

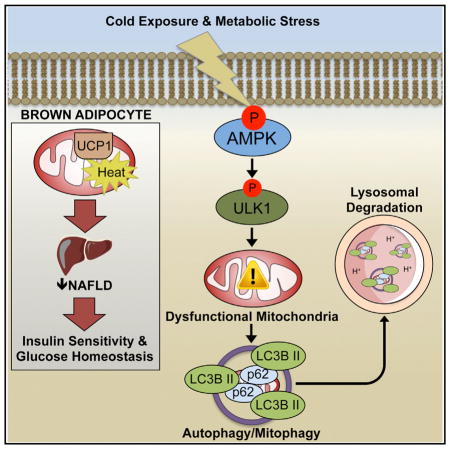
SUMMARY
Brown (BAT) and white (WAT) adipose tissues play distinct roles in maintaining whole-body energy homeostasis, and their dysfunction can contribute to non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. The AMP-activated protein kinase (AMPK) is a cellular energy sensor, but its role in regulating BAT and WAT metabolism is unclear. We generated an inducible model for deletion of the two AMPK β subunits in adipocytes (iβ1β2AKO) and found that iβ1β2AKO mice were cold intolerant and resistant to β-adrenergic activation of BAT and beiging of WAT. BAT from iβ1β2AKO mice had impairments in mitochondrial structure, function, and markers of mitophagy. In response to a high-fat diet, iβ1β2AKO mice more rapidly developed liver steatosis as well as glucose and insulin intolerance. Thus, AMPK in adipocytes is vital for maintaining mitochondrial integrity, responding to pharmacological agents and thermal stress, and protecting against nutrient-overload-induced NAFLD and insulin resistance.
In Brief
Mottillo et al. find mice lacking AMPK specifically in adipocytes are intolerant to cold and resistant to β-adrenergic stimulation of brown and beige adipose tissues. These defects, independent of lipolysis, are caused by impaired mitophagy, which results in defective BAT mitochondria, non-alcoholic fatty liver disease, and insulin resistance.
INTRODUCTION
Brown (BAT) and white (WAT) adipose tissue play distinct roles in maintaining whole-body energy homeostasis, and as such require an integrated system to match energy availability with demand in response to hormonal and nutritional cues. BAT functions to generate heat through lipid catabolism, and its activation has potent anti-obesity and anti-diabetic effects in rodents (Sidossis and Kajimura, 2015). Conversely, reduced activity or dysfunction in BAT can cause insulin resistance (Lowell et al., 1993). In humans, reduced metabolic activity of BAT is associated with insulin resistance (Chondronikola et al., 2014) and type 2 diabetes (Blondin et al., 2015). As human BAT may be targeted pharmacologically (Carey et al., 2013; Cypess et al., 2015), there is renewed interest in its therapeutic potential for treating metabolic diseases; however, the molecular mechanisms by which ATP sensing and use are integrated into this important metabolic circuit have yet to be fully defined.
AMP-activated protein kinase (AMPK) is a cellular sensor of energy homeostasis (Steinberg and Kemp, 2009), responding to various physiological, hormonal, and nutritional cues to balance ATP production with demand. AMPK regulates energy homeostasis by the phosphorylation of multiple substrates that alter most, if not all, branches of cellular metabolism (Ducommun et al., 2015; Hoffman et al., 2015; Schaffer et al., 2015; Steinberg and Kemp, 2009). AMPK is implicated in regulating multiple aspects of lipid synthesis and utilization (for review, see O’Neill et al., 2013). For example, AMPK-mediated inhibition of acetyl-CoA carboxylase (ACC) reduces malonyl-CoA levels and relieves inhibition on carnitine palmitoyltransferase 1 (CPT1) to promote fatty acid oxidation and suppress fatty acid synthesis (Carling et al., 1987; Fullerton et al., 2013). AMPK also indirectly regulates fatty acid oxidation by controlling mitochondrial homeostasis. AMPK regulates mitochondrial content, quality, and fission through peroxisome proliferator-activated receptor gamma co-activator 1α (PGC1α) (Jäger et al., 2007), unc-51 like autophagy activating kinase 1 (ULK1)-mediated mitophagy (Bujak et al., 2015; Egan et al., 2011; Tian et al., 2015), and mitochondrial fission factor (MFF) (Ducommun et al., 2015; Toyama et al., 2016), respectively. While AMPK has also been implicated in regulating lipolysis, its role is controversial (for review, see Watt and Steinberg, 2008), with groups reporting either inhibition (Daval et al., 2005; Sullivan et al., 1994), activation (Ahmadian et al., 2011; Yin et al., 2003), or no effect (Chakrabarti et al., 2011; Roepstorff et al., 2004). Despite the central role of AMPK in regulating multiple aspects of metabolism, our understanding of the role of AMPK in adipose tissue is largely based on studies conducted in cultured cells (Daval et al., 2005; Sullivan et al., 1994; Vila-Bedmar et al., 2010; Yin et al., 2003) or in mice genetically lacking single subunits of AMPK that retain substantial residual AMPK activity (Bauwens et al., 2011; Dzamko et al., 2010; Wan et al., 2014).
Obesity and insulin resistance are associated with reduced AMPK activity in both WAT and BAT (Lindholm et al., 2013; Ruderman et al., 2013). In addition, interventions in humans that reduce adiposity concomitantly reinstate AMPK levels within adipose tissue (Fritzen et al., 2015; Xu et al., 2015). However, the physiological role of AMPK in adipocytes in regulating whole-body energy expenditure, substrate utilization, and insulin sensitivity is not known. To address these questions, we created an inducible model for the deletion of AMPK activity in adipocytes by crossing AdipoQ-CreERT2 mice (Mottillo et al., 2014) with double floxed AMPK β1β2 mice (O’Neill et al., 2011). This model allows for the temporal and cell-specific deletion of AMPK without the consequences of removal in non-adipocyte cell types observed using other models (Jeffery et al., 2014; Lee et al., 2013) and also avoids any potential confounding effects that AMPK might have on adipocyte differentiation and development that occurs primarily over the first 3 weeks after birth (Habinowski and Witters, 2001; Vila-Bedmar et al., 2010; Wu et al., 2015). We found that adipocyte deletion of AMPK in adult mice did not alter lipolysis; however, it was vital for maintaining mitochondrial structure, function, and markers of mitophagy in BAT. AMPK deletion in adipocytes resulted in cold intolerance and reduced thermogenesis in response to acute and chronic β3-adrenoreceptor (β3-AR) stimulation, findings consistent with a defect in BAT and beige fat. Furthermore, the lack of adipocyte AMPK exacerbated the detrimental effects of a high-fat diet (HFD), including the development of liver steatosis and insulin resistance. Thus, adipocyte AMPK is a critical hub for integrating pharmacological, thermal and nutritional inputs, suggesting that targeted activation may be useful in treating non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes.
RESULTS
β-adrenergic stimulation, which mobilizes free fatty acids (FFAs) and stimulates thermogenesis, is known to activate AMPK in adipose tissue of rodents (Gauthier et al., 2008; Inokuma et al., 2005; Koh et al., 2007; Moule and Denton, 1998), but whether this occurs in human brown fat is not known. Treatment of human brown adipocyte cells, derived from the supraclavicular region of three different adult human subjects (Jespersen et al., 2013) with norepinephrine, increased activation of AMPK as detected by greater phosphorylation of AMPKα T172 and that of its downstream substrate ACC S79 (Figure 1A). To investigate the physiological role of adipocyte AMPK, we treated Control mice (floxed alleles, but no CreERT2) and mice deficient for AMPK in adipocytes (floxed alleles, with CreERT2, herein named iβ1-β2AKO) with tamoxifen at 8 weeks of age. Tamoxifen had a small effect on body weight within the first week, but mice returned to their previous weight within the second week (Figure S1A). Tamoxifen treatment removed AMPK β1 and β2 subunits from BAT and WAT of iβ1β2AKO mice, but not Control mice (Figure 1B). This deletion was specific to adipocytes, as AMPK β1 and β2 expression in other metabolic tissues (liver, muscle, and heart) or the stromal vascular cell fraction of adipose tissue was not altered (Figures 1B and S1B). As AMPK β1 and β2 subunits are essential for heterotrimer formation (Dzamko et al., 2010; Steinberg et al., 2010; Woods et al., 1996), this resulted in dramatic reductions in AMPKα expression and phosphorylation levels of AMPKα T172 and ACC S79 in adipocytes, but not other tissues (Figures 1B and S1B). Overall, these data indicate that efficient adipocyte deletion of AMPK was achieved, and it suggests that residual AMPKα expression in whole adipose tissue lysates is likely from resident stromal vascular cells in which AMPK expression was not affected.
Figure 1. Norepinephrine Activates AMPK in Human Brown Adipocytes but AMPK Does Not Regulate Lipolysis In Vivo.

(A) Primary human brown adipocytes were treated with 1 μM norepinephrine (NE) or vehicle for 45 min before measuring pAMPKa T172, total AMPKa, pACC S79, total ACC, and UCP1. *p < 0.05, **p < 0.01 denotes a NE effect as determined by Student’s t test.
(B) AMPKβ1 and β2, pAMPKα T172, total AMPKα, pACC S79, and total ACC levels in Control (CreERT2 −) or iβ1β2AKO mice (CreERT2 +) in BAT, iWAT, gWAT, liver, quadriceps muscle, and heart.
(C–J) Isolated adipocytes from Control (−) and iβ1β2AKO (+) mice were untreated (Basal) or treated with 10 μM isoproterenol (Iso) for 45 min and the amount of FFA (D) or glycerol (E) released into the media was quantified. Adipocytes were immunoblotted for total and phosphorylated ACC, HSL, ATGL, AMPKα, and total levels of AMPK β1 and β2 and β-tubulin (C). The ratios of phospho-AMPKαT172/AMPKα (F), phospho-ACCS79/ACC (G), phospho-HSLS565/HSL (H), phospho-HSLS660/HSL (I), and phospho-ATGLS406/ATGL (J) were quantified (n = 3 mice per genotype, from three independent experiments). Data are means ± SEM. ***p < 0.001 denotes a genotype effect within groups, and †p < 0.05, ††p < 0.01, and †††p < 0.001 denote an Iso effect as determined by Student’s t test (D) or two-way ANOVA and Bonferroni post hoc test. N.D., not determined. See also Figure S1.
AMPK Does Not Regulate Adipocyte Lipolysis In Vivo
We investigated the role of AMPK in lipolysis and found that neither basal nor isoproterenol-stimulated release of FFA or glycerol were different between genotypes (Figures 1D and 1E), despite greater phosphorylation of AMPKα by isoproterenol in adipocytes isolated from Control mice, compared to iβ1-β2AKO (Figures 1C and 1F). Protein levels of AMPKα, β1, and β2 (Figure 1C) and phosphorylation levels of AMPKα T172 and ACC S79 (Figures 1C and 1G) were almost completely abolished in fat cells from iβ1β2AKO mice; however, phosphorylation of the AMPK consensus site on hormone sensitive lipase (HSL), S565, was reduced by isoproterenol but not AMPK deletion (Figure 1H). The phosphorylation levels of the protein kinase A (PKA) site on HSL, S660, which stimulates HSL activity (Watt and Steinberg, 2008), was increased by isoproterenol treatment but was not different between control and AMPK-deficient adipocytes (Figure 1I). Phosphorylation of adipose triglyceride lipase (ATGL), the rate-limiting enzyme for lipolysis, on the putative AMPK site S406 (Ahmadian et al., 2011; Pagnon et al., 2012), was unaffected by deletion of AMPK and isoproterenol treatment (Figure 1J). One possibility for the lack of a detectable effect of AMPK on lipolysis could be that prior activation of AMPK is required (Watt et al., 2006). However, prior activation of AMPK with AICAR (as detected by increased phosphorylation levels of AMPKα T172 and ACC S79 in Control adipocytes) had no effect on the phosphorylation of HSL S565 (Figure S1C). In addition, AICAR reduced lipolysis in both Control and iβ1β2AKO adipocytes (Figure S1D), indicating that the effects of AICAR on lipolysis are likely AMPK independent. Similarly, there was no difference in serum FFA between Control and iβ1β2AKO mice treated in vivo with the β3-AR agonist CL-316,243 (CL; Figure S1E), nor in phospho-HSL S660 levels (Figure S1F). This was despite lower phosphorylation of the AMPK targets ACC, Raptor (Gwinn et al., 2008), and HSL S565 in iβ1β2AKO mice, which would be anticipated to increase lipolysis (Figure S1F). Collectively, these data indicate that lack of adipocyte AMPK in vivo does not affect lipolysis in response to β-adrenergic stimuli.
Adipocyte AMPK Is Critical for Brown Fat Function
A vital function for adipose tissue is to defend against cold exposure. Female Control and iβ1β2AKO mice of similar body weight were acclimatized at thermoneutrality (30°C) for 1 week (Figures 2A and S2A) and then challenged with cold exposure (4°C). iβ1β2AKO mice were unable to maintain core body temperature (Figure 2B) or interscapular BAT temperature (Figures 2C and 2D). Acute cold exposure significantly reduced triglyceride levels in BAT of Control mice; however, this effect was absent in iβ1-β2AKO mice (Figure 2E). Moreover, basal levels of UCP1 were lower in iβ1β2AKO mice (Figures 2F and 2I). The hypothermia in iβ1β2AKO mice was not due to a failure to mobilize FFA or glycerol into circulation (Figures S2B and S2C), further supporting a minimal role for adipocyte AMPK in regulating lipolysis in vivo. Similar cold intolerance (Figures S2D and S2E) and an inability to oxidize BAT derived lipid (Figure S2H) was also observed in male iβ1β2AKO mice housed at 23°C, then exposed to 4°C for 3 hr. Importantly, mice with double floxed β1 and β2 alleles behaved similarly to C57BL/6 mice when placed in the cold (Figures S2F and S2G). Cold exposure activated AMPK in BAT of Control mice (Figures 2F and 2G), as noted by greater phosphorylation of AMPKα T172 and a tendency for greater phosphorylation of the S79/212 site on ACC (Figures 2F and 2H).
Figure 2. Adipocyte AMPK Is Required for Acute BAT-Mediated Thermogenesis.

(A) Timeline, in weeks of age, for tamoxifen (TMX) treatment and cold exposure in female Control and iβ1β2AKO mice.
(B–D) Core body temperature (B), thermal images (C), and interscapular BAT temperature (D) in Control and iβ1β2AKO mice at 30°C, and 4°C for 1 and 2 hr (n = 6–9 per group).
(E) Triglyceride levels in BAT of mice kept at thermoneutrality (30°C) or exposed to cold (4°C) (n = 4–7 per group).
(F–I) Phosphorylation of AMPK ([F] and [G], pAMPKαT172/AMPKα) and ACC ([F] and [H], pACCS79/ACC), and total UCP1 protein levels ([F] and [I]) in response to cold (4°C) in BAT of Control and iβ1β2AKO mice (n = 4–6 per group). *p < 0.05 and ***p < 0.001 denotes a cold (4°C) effect within genotypes, and ††p < 0.01 and †††p < 0.001 denote a genotype effect as determined by two-way ANOVA and Bonferroni post hoc test.
(J) Timeline, in weeks of age, for tamoxifen (TMX) and acute CL-316,243 (CL, 0.033 nmol/kg) treatment in female Control and iβ1β2AKO mice (n = 8 mice per group).
(K–N) Oxygen consumption (VO2; [K]), representative thermal images of mice in given groups (L), interscapular BAT surface temperature (M), and plasma FFA (N) in response to a single injection of saline or CL (0.033 nmol/kg, 20 min; n = 8 per group). Data are means ± SEM. *p < 0.05 denotes a genotype effect within groups, and †††p < 0.001 denotes a CL effect as determined by two-way ANOVA and Bonferroni post hoc test. See also Figure S2.
We also probed BAT function using the β3-AR agonist CL (Figure 2J), which increases thermogenesis through a UCP1-dependent pathway (Crane et al., 2014). A single injection of CL increased AMPK activity as detected by greater phosphorylation of AMPKα T172 and Raptor S792 (Figure S2I). CL also increased oxygen consumption (Figure 2K) and interscapular BAT temperature in Control mice (Figures 2L and 2M), but this effect was diminished in iβ1β2AKO mice, despite normal FFA release (Figure 2N).
In liver (Fullerton et al., 2013) and resting skeletal muscle (O’Neill et al., 2014), AMPK regulates fatty acid oxidation via phosphorylation and inhibition of ACC, which reduces levels of malonyl-CoA, an allosteric inhibitor of CPT1 (Carling et al., 1987; Steinberg and Kemp, 2009). Thus, we tested whether AMPK-mediated phosphorylation of ACC and subsequent removal of CPT1 inhibition was involved in BAT thermogenesis in mice that have both an ACC1-S79A and ACC2-S212A knockin mutation (ACC DKI) (Fullerton et al., 2013). ACC DKI mice maintained core body temperature and interscapular BAT temperature at 4°C in a similar manner to wild-type mice (Figures S2J and S2K). Furthermore, when acutely challenged with CL, ACC DKI mice increased oxygen consumption equally to wild-type mice (Figure S2L). Overall, these results indicate that AMPK is required for acute BAT thermogenesis, but this effect does not depend on AMPK phosphorylation of ACC.
Adipocyte AMPK Regulates Mitochondrial Structure and Function in BAT
Since the defect in BAT of mice deficient for adipocyte AMPK was not due to the AMPK-ACC axis, we examined whether this was due to a defect in mitochondria. Assessment of mitochondrial morphology by electron microscopy revealed altered mitochondrial structure and greater disrupted cristae in iβ1β2AKO mice (Figures 3A and 3B), while total mitochondria did not differ (Figure 3C). Consistent with alterations in mitochondrial structure, respiration rates in isolated BAT mitochondria, which eliminates potential differences in mitochondrial number, were lower in iβ1β2AKO mice (Figure 3D). Non-UCP1-mediated respiration levels in the presence of GDP, the ATP synthase inhibitor oligomycin, or the chemical uncoupler FCCP, were also lower in iβ1-β2AKO mice (Figure S3A; significant difference for all but FCCP; p = 0.06). The alterations in mitochondrial structure and function suggested a defect in mitochondrial quality control but not mitochondrial content (biogenesis). In line with this observation, we found that protein levels of PGC1α, a master regulator of mitochondrial biogenesis, was not different between genotypes basally (Figure S3B). Recently, AMPK has been implicated in regulating mitochondrial morphology by phosphorylating MFF (Ducommun et al., 2015; Toyama et al., 2016). However, phosphorylation levels of MFF on the AMPK target site S129 were not different in BAT of Control and iβ1β2AKO mice (Figure S3C), nor did they change in response to cold challenge. Furthermore, the phosphorylation of MFF at S129 or S146 did not differ between brown adipocytes from Control or iβ1β2AKO mice under basal conditions or after pharmacological activation of AMPK (Figure S3D). This finding was in contrast to the reduction in the phosphorylation of ULK1 S555 (Figure S3D), which AMPK phosphorylates to increase mitophagy, a process that promotes the clearance of damaged mitochondria to maintain mitochondrial quality (Egan et al., 2011). The levels of phospho-ULK1 S555 were also lower in vivo, in BAT of mice deficient for adipocyte AMPK (Figure 3E). Consistent with a defect in mitochondrial clearance, the lipidation of the autophagy protein LC3B was lower (LC3B II/LC3B I; Figure 3F), with greater accumulation of the autophagy adaptor protein p62 in BAT of mice deficient for adipocyte AMPK (Figure 3G). These changes in markers of mitophagy were also observed in isolated mitochondria from the BAT of iβ1β2AKO mice (Figures S3E and S3F). In addition, cold exposure led to dynamic changes in pULK1 S555 and LC3B lipidation (LC3B II/LC3B I) in BAT of Control mice, effects not observed in iβ1β2AKO mice (Figure S3G). Overall, these data indicate that in BAT, AMPK maintains mitochondrial integrity and function through the regulation of mitophagy.
Figure 3. Adipocyte AMPK Is Important for Maintaining Mitochondrial Homeostasis in BAT.

(A–C) Representative electron micrographs of mitochondria at two different magnifications (A), quantification of percentage mitochondria with disrupted cristae (B) and total number of mitochondria per micrograph (C) from BAT of chow-fed Control and iβ1β2AKO mice (n = 3 to 4 mice per genotype).
(D) Respiration in isolated BAT mitochondria with malate and L-carnitine (M + L-Carn; 2 mM and 2.5 mM) and after the addition of palmitoyl-CoA (PCoA; 30 μM [n = 7 BAT mitochondrial isolations per genotype]).
(E) Phospho-ULK1S555 protein levels normalized to total ULK1 levels from BAT of Control and iβ1β2AKO mice (n = 11 to 12 mice per genotype).
(F and G) immunoblot analysis of the LC3BII/LCBI ratio (F) and p62 levels normalized to β-tubulin (G) in whole BAT tissue from chow-fed Control and iβ1β2AKO mice (n = 6 to 7 per group). Data are means ± SEM. *p < 0.05, **p < 0.01, and **p < 0.001 denote a genotype effect within groups as determined using a Student’s t test. See also Figure S3.
Adipocyte AMPK Is Important for the Catabolic Remodeling of BAT and WAT
Since mitochondrial function is critical for the catabolic remodeling of WAT and BAT, we examined remodeling in mice deficient for adipocyte AMPK in response to chronic β3-AR activation (Figure 4A). Daily treatment of mice with CL elevated oxygen consumption, carbon dioxide, and heat production in Control mice, but these effects were blunted in iβ1β2AKO mice (Figure 4B, S4A, and S4B). CL treatment had no effect on body weight, food or water intake, or activity levels (Figures S4C–S4F), suggesting that reductions in energy expenditure of iβ1β2AKO mice may have been due to a defect in adipose tissue metabolism. Mitochondrial cytochrome c oxidase (COX) activity was lower in BAT of iβ1β2AKO mice under control conditions and following 5 days of CL treatment (Figure 4C). Electron transport chain (ETC, OXPHOS) subunit content was similar between genotypes with saline; however, with CL treatment, the upregulation of OXPHOS subunit expression was attenuated in iβ1β2AKO mice (Figure 4D).
Figure 4. Adipocyte AMPK Is Required for the Adaptive Response to Thermogenesis and the Browning Program.

(A) Timeline, in weeks of age, for tamoxifen (TMX) and 5-day CL 316,243 (CL) treatment in female Control and iβ1β2AKO mice. Mice were treated with 0.5 mg/kg CL for four consecutive days and 1 mg/kg on the final day.
(B) Whole-body oxygen consumption (VO2) under basal conditions or 6 hr post-CL injection on indicated days (n = 6–9 per group).
(C and D) COX activity (C) and representative OXPHOS subunit immunoblotting with quantification (D) (n = 8–10 per group) in BAT of Control and iβ1β2AKO mice treated with saline or CL for 5 days (5D CL).
(E) Representative iWAT histological images (10× magnification) of Control and iβ1β2AKO mice treated with CL for 5 days.
(F) mRNA expression of browning markers (Cidea, Ppara, Pdk4) in iWAT of Control and iβ1β2AKO mice treated with saline or CL for 5 days (n = 6–9 per group).
(G and H) iWAT Ucp1 mRNA (G) and protein levels (H) in Control and iβ1β2AKO treated with saline or CL for 5 days (5D CL) (n = 6–9 per group). Data are means ± SEM. *p < 0.05 and **p < 0.01 denote a genotype effect within groups, and †p < 0.05, ††p < 0.01, and †††p < 0.001 denote a CL effect within genotype as determined by two-way ANOVA and Bonferroni post hoc test. See also Figure S4.
In addition to BAT, the blunted effects of the β3-AR agonist on increasing oxygen consumption in iβ1β2AKO mice may have also resulted from impaired catabolic remodeling of iWAT into beige or brown in white (brite) adipocytes (Rosen and Spiegelman, 2014). Control mice treated for 5 days of CL had a greater appearance of multilocular beige adipocytes in iWAT compared to iβ1β2AKO mice (Figure 4E). Furthermore, 5 days of CL increased the mRNA expression of browning markers (Cidea, Ppara, Pdk4; Figure 4F) and ETC complex I and II in Control mice (Figure S4G), but this effect was attenuated in iWAT of iβ1-β2AKO mice. Notably, chronic CL treatment increased mRNA and protein levels of UCP1 in Control mice, but not in iβ1β2AKO mice (Figures 4G and 4H). These results demonstrate that adipocyte AMPK is required for β-adrenergic-induced remodeling of BAT and iWAT.
Lack of Adipocyte AMPK Exacerbates HFD-Induced Insulin Resistance
Obesity is associated with reduced AMPK activity in both WAT and BAT (Lindholm et al., 2013; Ruderman et al., 2013). Moreover, dysfunctional BAT can contribute to diet-induced obesity and insulin resistance (Lowell et al., 1993). Therefore, to investigate the role of adipocyte AMPK in obesity-induced insulin resistance, mice were challenged with a HFD for 12 weeks (Figure 5A). As expected, the levels of phosphorylated AMPKα T172 in BAT were greatly reduced in HFD-fed iβ1β2AKO mice (Figure 5B). Mice lacking AMPK in adipocytes had greater weight gain after 5–8 weeks of the HFD treatment, but by 12 weeks body mass did not differ (Figure 5C) from controls. The histology of BAT, inguinal, and gonadal WAT (gWAT) did not show any gross differences under HFD (Figure S5A), and adiposity was not greater after 12 weeks of HFD (Figure 5D). Consistent with similar adiposity, whole-body metabolic parameters such as VO2, RER, food and water intake, and activity levels were also similar between genotypes (Figure S5B–S5F). Despite similar body mass and adiposity, HFD-fed iβ1β2AKO mice had higher fasting blood glucose, insulin (Figures 5E and 5F), and HOMA-IR levels (Figure S5G). Insulin sensitivity was not different between genotypes in chow-fed mice prior to starting the HFD intervention (Figure S5H), nor in mice fed an additional 11 weeks of chow diet (Figure S5I). However, on HFD, iβ1β2AKO mice were more glucose intolerant (Figure 5G) and less sensitive to an intraperitoneal insulin tolerance test compared to Control (Figure 5H). Control mice with AMPK β1β2-floxed alleles had similar glucose tolerance to C57BL/6 mice that received a HFD, but no tamoxifen (Figure S5J).
Figure 5. Adipocyte AMPK Protects against High-Fat-Diet-Induced Insulin Resistance.

(A) Timeline, in weeks of age, for tamoxifen (TMX) and chow or HFD treatments in male Control and iβ1β2AKO mice (n = 8 to 9 for chow fed, n = 11 to 12 for HFD).
(B) Total and phosphorylated (T172) levels of AMPKα in BAT of HFD-fed Control (−) and iβ1β2AKO (+) mice (n = 4 to 5).
(C and D) Body weight curves (C) and adiposity (D) in Control and iβ1β2AKO mice on indicated diet for 12 weeks.
(E and F) Fasting blood glucose (E) and plasma insulin concentrations (F) in Control and iβ1β2AKO mice on indicated diet for 10 weeks.
(G and H) Glucose tolerance test (GTT; [G]) and insulin tolerance test (ITT; [H]) performed after 10 to 11 weeks of HFD in Control and iβ1β2AKO mice. AUC, area under the curve. Data are means ± SEM. *p < 0.05 and **p < 0.01 denote a genotype effect within groups as determined by a Student’s t test ([B], AUC of [G] and [H]), two-way ANOVA (E, F), or two-way repeated-measures ANOVA ([C], glucose curves of [G] and [H]) and Bonferroni post hoc test. See also Figure S5.
Since differences in fasting blood glucose and insulin sensitivity were not observed under chow conditions, we further explored the tissue-specific metabolic defects in HFD-fed iβ1-β2AKO mice that promoted dysglycemia (Figure 6A). Mice deficient for adipocyte AMPK had a reduction in glucose uptake in response to insulin in BAT (Figure 6B) and liver (Figure S6A). Consistent with the defect in CL-mediated oxygen consumption under chow conditions (Figure 2K), change in oxygen consumption was lower in HFD-fed iβ1β2AKO mice after injection of CL (Figure 6C), but there was no difference in the change in serum FFA (Figure S6B). The levels of fasting and fed plasma FFA and glycerol between genotypes did not differ (Figures S6C and S6D). However, serum triglyceride levels were greater in the post-prandial state, but not under fasting conditions in iβ1-β2AKO mice (Figure 6D). An acute intraperitoneal injection of insulin increased AKT S473 phosphorylation in the liver, BAT, and gWAT of Control, but not iβ1β2AKO mice (Figure 6E). The increase in phospho-AKT S473 by insulin was similar between genotypes in quadriceps muscle (Figure 6E). The differences in whole-body insulin sensitivity between genotypes was likely not driven by inflammation in gWAT, as the expression of inflammatory and immune markers were increased similarly by HFD treatment in both genotypes (Figure S6E). We further explored the impairment in liver insulin sensitivity of iβ1β2AKO mice and found that their livers were heavier (Figure 6F) and displayed greater triglyceride accumulation (Figures 6G and 6H). Finally, levels of serum ALT, an indicator of liver damage, trended higher in iβ1β2AKO mice (p = 0.052; Figure 6I). Overall, these results indicate that AMPK deficiency in adipose tissue depresses energy utilization in BAT and thereby promotes the development of hepatic lipid accumulation and insulin resistance.
Figure 6. Deletion of Adipocyte AMPK Promotes Hepatic Lipid Accumulation.

(A) Timeline, in weeks of age, for tamoxifen (TMX) treatment and experimental procedures in male Control and iβ1β2AKO mice on HFD.
(B) 3H-2-deoxy-D-glucose (2-DG) uptake in BAT of Control and iβ1β2AKO mice on HFD, fasted for 6 hr and injected with 0.7 U/kg insulin (n = 3 to 4).
(C) Change in oxygen consumption in response to CL-316,243 (0.033 nmol/kg, β3-AR test) in Control and iβ1β2AKO mice on HFD (n = 7 to 8).
(D) Plasma triglyceride (TGs) levels in HFD-treated mice in fed and fasted (10 hr) states (n = 8 to 9 for fed, n = 11 to 12 for fasted).
(E) Levels of total AKT and phospho-AKTS473 (pAKTS473) under basal conditions (Basal) and in response to an IP injection of insulin (1.0 U/kg for 15 min) in the liver, BAT, gWAT, and quadriceps muscle (Quad) of 12 week HFD-treated Control (−) and iβ1β2AKO (+) mice (n = 5–7).
(F–H) Liver weights (F), representative H&E stains (20×; [G]) and liver triglyceride (TGs; [H]) levels in 12 weeks HFD-treated Control and iβ1β2AKO mice (n = 18–20 per group).
(I) Plasma ALT levels in HFD fed Control and iβ1β2AKO mice (n = 14 to 15). Data are means ± SEM. *p < 0.05 and **p < 0.01 denote a genotype effect within groups, and †p < 0.05 and ††p < 0.01 denote an insulin effect within genotypes as determined using a Student’s t test ([B], [C], [F], [H]) or, where appropriate, by two-way ANOVA and Bonferroni post hoc test. See also Figure S6.
DISCUSSION
Obesity and type 2 diabetes are characterized by reductions in the metabolic activity of BAT, suggesting that therapies that restore BAT function may be effective for treating these disorders. Despite intense interest, the molecular mechanisms contributing to reduced BAT activity that occur with obesity and diabetes are not fully understood. We found that deletion of AMPK in adipocytes leads to defects in BAT mitochondrial structure and function and reduced oxidative metabolism in response to cold exposure or β-adrenergic stimuli. Loss of adipocyte AMPK exacerbates the development of liver steatosis, liver insulin resistance, and whole-body glucose and insulin intolerance. These data indicate that reductions in adipose AMPK, as observed in obese humans (Ruderman et al., 2013), is an important contributing factor to reduced BAT activity, liver steatosis, insulin resistance, and dysglycemia.
The greater whole-body insulin resistance observed in iβ1β2AKO mice is similar to mice with a germline deletion of AMPK β2 (Steinberg et al., 2010), effects that at the time were attributed primarily to impairments in skeletal muscle function. Interestingly, loss of both AMPK β subunits specifically in skeletal muscle does not affect insulin sensitivity when mice are fed a HFD (Marcinko et al., 2015), suggesting that reductions in BAT metabolic activity may have been a contributing factor to the insulin resistance observed in germline AMPK β2 null mice. While we previously observed very low levels of AMPK β2 in WAT of AMPK β1 KO mice (Dzamko et al., 2010), these low levels of detection compared to the current study may have been the result of using a β pan antibody that had a much greater affinity for detecting the β1 compared to β2 subunit. Importantly, similar to iβ1β2AKO mice, whole-body deletion of AMPK β1 also did not alter adipocyte lipolysis (Dzamko et al., 2010).
Studies using a variety of pharmacological agents have suggested that AMPK may regulate lipolysis through direct phosphorylation of ATGL and HSL (Ahmadian et al., 2011; Pagnon et al., 2012; Watt et al., 2006). ATGL has been shown to be phosphorylated at S406 by both AMPK and PKA (Ahmadian et al., 2011; Pagnon et al., 2012); however, we found no difference in iβ1β2AKO mice, making it unlikely that AMPK is the primary kinase that phosphorylates this residue. We found that levels of phospho-HSL S565 were not reduced in isolated adipocytes of mice lacking AMPK, supporting the importance of other kinases in maintaining phosphorylation of this residue as previously suggested (Garton and Yeaman, 1990). Consistent with minimal effects of AMPK on the phosphorylation of these residues, we found no changes in adipose tissue lipolysis basally or following β-adrenergic stimulation. Interestingly, the non-specific pharmacological AMPK activator AICAR impaired lipolysis independent of AMPK—an effect that is likely related to observations that AICAR and biguanides can supress cAMP production in cells independent of AMPK (Kalderon et al., 2012; Miller et al., 2013). Collectively, these studies suggest that AMPK has a minimal role in regulating adipocyte lipolysis.
We found that β-adrenergic stimuli activate AMPK in human brown adipocytes and BAT of mice. Previous studies have indicated that this is likely a consequence of increases in lipolysis (Gauthier et al., 2008) and mitochondrial uncoupling (Inokuma et al., 2005), which collectively cause reductions in the AMP/ADP: ATP ratio and activation of AMPK. The activation of AMPK in BAT by stimuli that increase energy demand is in contrast to AMPK’s role in the hypothalamus, where it prevents negative energy balance by reducing sympathetic outflow (López et al., 2010). These opposing functions of AMPK in peripheral and central circuits are consistent with hormonal regulators of energy balance such as leptin and ghrelin, which differentially regulate AMPK activity in the periphery and CNS (Dzamko and Steinberg, 2009).
The role of AMPK in adipose tissue thermogenesis does not appear to involve acute regulation of fatty acid oxidation, as we found that mice lacking an important AMPK phosphorylation site on ACC, which controls fatty acid oxidation in the liver and resting skeletal muscle (Fullerton et al., 2013; O’Neill et al., 2014), responded normally to both cold and β-adrenergic stimuli. Instead, the primary role of AMPK in BAT involves the maintenance of mitochondrial quality. Our deletion of AMPK in adipocytes occurs in adult mice, at a time when BAT is completely developed and has a full complement of mitochondria, which may explain why there were no differences in PGC1α protein and mitochondrial number. However, BAT of iβ1β2AKO mice had greatly altered mitochondrial morphology and function, along with a defect in autophagy signaling, marked by an elevated LC3BII/LC3BI ratio, accumulation of p62, and reduced phosphorylation of ULK1. These findings are consistent with recent work that suggests a role for autophagy in regulating lipid metabolism in adipocytes (Martinez-Lopez et al., 2016) and adds to these findings by indicating that AMPK is important for regulating this pathway and controlling mitochondrial quality.
Both cold and β3-AR agonists activated AMPK and have been shown to chronically increase BAT metabolic activity in humans; however, discomfort and tachycardia, respectively, will likely limit the widespread adoption of these therapies (Chondronikola et al., 2014; Cypess et al., 2015). On the other hand, as AMPK activity is reduced in adipose tissue of diabetic patients (Ruderman et al., 2013) and AMPK activators such as metformin and salicylate are well tolerated in humans (Hawley et al., 2012; Zhou et al., 2001), the development of direct AMPK activators (Xiao et al., 2013) that target BAT may be an effective means to improve mitochondrial function, reduce liver lipids, and improve insulin sensitivity.
EXPERIMENTAL PROCEDURES
Animals
Mice deficient for AMPK β1β2 in adipocytes were generated by crossing AMPK β1flox/flox β2flox/flox mice (O’Neill et al., 2011) with mice harboring the tamoxifen-sensitive Cre recombinase (CreERT2) under the control of the adiponectin promoter B6N.129S-Tg (Adipoq-CreERT2)tm1Jgg (Mottillo et al., 2014) that had been backcrossed on C57BL/6 background. All experiments were sanctioned by the McMaster University Animal Ethics Committees. Details on the generation, and maintenance of mouse strains are in the Supplemental Experimental Procedures.
Metabolic and Blood Measurements
Metabolic monitoring was performed in a Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments) as described (O’Neill et al., 2011). To assess UCP1-mediated thermogenesis, CL-316,243 (CL; 0.033 nmol/g body weight) treatment was performed using CLAMS, and infrared thermography as previously described (Crane et al., 2014). A rectal thermal probe was used to determine core body temperature in mice. All metabolic monitoring was performed in a room kept between 26°C and 28°C. Details on metabolic and blood measurements are in the Supplemental Experimental Procedures.
Adipocyte Isolations and Lipolysis
Adipose tissue depots from three mice per genotype were pooled and digested with collagenase Type II (Sigma) and performed as detailed in the Supplemental Experimental Procedures.
Immunoblotting and Immunoprecipitation
Antibodies, immunoblotting, and immunoprecipitation are described in detail in the Supplemental Experimental Procedures.
Tissue Triglyceride Determination
Determination of tissue triglyceride content is described in detail in the Supplemental Experimental Procedures.
COX Activity Assay
COX activity was measured using homogenates that were frozen and thawed three times to disrupt membranes. Details on COX activity assay are in the Supplemental Experimental Procedures.
Tissue Processing and Transmission Electron Microscopy
Detailed procedures for tissue processing and transmission electron microscopy are in the Supplemental Experimental Procedures.
Cell Culture
Generation and culture of human primary and murine immortalized brown adipocyte cell lines is detailed in the Supplemental Experimental Procedures.
Isolation of BAT Mitochondria and Respirometry
BAT from the interscapular and axillary regions was combined from individual mice, and mitochondria were isolated and respirometry performed as described in detail in the Supplemental Experimental Procedures.
RNA Isolation and Real-Time Quantitative PCR (RT-qPCR)
RT-qPCR was carried out as previously described to determine mRNA expression levels (Galic et al., 2011; O’Neill et al., 2011). Briefly, adipose tissues were lysed in TRIzol reagent (Invitrogen) to remove lipid and the aqueous phase was applied to an RNeasy kit (QIAGEN) column for subsequent purification. Relative gene expression was calculated using the comparative Ct (2−ΔΔCt) method, where values were normalized to a housekeeping gene (Ppia). All Taqman primers were purchased from Invitrogen (see Supplemental Experimental Procedures).
Statistical Analysis
Results were analyzed using Student’s t test or ANOVA where appropriate, using GraphPad Prism software. A repeated-measures ANOVA was used for all body weight plots, thermography measurements, and GTT and ITT data. A Bonferroni post hoc test was used to test for significant differences as determined by the ANOVA. Significance was accepted at p ≤ 0.05. Data are presented as mean ± SEM.
Supplementary Material
Highlights.
Adipocyte AMPK is required for cold and β-adrenergic-stimulated thermogenesis
AMPK is required for the browning of white adipose tissue (beige/brite fat)
AMPK is critical for mitophagy and maintaining BAT mitochondrial quality
Adipocyte AMPK is required to prevent NAFLD and insulin resistance
Acknowledgments
We thank Marcia Reid for technical assistance in sample preparation for EM and Rick McKenzie for advice in denoising filters. We acknowledge Dr. John Rubinstein and the Hospital for Sick Children in Toronto for allowing access to the EM Facility. We thank Julian Yabut for technical assistance with genotyping and cell culture and Dr. Matthew J. Watt (Monash University) for providing ATGL antibodies. These studies were supported by grants from the Canadian Diabetes Association (G.R.S.), the Canadian Institutes of Health Research (G.R.S.), the Natural Sciences and Engineering Research Council of Canada (G.R.S.), and the National Health and Medical Research Council of Australia (G.R.S. and B.E.K.). E.P.M. is a Canadian Diabetes Association Post-doctoral Fellow. E.M.D. is a recipient of an Ontario Graduate Scholarship and Queen Elizabeth II Graduate Scholarship in Science and Technology. B.K.S. is a recipient of a CIHR post-doctoral fellowship. C.S. was supported by the Novo Nordisk Foundation. T.I.H. was supported by a PhD scholarship from Faculty of Health Sciences, University of Copenhagen. CFAS is supported by Trygfonden. The Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) is supported by an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen. A.E.G. is a recipient of a CIHR/MitoCanada Doctoral Research Award. G.R.S. is a Canada Research Chair in Metabolism and Obesity and the J. Bruce Duncan Chair in Metabolic Diseases.
Footnotes
Supplemental Information includes six figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.06.006.
AUTHOR CONTRIBUTIONS
E.P.M., E.M.D., B.K.S., J.D.C., and G.R.S. designed the experiments. E.P.M., E.M.D., J.D.C., A.E.G., B.K.S., S.D., I.A.R., and T.I.H. performed experiments and testing. E.P.M., E.M.D., J.D.C., B.K.S., J.G.G., K.S., T.J.H., C.S., A.R., J.O., and B.E.K. provided technical expertise and performed data analyses. E.P.M. and G.R.S. wrote the manuscript. All authors edited the manuscript and provided comments.
References
- Ahmadian M, Abbott MJ, Tang T, Hudak CSS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauwens JD, Schmuck EG, Lindholm CR, Ertel RL, Mulligan JD, Hovis I, Viollet B, Saupe KW. Cold tolerance, cold-induced hyperphagia, and nonshivering thermogenesis are normal in α1-AMPK−/− mice. Am J Physiol Regul Integr Comp Physiol. 2011;301:R473–R483. doi: 10.1152/ajpregu.00444.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondin DP, Labbé SM, Noll C, Kunach M, Phoenix S, Guérin B, Turcotte ÉE, Haman F, Richard D, Carpentier AC. Selective Impairment of Glucose but Not Fatty Acid or Oxidative Metabolism in Brown Adipose Tissue of Subjects With Type 2 Diabetes. Diabetes. 2015;64:2388–2397. doi: 10.2337/db14-1651. [DOI] [PubMed] [Google Scholar]
- Bujak AL, Crane JD, Lally JS, Ford RJ, Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD, Steinberg GR. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015;21:883–890. doi: 10.1016/j.cmet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AL, Formosa MF, Van Every B, Bertovic D, Eikelis N, Lambert GW, Kalff V, Duffy SJ, Cherk MH, Kingwell BA. Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia. 2013;56:147–155. doi: 10.1007/s00125-012-2748-1. [DOI] [PubMed] [Google Scholar]
- Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, Luo Z, Farmer SR, Kandror KV. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J Lipid Res. 2011;52:1693–1701. doi: 10.1194/jlr.M014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondronikola M, Volpi E, Børsheim E, Porter C, Annamalai P, Enerbäck S, Lidell ME, Saraf MK, Labbe SM, Hurren NM, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63:4089–4099. doi: 10.2337/db14-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JD, Mottillo EP, Farncombe TH, Morrison KM, Steinberg GR. A standardized infrared imaging technique that specifically detects UCP1-mediated thermogenesis in vivo. Mol Metab. 2014;3:490–494. doi: 10.1016/j.molmet.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elía E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, Kolodny GM. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 2015;21:33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet B, Vaulont S, Hajduch E, Ferré P, Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem. 2005;280:25250–25257. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- Ducommun S, Deak M, Sumpton D, Ford RJ, Núñez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27:978–988. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Dzamko NL, Steinberg GR. AMPK-dependent hormonal regulation of whole-body energy metabolism. Acta Physiol (Oxf) 2009;196:115–127. doi: 10.1111/j.1748-1716.2009.01969.x. [DOI] [PubMed] [Google Scholar]
- Dzamko N, van Denderen BJW, Hevener AL, Jørgensen SB, Honeyman J, Galic S, Chen ZP, Watt MJ, Campbell DJ, Steinberg GR, Kemp BE. AMPK β1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem. 2010;285:115–122. doi: 10.1074/jbc.M109.056762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzen AM, Lundsgaard AM, Jordy AB, Poulsen SK, Stender S, Pilegaard H, Astrup A, Larsen TM, Wojtaszewski JFP, Richter EA, Kiens B. New Nordic Diet-Induced Weight Loss Is Accompanied by Changes in Metabolism and AMPK Signaling in Adipose Tissue. J Clin Endocrinol Metab. 2015;100:3509–3519. doi: 10.1210/jc.2015-2079. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, Izon D, Honeyman J, Chen ZP, van Denderen BJ, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton AJ, Yeaman SJ. Identification and role of the basal phosphorylation site on hormone-sensitive lipase. Eur J Biochem. 1990;191:245–250. doi: 10.1111/j.1432-1033.1990.tb19116.x. [DOI] [PubMed] [Google Scholar]
- Gauthier MS, Miyoshi H, Souza SC, Cacicedo JM, Saha AK, Greenberg AS, Ruderman NB. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J Biol Chem. 2008;283:16514–16524. doi: 10.1074/jbc.M708177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habinowski SA, Witters LA. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun. 2001;286:852–856. doi: 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015;22:922–935. doi: 10.1016/j.cmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuma K, Ogura-Okamatsu Y, Toda C, Kimura K, Yamashita H, Saito M. Uncoupling protein 1 is necessary for norepinephrine-induced glucose utilization in brown adipose tissue. Diabetes. 2005;54:1385–1391. doi: 10.2337/diabetes.54.5.1385. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery E, Berry R, Church CD, Yu S, Shook BA, Horsley V, Rosen ED, Rodeheffer MS. Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte. 2014;3:206–211. doi: 10.4161/adip.29674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homøe P, Loft A, de Jong J, Mathur N, Cannon B, Nedergaard J, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supra-clavicular region of adult humans. Cell Metab. 2013;17:798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Kalderon B, Azazmeh N, Azulay N, Vissler N, Valitsky M, Bar-Tana J. Suppression of adipose lipolysis by long-chain fatty acid analogs. J Lipid Res. 2012;53:868–878. doi: 10.1194/jlr.M022673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh HJ, Hirshman MF, He H, Li Y, Manabe Y, Balschi JA, Goodyear LJ. Adrenaline is a critical mediator of acute exercise-induced AMP-activated protein kinase activation in adipocytes. Biochem J. 2007;403:473–481. doi: 10.1042/BJ20061479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem. 2013;69:165–175. doi: 10.1007/s13105-012-0199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López M, Varela L, Vázquez MJ, Rodríguez-Cuenca S, Gonzólez CR, Velagapudi VR, Morgan DA, Schoenmakers E, Agassandian K, Lage R, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16:1001–1008. doi: 10.1038/nm.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- Marcinko K, Sikkema SR, Samaan MC, Kemp BE, Fullerton MD, Steinberg GR. High intensity interval training improves liver and adipose tissue insulin sensitivity. Mol Metab. 2015;4:903–915. doi: 10.1016/j.molmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ, Singh R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottillo EP, Balasubramanian P, Lee YH, Weng C, Kershaw EE, Granneman JG. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic β3-adrenergic receptor activation. J Lipid Res. 2014;55:2276–2286. doi: 10.1194/jlr.M050005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moule SK, Denton RM. The activation of p38 MAPK by the β-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett. 1998;439:287–290. doi: 10.1016/s0014-5793(98)01392-1. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol. 2013;366:135–151. doi: 10.1016/j.mce.2012.06.019. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Lally JS, Galic S, Thomas M, Azizi PD, Fullerton MD, Smith BK, Pulinilkunnil T, Chen Z, Samaan MC, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 2014;57:1693–1702. doi: 10.1007/s00125-014-3273-1. [DOI] [PubMed] [Google Scholar]
- Pagnon J, Matzaris M, Stark R, Meex RCR, Macaulay SL, Brown W, O’Brien PE, Tiganis T, Watt MJ. Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology. 2012;153:4278–4289. doi: 10.1210/en.2012-1127. [DOI] [PubMed] [Google Scholar]
- Roepstorff C, Vistisen B, Donsmark M, Nielsen JN, Galbo H, Green KA, Hardie DG, Wojtaszewski JFP, Richter EA, Kiens B. Regulation of hormone-sensitive lipase activity and Ser563 and Ser565 phosphorylation in human skeletal muscle during exercise. J Physiol. 2004;560:551–562. doi: 10.1113/jphysiol.2004.066480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, Benayoun BA, Banko MR, Shaw RJ, Shokat KM, Brunet A. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 2015;22:907–921. doi: 10.1016/j.cmet.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidossis L, Kajimura S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest. 2015;125:478–486. doi: 10.1172/JCI78362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, O’Neill HM, Dzamko NL, Galic S, Naim T, Koopman R, Jørgensen SB, Honeyman J, Hewitt K, Chen Z-P, et al. Whole body deletion of AMP-activated protein kinase beta2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285:37198–37209. doi: 10.1074/jbc.M110.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, Zhong W, Chen Y, Wu W, Lin C, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589:1847–1854. doi: 10.1016/j.febslet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Bedmar R, Lorenzo M, Fernández-Veledo S. Adenosine 5′-monophosphate-activated protein kinase-mammalian target of rapamycin cross talk regulates brown adipocyte differentiation. Endocrinology. 2010;151:980–992. doi: 10.1210/en.2009-0810. [DOI] [PubMed] [Google Scholar]
- Wan Z, Root-McCaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring) 2014;22:730–738. doi: 10.1002/oby.20605. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Steinberg GR. Regulation and function of triacylglycerol lipases in cellular metabolism. Biochem J. 2008;414:313–325. doi: 10.1042/BJ20080305. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Holmes AG, Pinnamaneni SK, Garnham AP, Steinberg GR, Kemp BE, Febbraio MA. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2006;290:E500–E508. doi: 10.1152/ajpendo.00361.2005. [DOI] [PubMed] [Google Scholar]
- Woods A, Cheung PCF, Smith FC, Davison MD, Scott J, Beri RK, Carling D. Characterization of AMP-activated Protein Kinase and Subunits: ASSEMBLY OF THE HETEROTRIMERIC COMPLEX IN VITRO. J Biol Chem. 1996;271:10282–10290. doi: 10.1074/jbc.271.17.10282. [DOI] [PubMed] [Google Scholar]
- Wu Y, Song P, Zhang W, Liu J, Dai X, Liu Z, Lu Q, Ouyang C, Xie Z, Zhao Z, et al. Activation of AMPKα2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. 2015;21:373–382. doi: 10.1038/nm.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017. doi: 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Apovian C, Hess D, Carmine B, Saha A, Ruderman N. Improved insulin sensitivity 3 months after RYGB surgery is associated with increased subcutaneous adipose tissue AMPK activity and decreased oxidative stress. Diabetes. 2015;64:3155–3159. doi: 10.2337/db14-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Mu J, Birnbaum MJ. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-L1 adipocytes. J Biol Chem. 2003;278:43074–43080. doi: 10.1074/jbc.M308484200. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.