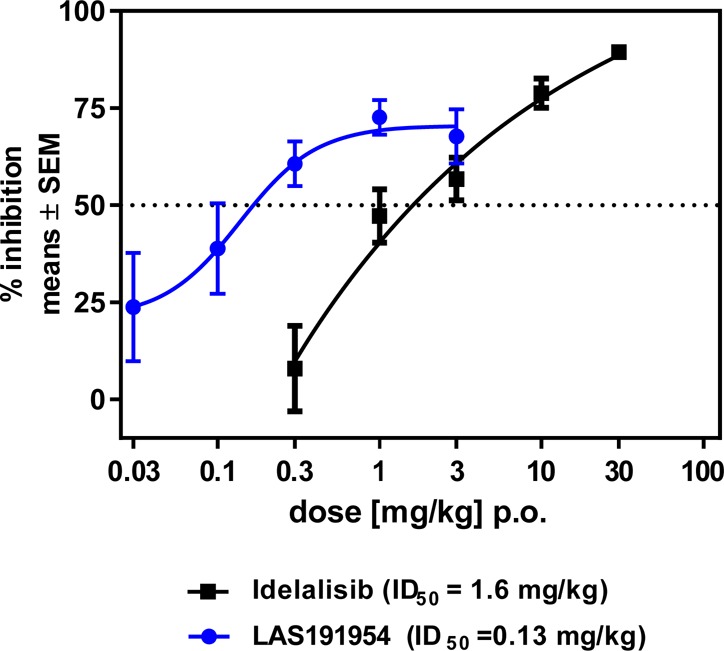
Inhibition of ConA induced increase in IL-2 levels in rat plasma of LAS191954 vs reference compound Idelalisib.
PI3Kδ participates in the proliferation, differentiation, migration, and cytokine production of T lymphocytes, in the migration and release of ROS and proteases by neutrophils, and in the migration, alternative M2 activation, and loss of steroid sensitivity of macrophages. PI3Kδ additionally regulates Fcε mediated mast cell and basophil degranulation. Thus, the anti-inflammatory efficacy of LAS191954 was assessed in an airway allergic inflammation model characterized by infiltration of eosinophils to the alveolar compartment in response to ovalbumin challenge in previously sensitized Brown Norway rats.
A twice daily administration of LAS191954 1 h prior to and 6 h post-ovalbumin (OVA) challenge dose-dependently reduced the number of eosinophils in the bronchoalveolar lavage (BAL) at 24 h in rat with an ID50 of 0.16 mg/kg. The reference compound Idelalisib, administered also twice daily, displayed a considerably lower potency with an ID50 of 15 mg/kg (Figure 3).
Figure 3.
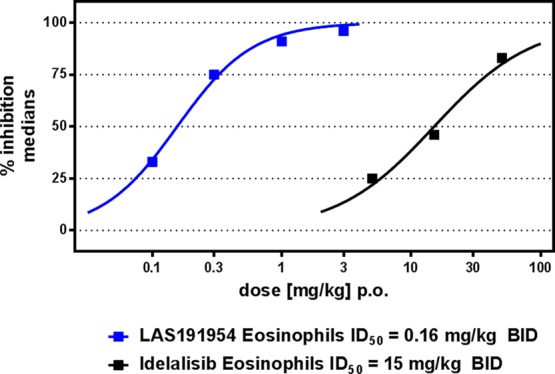
Inhibition of OVA BAL cell accumulation in BN rats at 24 h by twice daily doses of LAS191954 and Idelalisib.
In both in vivo models LAS191954 showed efficacy at significantly lower doses than Idelalisib. These results could not be explained by differences in in vitro potency as Idelalisib exhibited a similar cellular PI3Kδ inhibitor potency with an IC50 of 7.6 nM, but they are in accordance with a superior PK profile (higher systemic exposure and longer half-life) of LAS191954 in rat.
In summary, a new series of potent and selective PI3Kδ inhibitors has been identified. The SAR exercise focused on optimizing both the in vitro potency and the ADME profile of the compounds in order to find a potentially once-a-day drug. The pyrrolotriazinone scaffold enabled us to build in better pharmacokinetic properties in preclinical species, which may ultimately translate in a compound with reduced dosing in humans, and culminated in the identification of LAS191954 as a candidate for clinical development.
Acknowledgments
The authors wish to thank Proteros Biostructures GmbH for X-ray structure determination of human PI3Kδ in complex with compound 11 (PDB code: 5M6U). We also acknowledge the significant technical support provided by Jordi Sanahuja, Gaspar Casals, and Antonia Molina. We thank Sonia Espinosa and Josep M. Huerta for their support on physicochemical properties analysis and structural characterization of compounds.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00438.
Characterization of all compounds. Experimental procedures for the sequence leading to key compound 26. Description of all biological assays and in vivo studies. Crystallographic data collection and refinement statistics for crystal structure of compound 11 (PDF)
Accession Codes
PDB code for X-ray crystal structure described in this study has been deposited in the Protein Data Bank under the following accession codes: 5M6U (compound 11 in complex with PI3Kδ).
Author Present Address
⊥ Pharmaxis Ltd., 20 Rodborough Road, Frenchs Forest, NSW 2086, Australia.
Author Present Address
# Bionorica SE, Kerschensteinerstraße 11–15, 92318 Neumarkt, Germany.
Author Present Address
∇ Fundació ACE, Marqués de Sentmenat, 57, 08029 Barcelona, Spain.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Stark A.; Sriskantharajah S.; Hessel E. M.; Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. 10.1016/j.coph.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri K. D.; Gold M. R. Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Front. Immunol. 2012, 3, 256. 10.3389/fimmu.2012.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah C. Y.; Fowler N. H. Idelalisib in the management of lymphoma. Blood 2016, 128 (3), 331–6. 10.1182/blood-2016-02-702761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand C. A.; Richer M. J.; Brenker K.; Graves M.; Shanina I.; Choi K.; Horwitz M. S.; Puri K. D.; Gold M. R. Selective Pharmacological Inhibition of Phosphoinositide 3-Kinase p110delta Opposes the Progression of Autoimmune Diabetes in Non-Obese Diabetic (NOD) Mice. Autoimmunity 2013, 46 (1), 62–73. 10.3109/08916934.2012.732130. [DOI] [PubMed] [Google Scholar]
- Horak F.; Puri K. D.; Steiner B. H.; Holes L.; Xing G.; Zieglmayer P.; Zieglmayer R.; Lemell P.; Yu A. Randomized Phase 1 study of the phosphatidylinositol 3-kinase δ inhibitor Idelalisib in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2016, 137 (6), 1733–41. 10.1016/j.jaci.2015.12.1313. [DOI] [PubMed] [Google Scholar]
- Rowan W. C.; Smith J. L.; Affleck K.; Amour A. Targeting phosphoinositide 3-kinase δ for allergic asthma. Biochem. Soc. Trans. 2012, 40 (1), 240–5. 10.1042/BST20110665. [DOI] [PubMed] [Google Scholar]
- Sriskantharajah S.; Hamblin N.; Worsley S.; Calver A. R.; Hessel E. M.; Amour A. Targeting phosphoinositide 3-kinase δ for the treatment of respiratory diseases. Ann. N. Y. Acad. Sci. 2013, 1280, 35–9. 10.1111/nyas.12039. [DOI] [PubMed] [Google Scholar]
- Soond D. R.; Bjørgo E.; Moltu K.; Dale V. Q.; Patton D. T.; Torgersen K. M.; Galleway F.; Twomey B.; Clark J.; Gaston J. S.; Taskén K.; Bunyard P.; Okkenhaug K. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood 2010, 115 (11), 2203–2213. 10.1182/blood-2009-07-232330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali K.; Camps M.; Pearce W. P.; Ji H.; Rückle T.; Kuehn N.; Pasquali C.; Chabert C.; Rommel C.; Vanhaesebroeck B. Isoform-Specific Functions of Phosphoinositide 3-Kinases: p110δ Not p110γ Promotes Optimal Allergic Responses In Vivo. J. Immunol. 2008, 180 (4), 2538–2544. 10.4049/jimmunol.180.4.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung-Leung W. P. Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function. Cell. Signalling 2011, 23 (4), 603–8. 10.1016/j.cellsig.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Marwick J. A.; Caramori G.; Casolari P.; Mazzoni F.; Kirkham P. A.; Adcock I. M.; Chung K. F.; Papi A. A role for phosphoinositol-3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2010, 125 (5), 1146–53. 10.1016/j.jaci.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Berndt A.; Miller S.; Williams O.; Le D. D.; Houseman B. T.; Pacold J. I.; Gorrec F.; Hon W.-C.; Liu Y.; Rommel C.; Gaillard P.; Ruckle T.; Schwarz M. K.; Shokat K. M.; Shaw J. P.; Williams R. L. The p110δ structure: mechanisms for selectivity and potency of new PI(3)k inhibitors. Nat. Chem. Biol. 2010, 6 (2), 117–124. 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evarts J. B.; Ulrich R. G.. Atropisomers of PI3K-inhibiting compounds. WO2012/040634.
- Somoza J. R.; Koditek D.; Villaseñor A. G.; Novikov N.; Wong M. H.; Liclican A.; Xing W.; Lagpacan L.; Wang R.; Shultz B. E.; Papalia G. A.; Samuel D.; Lad L.; McGrath M. E. Structural, Biochemical, and Biophysical Characterization of Idelalisib Binding to Phosphoinositide 3-Kinase δ. J. Biol. Chem. 2015, 290 (13), 8439–8446. 10.1074/jbc.M114.634683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler D. G.; Faia K. L.; DiNitto J. P.; Ali J. A.; White K. F.; Brophy E. E.; Pink M. M.; Proctor J. L.; Lussier J.; Martin C. M.; Hoyt J. G.; Tillotson B.; Murphy E. L.; Lim A. R.; Thomas B. D.; Macdougall J. R.; Ren P.; Liu Y.; Li L.; Jessen K. A.; Fritz C. C.; Dunbar J. L.; Poter J. R.; Rommel C.; Palombella V. J.; Changelian P. S.; Kutok J. L. PI3K-δ and PI3K-γ inhibiton by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem. Biol. 2013, 20 (11), 1364–74. 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- McGinnity D. F.; Soars M. G.; Ubanowicz R. A.; Riley R. J. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab. Dispos. 2004, 32, 1247–1253. 10.1124/dmd.104.000026. [DOI] [PubMed] [Google Scholar]
- Heller R.; Chang Q.; Ehrlich G.; Hsieh S. N.; Schoenwaelder S. M.; Kuhlencordt P. J.; Preissner K. T.; Hirsch E.; Wetzker R. Overlapping and distinct roles for PI3Kβ and γ isoforms in S1P-induced migration of human and mouse endothelial cells. Cardiovasc. Res. 2008, 80, 96–105. 10.1093/cvr/cvn159. [DOI] [PubMed] [Google Scholar]
- Dal Porto J. M.; Gauld S. B.; Merrel K. T.; Mills D.; Pugh-Bernard A. E.; Cambier J. B cell antigen receptor signaling 101. Mol. Immunol. 2004, 41 (6–7), 599–613. 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Banker M. J.; Clark T. H.; Williams J. A. Development and validation of 96Well Equilibrium dialysis apparatus for measuring plasma protein binding. J. Pharm. Sci. 2003, 92 (5), 967–974. 10.1002/jps.10332. [DOI] [PubMed] [Google Scholar]
- Wohnsland F.; Faller B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 2001, 44 (6), 923–30. 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- Soond D. R.; Slack E. C. M.; Garden O. A.; Patton D. T.; Okkenhaug K. Does the PI3K pathway promote or antagonize regulatory T cell development and function?. Front. Immunol. 2012, 3, 244. 10.3389/fimmu.2012.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.