

Abstract
Chemical and enzymatic probing of RNA secondary structure and RNA/protein interactions provides the basis for understanding the functions of structured RNAs. However, the ability to rapidly perform such experiments using capillary electrophoresis has been hampered by relatively labor-intensive data analysis software. While these computationally robust programs have been shown to calculate residue-specific reactivities to a high degree of accuracy, they often require time-consuming manual intervention and lack the ability to be easily modified by users. To alleviate these issues, RiboCAT (Ribonucleic acid capillary-electrophoresis analysis tool) was developed as a user-friendly, Microsoft Excel–based tool that reduces the need for manual intervention, thereby significantly shortening the time required for data analysis. Features of this tool include (i) the use of an Excel platform, (ii) a method of intercapillary signal alignment using internal size standards, (iii) a peak-sharpening algorithm to more accurately identify peaks, and (iv) an open architecture allowing for simple user intervention. Furthermore, a complementary tool, RiboDOG (RiboCAT data output generator) was designed to facilitate the comparison of multiple data sets, highlighting potential inconsistencies and inaccuracies that may have occurred during analysis. Using these new tools, the secondary structure of the HIV-1 5′ untranslated region (5′UTR) was determined using selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE), matching the results of previous work.
Keywords: RNA structure, SHAPE, secondary structure, capillary electrophoresis
INTRODUCTION
Once thought to be noncatalytic messengers and scaffolds for use in translation, RNA has been shown to play active roles in such diverse processes as mRNA splicing (Kruger et al. 1982; McNeil et al. 2016), regulation of transcription (Grundy et al. 1994; Henkin 1994; Serganov and Nudler 2013; Furtig et al. 2015) and translation (Lee et al. 1993; Wightman et al. 1993; Filipowicz et al. 2008), viral assembly (Zeffman et al. 2000; Cantara et al. 2014; Sardo et al. 2015; Stockley et al. 2016), and immunity (Brouns et al. 2008; Zhang et al. 2010; Dhahbi 2015; Cavalieri et al. 2016). Many of these functions result from the ability of RNA to fold into complex secondary and tertiary structures. The folding of RNA is hierarchical in that secondary structure features form first, followed by specific tertiary folds (Brion and Westhof 1997; Schroeder et al. 2004; Woodson 2010). Proximal complementary residues tend to form Watson-Crick (WC) base pairs resulting in stable A-form helices. These helices can then orient into complex tertiary structures stabilized either by WC interactions between complementary single-stranded bases or noncanonical interactions. Additionally, non-A-form motifs can also be formed in loops and bulges that are stabilized by base-stacking or noncanonical internucleotide pairings. Therefore, important clues with regard to the structure and function of a particular RNA construct are revealed by determining the secondary structure. Moreover, secondary structure information is a key first step to solving three-dimensional structure by enabling design of stable constructs for analysis using NMR, crystallography, or other techniques (Cantara et al. 2014).
While computational analysis of phylogenetic data is an important method of identifying secondary structural features of many RNAs, this technique is less useful in regions where sequence conservation is high or if few primary sequences are available. Numerous methods of chemical and enzymatic probing have been developed for high-throughput data collection, which have allowed empirical determination of RNA secondary structure at a much faster rate and to a higher level of confidence (Mitra et al. 2008; Weeks 2010; Kenyon et al. 2014; Rice et al. 2014; Ge and Zhang 2015). As an example, the chemical probing method of selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) is useful for probing the secondary structure and flexibility of complex RNAs and has been used in a high-throughput format (Merino et al. 2005; Wilkinson et al. 2006, 2008; Watts et al. 2009; McGinnis et al. 2012; Rice et al. 2014). Briefly, electrophilic reagents such as 1-methyl-6-nitroisatoic anhydride (1M6), 1-methyl-7-nitroisatoic anhydride (1M7), and N-methylisatoic anhydride (NMIA) preferentially form covalent adducts with the 2′-hydroxyl of flexible residues that are therefore more likely to be unpaired (Merino et al. 2005; McGinnis et al. 2012). When subjected to a primer-extension reaction from a fluorescently labeled primer, reverse transcriptase will halt at modified residues resulting in labeled DNA fragments with lengths corresponding to the site of adduct formation. These fluorescently labeled fragments are then separated by capillary electrophoresis (CE) using a standard DNA sequencing instrument.
Despite the high-throughput capability of RNA probing methods using CE, a bottleneck occurs when it comes to data analysis. Many tools are available to aid in RNA probing CE data analysis such as ShapeFinder (Vasa et al. 2008), CAFA (Mitra et al. 2008), HiTRACE (Yoon et al. 2011; Kim et al. 2013), and FAST (Pang et al. 2011), but the most recent and widely used analysis software is QuSHAPE (Karabiber et al. 2013). Despite the many successes of these tools, there remains room for advancement through functional improvements to key algorithms such as signal alignment, peak identification, sequence alignment, and simplicity of error correction. The particular shortcomings of these programs can impose appreciable time requirements on the user, reducing the throughput of CE-based RNA probing experiments.
RiboCAT (Ribonucleic acid capillary-electrophoresis analysis tool), a newly developed tool described herein, was designed for simplicity of use, reduced alignment and peak identification errors due to new automated features, ease of error correction, and thus, reduced overall data analysis time. The Microsoft Excel platform was chosen for its familiarity to most users. The underlying functions of Excel are also not altered, allowing the user to modify the data as necessary and troubleshoot each step in the process. As a proof-of-principle, this new tool was used to analyze SHAPE data collected on the HIV-1 5′-untranslated region (5′UTR). The analyzed data show good agreement with published reactivity data and reproduce the previously determined secondary structure (Wilkinson et al. 2008; Kenyon et al. 2013), but required significantly less user intervention and data analysis time than previous tools.
RESULTS
Although existing algorithms for analyzing CE RNA probing data are time-efficient, a major shortcoming of previous programs is the time commitment required for manual adjustment of incorrectly picked peaks, which scales with the size of the RNA. In RiboCAT, the time required for analysis of RNA probing data was significantly reduced by implementing a revised signal alignment protocol and peak identification functions, resulting in less input from the user and faster correction mechanisms than previous analysis tools (Fig. 1A). Specifically, signal alignment in QuSHAPE is based on the use of a similarity matrix, which often misaligns the electropherograms from different capillaries, requiring the user to perform manual adjustments (Fig. 1B, top). The peak identification in QuSHAPE also generally requires user intervention, which increases time for data analysis (Fig. 1C, top). The new functions effectively eliminate errors associated with sequence alignment (Fig. 1B, bottom) and minimize the number of user-adjustments required after peak identification (Fig. 1C, bottom).
FIGURE 1.
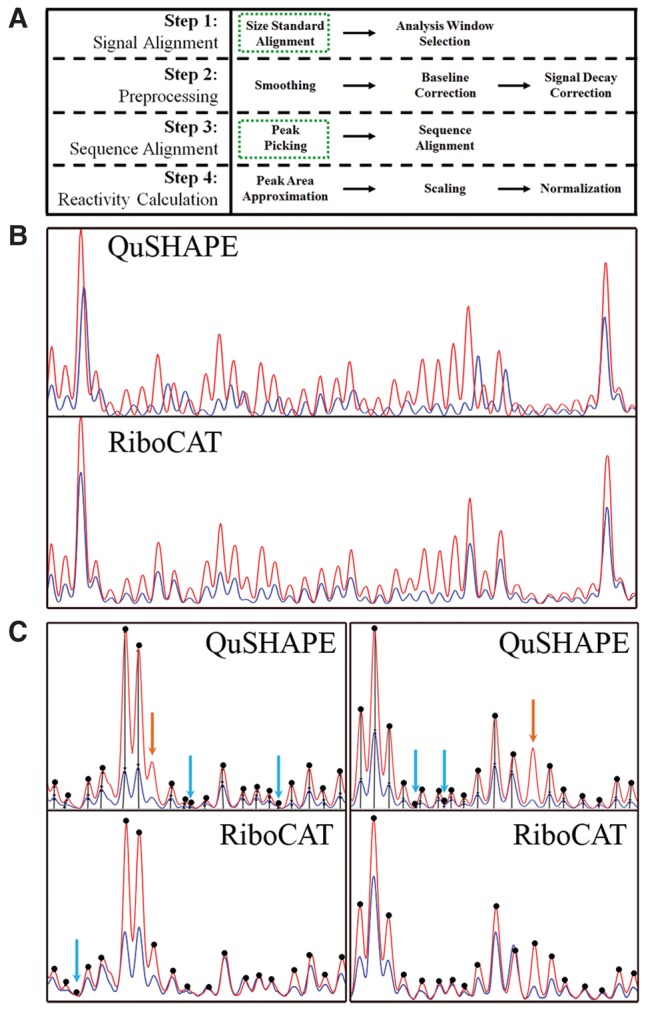
Improvements to CE processing steps reduce data analysis errors. (A) Four primary steps are carried out during analysis of CE data; signal alignment, data preprocessing, sequence alignment, and reactivity calculations. The two main areas of improvement, signal alignment and peak picking, are highlighted in green dashed boxes. (B) Errors associated with signal alignment in QuSHAPE are eliminated using an improved alignment strategy that utilizes internal size standards. Blue and red lines indicate the minus and plus traces, respectively, (same in C). (C) The frequency of misidentified peaks in QuSHAPE is significantly reduced in RiboCAT via introduction of a peak sharpening algorithm. Dots indicate picked peaks. Orange and blue arrows denote missed or incorrectly picked peaks, respectively.
Single-fluorophore SHAPE method
SHAPE experiments were carried out using a single-fluorophore/three-capillary method with an internal LIZ600 size standard (Mitra et al. 2008; Pang et al. 2011). Although other setups are possible (e.g., multiple fluorophores using a single capillary for the minus, plus, and sequencing reactions), advantages of the single-fluorophore method include (i) reduced costs related to only requiring one fluorophore-labeled primer and only a single sequencing data set per RNA region, (ii) less spectral overlap between different fluorophores, (iii) no requirement for fluorophore-specific x-axis mobility shift corrections, and (iv) y-axis scaling is not influenced by spectral differences in the fluorophores. Additionally, this method significantly simplifies analysis and allows for less ambiguity in such error-prone steps as intercapillary signal alignment and sequence-peak matching during sequence alignment.
Size-standard signal alignment
The first step to analyzing CE data is to align the signals from the sequencing reactions with the minus (no probing reagent control) and plus (probing experiment) CE runs. To accomplish this, size standards are run in each capillary along with the experimental samples (see Materials and Methods for detailed protocol). These size standards are used to relate the migration time axis (Xo) to a nucleotide (nt)-based axis (Xnt). Size-standard peaks are picked using a moving-window linear approximation. In this calculation, two user-input peaks calculate an initial slope relating Xo to Xnt and identify the peaks corresponding to the two highest molecular weight size standards. The remaining peaks are picked using the approximately linear relationship between nucleotide length and migration time of any three size standard peaks according to Equation 1:
| (1) |
where the x-axis location of size standard peak i (Xo,i) is approximated using the slope between the next two larger fragment peaks multiplied by the difference between the nucleotide lengths of peak i (Xnt,i) and peak i + 1 (Xnt,i+1). This product is then added to the x-axis location of peak i + 1 (Xo,i+1). The amplitudes of size-standard peaks vary significantly making it difficult to set the minimum and maximum constraints defining what constitutes a peak. However, peaks in a similar region are generally close to the same amplitude. To account for this, a moving threshold is used where the minimum (Equation 2) and maximum (Equation 3) constraints are set based on the average amplitude of the next two larger molecular weight peaks (〈Ai+1, Ai+2〉):
| (2) |
| (3) |
Exploiting amplitude and migration time patterns allows for tight constraints to be applied to size-standard peak picking, thus minimizing error. Peaks are identified as local maxima found within each calculated window. This size-standard peak-picking algorithm picked peaks within 0.046 ± 1.07 data points of those picked by Peak Scanner (Applied Biosystems) for the six different experimental data sets described herein.
The resulting size standard peaks were used to align the traces from the three different capillaries by calculating a polynomial that approximates the relationship between the capillary-specific Xo and the capillary-independent Xnt. Increasing the order of the polynomial fit equation resulted in reduced average alignment error as measured by the root-mean-square deviation (RMSD) in the aligned size-standard peaks in six independent CE experiments. A plateau was reached at a polynomial order value of 9 (Fig. 2A), which was used for all subsequent analyses resulting in well-aligned size-standard (Fig. 2B) and experimental spectra (Fig. 2C) between capillaries. Thus, the capillary-independent x-axis value (Equation 4) is calculated by fitting a ninth order polynomial with coefficients B and Mj:
| (4) |
FIGURE 2.

Signal alignment using an internal size standard. (A) Optimization of the polynomial order used for signal alignment. The inset shows a zoomed region encompassing orders 5–10 with RMSD values reaching a plateau at an average of X0 = 1.2 at order 9. (B) The unaligned (top) and aligned (bottom) size standards for three separate CE experiments of sequencing (black), no reagent control (blue), and SHAPE reaction (red). (C) Aligned SHAPE reaction traces for no reagent control (blue) and SHAPE reagent (red) show highly accurate signal alignment based on the internal size standard.
Reaction peak picking
After aligned electropherograms were preprocessed as described in the Materials and Methods (Fig. 1A), peaks were identified in the three reaction traces (sequencing, minus, and plus). Peaks in the reaction capillaries correspond to different lengths of primer extension products, each separated by a single nucleotide. Ideally, each peak can be identified by the x-axis position of local amplitude maxima, separated by a single nucleotide. However, identification of local maxima can be complicated by poorly separated peaks or shoulders in the data. To improve the fidelity of peak picking, a peak-sharpening (i.e., signal-enhancement) algorithm was applied (Fig. 3). The sharpened amplitude (YSharp) is calculated as the difference between the amplitude of preprocessed data (YPreproc) and the second derivative of the preprocessed data (Y″Preproc), as described in the following equation:
| (5) |
FIGURE 3.
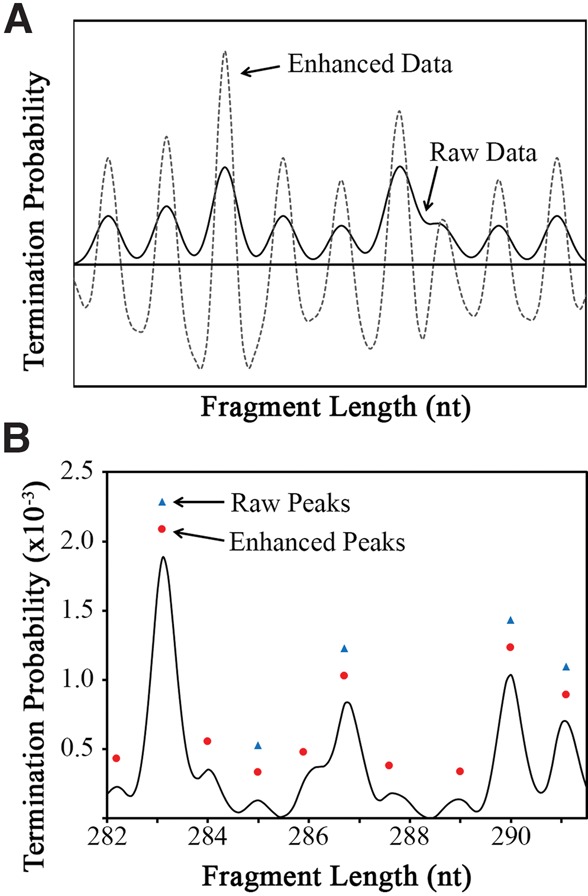
Improved peak picking using a peak-sharpening protocol. (A) The peak-sharpening algorithm computes an enhanced data trace (dashed gray line) that has significantly exaggerated the peaks and troughs compared to the raw data trace (solid black line). (B) The enhanced data allow for more robust peak assignment. In the example shown, five out of 10 peaks can be assigned using the raw data (blue triangles), whereas all 10 peaks are properly assigned when using the peak-sharpened, enhanced data (red circles).
Peaks are then identified as the local maxima within a moving window of ∼1.1 nt on the x-axis of the peak-sharpened data. Importantly, the peak-sharpened data are used only to identify the x-axis position of each peak. All further analysis is based on the unsharpened, preprocessed data.
Despite the improved robustness of the peak-picking algorithm, a small number of errors are common for each data set. RiboCAT has been designed to allow the user to easily add or remove peaks based on manual inspection of the data without altering other, already identified peaks. Furthermore, although peak separation will vary throughout the length of the capillary, analysis of many data sets revealed that peaks are rarely separated by less than 0.6 or more than 1.5 x-axis units. Therefore, a checking system was implemented to alert the user of locations where the identified peaks are not within 0.6–1.5 units of separation. Additionally, peaks are occasionally identified in the minus trace that are not identified in the plus. A check was also incorporated to notify the user of peaks in one trace that have no match in the other. A convenient peak-editing user form has been implemented that displays the peaks identified by this check, automatically takes the user to the regions of the electropherograms that are in question, and contains fields for the user to add and remove peaks. Users then have the choice of whether to make a manual correction.
Sequence alignment
Aligning the RNA sequence to the calculated reactivity of each peak is not always an unambiguous process due to subtle differences between the migration times in the reaction and sequencing capillaries, especially for shorter fragments (<50 nt). However, migration-time differences between the reaction and sequencing peaks approach zero as more peaks corresponding to longer fragments (>50 nt) are included. This is algorithmically accounted for by incrementally shifting the nucleotide numbers assigned to the reaction peaks, and selecting the alignment that minimizes the RMSD between the sequencing values (Xnt,seq) and their corresponding reaction values (Xnt,rxn) (Fig. 4A). The incorporation of every sequencing peak into the alignment algorithm results in the most consistent alignment of sequence to reactivity values.
FIGURE 4.
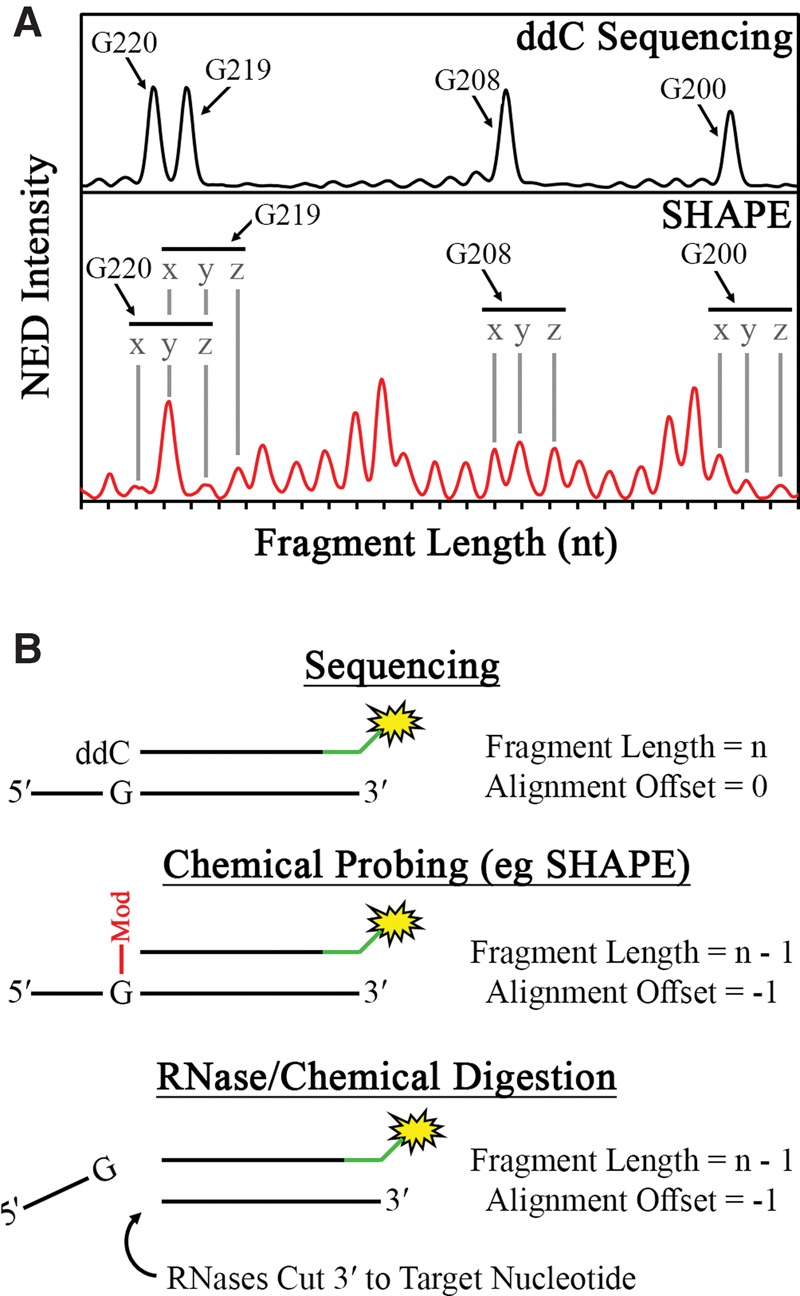
Schematic description of sequence alignment in RiboCAT. (A) To optimize the assignment of peaks to their corresponding nucleotides in the RNA sequence, an initial guess of the alignment is made (peaks labeled “y”) based on the x-axis similarities in peaks from the sequencing (top) and experimental (bottom) traces. The alignment is then shifted incrementally to the left (peaks labeled “x”) or incrementally to the right (peaks labeled “z”), and the RMSD between the x-axis values are calculated. The minimum RMSD over the entire trace is chosen as the correct alignment. (B) In standard Sanger sequencing, the DNA fragment corresponding to a particular residue will include the cognate dideoxy nucleotide (top); however, in both chemical probing methods (middle) and RNase/chemical digestion (bottom), the cognate nucleotide is prohibited from being incorporated by either a chemical modification or backbone cleavage, respectively, leading to an offset of −1.
Finally, reverse transcriptase termination in probing and sequencing reactions does not necessarily result in fragments of the same length for the same nucleotide. This stems from the fact that sequencing termination always occurs with the addition of a 2′,3′-dideoxynucleotide, while many RNA probing methods, such as SHAPE, result in termination at the nucleotide prior to the reactive nucleotide (Fig. 4B). This is corrected by offsetting the sequence alignment by this difference in length, and this offset may be different for various RNA probing methods (Fig. 4B). Users can input an offset factor specific to the experimental method being used.
RiboDOG: combined analysis of multiple data sets
After analysis of an RNA probing experiment, it is important to ensure that the migration time (Xnt) of each nucleotide is consistent throughout each replicate prior to comparing the reactivity. As a result of the size-standard signal alignment, the Xnt of each nucleotide should be highly repeatable. Therefore, comparing Xnt values between replicates allows for the identification and correction of erroneously picked or missing peaks. The primary function of RiboDOG (RiboCAT data output generator) is to align the data from multiple replicates to facilitate this type of analysis. However, this program also contains a form that allows the user to view and compare traces from these data sets, and add and remove peaks in regions of disagreement between replicates. Additionally, the program will recalculate the Gaussian fitting, scaling, normalization, and sequence alignment algorithms for any alterations made to the peak list. Finally, RiboDOG will generate “.shape” files based on the summarized data from multiple primers and replicates that are compatible with secondary structure prediction programs such as RNAstructure.
Prediction of the HIV-1 5′UTR secondary structure
As a validation of CE data analysis using RiboCAT, the HIV-1 5′UTR secondary structure was determined using SHAPE and results were compared to published data (Wilkinson et al. 2008). A 352-nt construct including the untranslated region in addition to 21-nt of the Gag coding region was probed using both NMIA and 1M6, followed by primer extension and analysis by CE. This RNA construct contains an A34U mutation in the TAR loop and a GAGA tetraloop replacing the dimerization initiation site (ΔDIS) to inhibit dimerization (Skripkin et al. 1994; Helga-Maria et al. 1999; Andersen et al. 2004). The ΔDIS mutation also negates a potential alternative conformation involving a tertiary interaction between the DIS and U5 sequences (Lu et al. 2011), resulting in a conformationally homogenous sample (data not shown). The data showed a high degree of inter–data set reproducibility with pairwise Pearson's R-values of 0.79 and 0.91 for NMIA and 1M6 data sets, respectively.
Reactivity data from three independent experiments using NMIA were averaged and entered into RNA Structure (Reuter and Mathews 2010; Mathews 2014) as pseudoenergy restraints for secondary structure calculation. The lowest energy structure was an exact match to the one previously calculated (Fig. 5A; Wilkinson et al. 2008). Additionally, plotting the average reactivity values from three independent experiments using 1M6 revealed a very good match to the NMIA-derived structure (Fig. 5B). The next two predicted lowest-energy structures for both NMIA and 1M6 only showed structural variations in the poly(A) hairpin and at the base of the TAR stem (Supplemental Fig. S1). Moreover, 74% of the residues deemed to be medium-high reactivity by NMIA were found to be in the same range for the 1M6 experiments; differences in the reactivities for these two reagents may be indicative of different rates of nucleotide flexibility (Rice et al. 2014).
FIGURE 5.

RiboCAT replicates QuSHAPE-derived reactivities in the HIV-1 5′UTR. The lowest energy secondary structures calculated by RNAStructure using reactivity values derived using NMIA (A) and 1M6 (B). Sites of mutation, A34U and ΔDIS, are noted with the WT DIS shown. The reactivities at each nucleotide are depicted as colored circles matching the legend in the middle. (C) SHAPE reactivities calculated using QuSHAPE (red) and RiboCAT (black outline) plotted for each nucleotide show a high degree of similarity with regions of high and low reactivity matching in both plots. (D) The SHAPE reactivities calculated using RiboCAT and QuSHAPE are very comparable with a Pearson's R-value of 0.96.
Comparison of RiboCAT and QuSHAPE
To further demonstrate the utility of this method, data were collected and analyzed in parallel with both QuSHAPE and RiboCAT. In order to accommodate QuSHAPE analysis, an additional sequencing reaction was performed using a primer labeled with the standard 5′-VIC fluorophore and analyzed in the same capillaries as the NMIA reactions as previously described (Wilkinson et al. 2006, 2008; Watts et al. 2009; Karabiber et al. 2013). Comparison of the SHAPE reactivity values for each nucleotide reveals a high correlation between RiboCAT and QuSHAPE (Fig. 5C). Averaged results from both programs showed very close agreement with a Pearson's R-value of 0.96, a slope of 1.06, and intercept of 0.06 (Fig. 5D). Moreover, 84% of the nucleotides with reactivities >0.5 identified by RiboCAT were also identified by QuSHAPE. Processing of the data using RiboCAT was very efficient, taking approximately one-fourth to one-fifth the amount of analysis time in comparison to QuSHAPE, due mostly to the requirement for significant manual adjustments during the sequence alignment stage in the latter. To demonstrate the improvement to this step, the data analyzed in QuSHAPE required the manual addition of four and deletion of 67 peaks, compared to only an addition of three and deletion of two peaks in RiboCAT. Misalignment of the electropherograms also needed to be manually corrected in QuSHAPE, but this was not as time consuming as correcting the peak misidentification.
DISCUSSION
Chemical and enzymatic RNA probing methods are useful tools for understanding RNA structure. Although many of these biochemical techniques are high-throughput in nature, data analysis remains a time-consuming and sometimes ambiguous process. Microsoft Excel was selected for RiboCAT as it allows the data to be completely visible and adjustable at each processing step while also providing a familiar platform for most scientific users. Additionally, the numerical format is very conducive to accurate adjustment of high-error steps such as peak picking. Improvements to signal alignment, peak picking, and sequence alignment processes reduce the need for these types of manual corrections, further speeding up analysis.
Using both NMIA and 1M6, the previous SHAPE-derived secondary structure of the HIV-1 5′UTR (Wilkinson et al. 2008; Kenyon et al. 2013) was replicated using this analysis method. The only variation is the lack of two A–U base pairs toward the top of the poly(A) stem in the prediction based on the 1M6 data. However, as 1M6 is known to react with a more rapidly flexible nucleotide than NMIA (Rice et al. 2014), the detection of this reactivity with 1M6 and not NMIA could result from these two base pairs undergoing a rapid transition between a paired and unpaired state. Direct comparison between the SHAPE reactivities calculated in RiboCAT with those from QuSHAPE reveal a strong correlation (R = 0.96).
An interesting conclusion from the initial SHAPE study of the HIV-1 5′UTR was the prediction of a long-range pseudoknot interaction between the 3′ side of the poly(A) loop (nt 79–85) and a loop downstream from the Gag translation start site (nt 443–449) (Wilkinson et al. 2008). The major splice donor (SD) site is located in stem–loop 2 (SL2), causing this interaction to be absent in spliced RNAs. Even though the RNA used in the present study does not contain nt 443–449 of the pseudoknot, the 3′ side of poly(A) lacks NMIA reactivity entirely, and only two loop residues, G84 and C86, had 1M6 reactivities of ∼0.3, suggesting a different interaction in this context (Fig. 5A,B). This reactivity pattern in the poly(A) loop, along with the presence of multiple medium- and highly reactive residues in the stem imply instability in this hairpin, which is reflected in the second and third lowest-energy SHAPE-derived secondary structures (Supplemental Fig. S1).
In conclusion, RiboCAT has been demonstrated to reproduce final reactivity data calculated with the commonly used QuSHAPE program with high efficiency, reducing the time requirements of the user and allowing a friendlier interface that allows manual correction of calculated values for users who lack in-depth knowledge of computer programming. In addition to improvements in signal alignment, peak picking and sequence alignment, RiboCAT uses the common Microsoft Excel platform and does not require installation of additional software. Furthermore, the accessory program, RiboDOG, has been designed to facilitate the combined analysis of multiple SHAPE experiments and assess the consistency between trials. Analysis of primer extension products from the many different chemical and enzymatic probing strategies is fundamentally the same in that the reactivity at a specific residue is a function of the number of DNA fragments of the corresponding size. Therefore, RiboCAT should be broadly applicable to analysis of data from many different RNA probing methods, provided CE is used for detection of primer extension products.
MATERIALS AND METHODS
RNA preparation
The UTR:ΔDIS:A34U construct used in this work is derived from the first 356 nt of the HIV-1 NL4-3 isolate cloned into a pUC19 parent plasmid. Both the ΔDIS (replacement of the SL1 palindromic loop with a GAGA tetraloop) and A34U mutations prevent genomic dimerization and facilitate homogeneous RNA preparation (Skripkin et al. 1994; Helga-Maria et al. 1999; Andersen et al. 2004). The final construct size, with mutations is 352 nt. The transcription template was generated by digestion of pUC19-UTR:ΔDIS:A34U plasmid with FokI (New England Biolabs). RNAs were prepared via in vitro transcription with T7 RNA polymerase (Milligan et al. 1987) and purified using 8 M urea (denaturing) polyacrylamide gel electrophoresis (PAGE). Desired bands were excised, crushed, and soaked in RNA elution buffer (0.5 mM NH4OAc, 1 mM EDTA) overnight at 37°C. Eluted RNA was butanol extracted, ethanol precipitated, and resuspended in diethylpyrocarbonate (DEPC)-treated water. Purified RNA was folded in 50 mM HEPES (pH 7.4) buffer by heating at 80°C for 2 min, cooling to 60°C for 2 min, adding 1 M MgCl2 to a final concentration of 1 mM, incubating at 37°C for 30 min, and cooling on ice for 30 min. Different durations of the 37°C incubation step were tested for optimal sample homogeneity (data not shown).
SHAPE probing experiments
For SHAPE experiments, two different reagents were used: NMIA (Sigma-Aldrich) and 1M6 (Sigma-Aldrich). NMIA has a relatively slow reaction rate making it useful for analyzing regions of RNA with slow dynamics (Rice et al. 2014). In contrast, 1M6 reacts and deactivates in water more rapidly, allowing the study of nucleotides with faster dynamics (Rice et al. 2014). Prior to experimental data collection, probing reaction times at 37°C were optimized to ensure single-hit kinetics. Reaction times of 3, 5, and 10 min for 1M6 and 22, 30, and 45 min for NMIA were tested. The optimum time was selected based on the lowest 3′ end reactivity bias (3 min for 1M6 and 45 min for NMIA) (data not shown).
SHAPE experiments were performed as previously described (Wilkinson et al. 2006, 2008; Watts et al. 2009; Karabiber et al. 2013) with minor variations. All primer extension products were generated using a primer containing a 5′-NED fluorophore (ThermoFisher Scientific). A (−) reaction that lacked the SHAPE reagent served as a negative control to account for spontaneous primer-extension termination and other background effects. Each reaction contained ∼8 pmol of RNA, and reactions were initiated with either 1 µL of 80 mM SHAPE reagent (in DMSO) or 1 µL of DMSO for the (+)-reaction and (−)-reaction, respectively (final reaction volume of 10 µL). Reactions were incubated at 37°C for the optimized time duration and quenched by ethanol precipitation. RNA pellets were resuspended in DEPC-treated water, and annealed NED-labeled primers were extended using Superscript III reverse transcriptase following the manufacturer's protocol (Invitrogen).
Sanger-style sequencing reactions were performed on the transcription template plasmid (pUC19-UTR:ΔDIS:A34U) using the Thermo Sequenase Cycle Sequencing Kit (Affymetrix) and the same NED-labeled primer used above for primer extension. The amount of SHAPE and sequencing reactions analyzed via CE was optimized by precipitating different volumes of each type of sample, and selecting the amount that exhibited the least detector saturation while maintaining a high signal-to-noise ratio. In each individual capillary, GeneScan 600 LIZ Size Standard (Applied Biosystems) was included for intercapillary alignment. Samples were analyzed using an Applied Biosystems 3730 DNA Analyzer (Plant-Microbe Genomics Facility, The Ohio State University).
Preprocessing
Prior to peak-picking, the raw electropherograms were preprocessed with smoothing, baseline correction, and signal decay correction algorithms essentially as described previously (Pang et al. 2011; Karabiber et al. 2013). Briefly, in order to eliminate high frequency noise in the data, which may complicate peak identification, a moving triangular average smoothing was performed. All data described herein were smoothed using a window size of one, indicating that the smoothed value of a particular data point was calculated by accounting for the intensities of one data point before (Ai−1) and one data point after (Ai+1), as shown in Equation 6. Trials of different window sizes were tested with results indicating that window sizes of >2 cause significant loss of information and should not be used in most cases. The calculation for a window size of two is shown in Equation 7 where SAi refers to the smoothed intensity at point i.
| (6) |
| (7) |
Following data smoothing, low frequency errors and baseline offset originating from load error were eliminated by performing a baseline correction. Here, the minimum value within a user-defined window is subtracted from each data point, as described in the following equation:
| (8) |
where BAi is the baseline corrected intensity for point i. A standard window size of 25, based on a previously published default (Karabiber et al. 2013), was used for all data analysis.
The final step of preprocessing corrects for the imperfect processivity of primer extension reactions, which leads to decay in signal across a CE electropherogram. For this step, a previously described heuristic approach was implemented based on the premise that the average probability of primer extension termination over the first half of the RNA molecule should be equal to that of the second half (Pang et al. 2011; Karabiber et al. 2013). Equations 9 and 10 describe the calculations for determining the probability of termination at each point in the data:
| (9) |
| (10) |
Here, the probability of termination (Pterm) at a given nucleotide is equal to the intensity of that nucleotide divided by the sum of intensities for every nucleotide in the RNA. This calculation must include the probabilities for the data within the user-specified range up to nt k (), as well as the unknown probabilities for the data beyond nt k (Punk). The algorithm determines the value for Punk that minimizes the difference between the sum of probabilities in the first () and second halves () of the electropherogram.
Reactivity calculation
The final step in CE RNA probing data analysis is reactivity calculation, which includes three processes: peak area approximation, scaling, and normalization. Peak areas are approximated by algorithmically fitting Gaussian functions resulting in an integrable curve representing each peak:
| (11) |
The parameters A, P, and σ of the Gaussian function are unique to each peak of a CE trace and represent the amplitude, x-axis position, and width of the peak at half-height, respectively. Values for these parameters are calculated using a moving window that includes the peak of interest, as well as one peak to either side. The A, P, and σ parameters are varied for the peak of interest, and the y-values of the three peaks are summed at every data point within the window and compared to the corresponding preprocessed data. The optimum value for a parameter is selected as the value that gives the lowest error between Gaussian approximations and the preprocessed data within the window. Peak areas are then calculated by integrating the Gaussian functions.
The amount of primer extension product loaded into plus and minus capillaries is not exactly equal. To correct for this, the trace from one capillary must be scaled to the other. Scaling is done under the assumption that low-area peaks in the plus reaction represent nucleotides with approximately zero reactivity. Therefore, the areas of these peaks should be made equal to the corresponding peaks in the minus reaction. This is done using a scaling factor (α) that is calculated by dividing the average of the lowest 20% (A(20%,+)) of plus peaks by the average of the lowest 20% of minus peaks (A(20%,−)):
| (12) |
All minus-peak areas can then be scaled to the plus by multiplying by α. Raw areas cannot be compared between replicates or RNAs and must be normalized. The normalized reactivity values are determined by first subtracting the minus-peak areas from the plus, and then dividing the resulting background-subtracted values [Abs (nt)] by the average of the top 10% of these values (<Top 10% Abs>):
| (13) |
Outliers are excluded from the top 10% calculation if they are greater than 1.5 times the interquartile range. This normalizes the data by setting the value of the average high-reactivity nucleotide to one. All reactivity calculation processes are performed automatically by the RiboCAT tool and are performed using the same criteria as QuSHAPE (Karabiber et al. 2013).
Data output and secondary structure determination
Following analysis by RiboCAT, it is important to have a standard method of comparing the results of multiple data sets to allow for inconsistencies to be corrected or for reproducibility to be measured. A support tool, RiboDOG (RiboCAT data output generator) was designed to automate this task, as well as to allow for export of reactivity files that are properly formatted for use in RNAStructure software for secondary structure determination. Using this tool, all data from the HIV-1 5′UTR experiments were compared for consistency between each trial in terms of reactivities and Xnt values of identified peaks. Additionally, the final reactivity values were exported for secondary structure determination using the freely available RNAStructure software (Reuter and Mathews 2010; Mathews 2014). Both NMIA- and 1M6-derived reactivity values were used separately for secondary structure determination. Secondary structure depictions were prepared using XRNA (http://rna.ucsc.edu/rnacenter/xrna/xrna_faq.html).
RiboCAT and RiboDOG along with user guides are freely available at https://research.cbc.osu.edu/musier-forsyth.1/tools/ under the Lesser GNU General Public License, version 3. Test data along with a video tutorial, user manual, and step-by-step guide are also available.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Janie Frandsen and Roopa Comandur for testing and bug reporting, Alex Getz for logo design, and the Plant Microbe Genomics Facility at The Ohio State University for collecting capillary electrophoresis data. We acknowledge the support of National Institute of General Medical Sciences (National Institutes of Health) grant P50 GM103368 (HIVE Center) for experimental aspects of this work. This work was also supported by National Institute of General Medical Sciences (National Institutes of Health) grants R01 GM065056 and R01 GM113887.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.058404.116.
REFERENCES
- Andersen ES, Contera SA, Knudsen B, Damgaard CK, Besenbacher F, Kjems J. 2004. Role of the trans-activation response element in dimerization of HIV-1 RNA. J Biol Chem 279: 22243–22249. [DOI] [PubMed] [Google Scholar]
- Brion P, Westhof E. 1997. Hierarchy and dynamics of RNA folding. Annu Rev Biophys Biomol Struct 26: 113–137. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, Olson ED, Musier-Forsyth K. 2014. Progress and outlook in structural biology of large viral RNAs. Virus Res 193: 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri D, Rizzetto L, Tocci N, Rivero D, Asquini E, Si-Ammour A, Bonechi E, Ballerini C, Viola R. 2016. Plant microRNAs as novel immunomodulatory agents. Sci Rep 6: 25761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM. 2015. 5′ tRNA halves: the next generation of immune signaling molecules. Front Immunol 6: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. 2008. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114. [DOI] [PubMed] [Google Scholar]
- Furtig B, Nozinovic S, Reining A, Schwalbe H. 2015. Multiple conformational states of riboswitches fine-tune gene regulation. Curr Opin Struct Biol 30: 112–124. [DOI] [PubMed] [Google Scholar]
- Ge P, Zhang S. 2015. Computational analysis of RNA structures with chemical probing data. Methods 79–80: 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy FJ, Rollins SM, Henkin TM. 1994. Interaction between the acceptor end of tRNA and the T box stimulates antitermination in the Bacillus subtilis tyrS gene: a new role for the discriminator base. J Bacteriol 176: 4518–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helga-Maria C, Hammarskjold ML, Rekosh D. 1999. An intact TAR element and cytoplasmic localization are necessary for efficient packaging of human immunodeficiency virus type 1 genomic RNA. J Virol 73: 4127–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkin TM. 1994. tRNA-directed transcription antitermination. Mol Microbiol 13: 381–387. [DOI] [PubMed] [Google Scholar]
- Karabiber F, McGinnis JL, Favorov OV, Weeks KM. 2013. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA 19: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon JC, Prestwood LJ, Le Grice SF, Lever AM. 2013. In-gel probing of individual RNA conformers within a mixed population reveals a dimerization structural switch in the HIV-1 leader. Nucleic Acids Res 41: e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon J, Prestwood L, Lever A. 2014. Current perspectives on RNA secondary structure probing. Biochem Soc Trans 42: 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Cordero P, Das R, Yoon S. 2013. HiTRACE-Web: an online tool for robust analysis of high-throughput capillary electrophoresis. Nucleic Acids Res 41: W492–W498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. 1982. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell 31: 147–157. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, Dorjsuren B, Kulandaivel G, Jones S, Hiremath A, et al. 2011. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH. 2014. RNA secondary structure analysis using RNA structure. Curr Protoc Bioinformatics 46: 12.16.11–12.16.25. [DOI] [PubMed] [Google Scholar]
- McGinnis JL, Dunkle JA, Cate JH, Weeks KM. 2012. The mechanisms of RNA SHAPE chemistry. J Am Chem Soc 134: 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil BA, Semper C, Zimmerly S. 2016. Group II introns: versatile ribozymes and retroelements. Wiley Interdiscip Rev RNA 7: 341–355. [DOI] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. 2005. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127: 4223–4231. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A. 2008. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res 36: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PS, Elazar M, Pham EA, Glenn JS. 2011. Simplified RNA secondary structure mapping by automation of SHAPE data analysis. Nucleic Acids Res 39: e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. 2010. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GM, Leonard CW, Weeks KM. 2014. RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA 20: 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardo L, Hatch SC, Chen J, Nikolaitchik O, Burdick RC, Chen D, Westlake CJ, Lockett S, Pathak VK, Hu WS. 2015. Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J Virol 89: 10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R, Barta A, Semrad K. 2004. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol 5: 908–919. [DOI] [PubMed] [Google Scholar]
- Serganov A, Nudler E. 2013. A decade of riboswitches. Cell 152: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. 1994. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci 91: 4945–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley PG, White SJ, Dykeman E, Manfield I, Rolfsson O, Patel N, Bingham R, Barker A, Wroblewski E, Chandler-Bostock R, et al. 2016. Bacteriophage MS2 genomic RNA encodes an assembly instruction manual for its capsid. Bacteriophage 6: e1157666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. 2008. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 14: 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM. 2010. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol 20: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. 1993. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75: 855–862. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1: 1610–1616. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson SA. 2010. Compact intermediates in RNA folding. Annu Rev Biophys 39: 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kim J, Hum J, Kim H, Park S, Kladwang W, Das R. 2011. HiTRACE: high-throughput robust analysis for capillary electrophoresis. Bioinformatics 27: 1798–1805. [DOI] [PubMed] [Google Scholar]
- Zeffman A, Hassard S, Varani G, Lever A. 2000. The major HIV-1 packaging signal is an extended bulged stem loop whose structure is altered on interaction with the Gag polyprotein. J Mol Biol 297: 877–893. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, Sun F, Lu J, Yin Y, Cai X, et al. 2010. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell 39: 133–144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.