

Abstract
Background
Reduction in the cost of genomic assays has generated large amounts of biomedical-related data. As a result, current studies perform multiple experiments in the same subjects. While Bioconductor’s methods and classes implemented in different packages manage individual experiments, there is not a standard class to properly manage different omic datasets from the same subjects. In addition, most R/Bioconductor packages that have been designed to integrate and visualize biological data often use basic data structures with no clear general methods, such as subsetting or selecting samples.
Results
To cover this need, we have developed MultiDataSet, a new R class based on Bioconductor standards, designed to encapsulate multiple data sets. MultiDataSet deals with the usual difficulties of managing multiple and non-complete data sets while offering a simple and general way of subsetting features and selecting samples. We illustrate the use of MultiDataSet in three common situations: 1) performing integration analysis with third party packages; 2) creating new methods and functions for omic data integration; 3) encapsulating new unimplemented data from any biological experiment.
Conclusions
MultiDataSet is a suitable class for data integration under R and Bioconductor framework.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-1455-1) contains supplementary material, which is available to authorized users.
Keywords: Omics data, Data integration, Data infrastructure, Data organization, R
Background
Projects in biotechnology are constantly generating new types of data. As the reduction of costs of genomic assays drops, volume as well as data diversity greatly increase. While advances in knowledge of personalized medicine are expected, scientists are continuously challenged on data management and analysis [1, 2].
Bioconductor started in 2001 as an initiative to provide free software, written in R and implemented in standardized methods and classes, to analyze high-throughput biological data [3, 4]. Currently, Bioconductor is a public repository where scientists can find R packages to analyze all kinds of high-throughput omic data (e.g. genomic, transcriptomic, epigenomic, proteomic…). A standard infrastructure has been created to represent biological data comprising, amongst others, two basic R classes to load experiment’s information: eSet and SummarizedExperiment. The main objective of these classes is that biological data and phenotypic descriptions are well coordinated. In particular, methods such as subsetting are easily applied simultaneously to experiment and phenotypic data. While eSet is designed for microarray data, SummarizedExperiment is for next generation sequencing data.
Major public projects have performed experiments to a group of individuals generating different types of datasets [5]. For instance, the Cancer Genome Atlas (TCGA) [6, 7], is the largest resource available for multi-assay cancer genomics data; the 1000 Genome Project [8, 9] aims to provide a comprehensive resource for human genetic variants and gene-expression across populations and; the International Cancer Genome Consortium (ICGC) [10, 11] coordinates 55 research projects to characterize the genome, transcriptome and epigenome of multiple tumors. In addition, large repositories collect data of several smaller projects allowing unified storage and stimulating data sharing. Gene Expression Omnibus (GEO) [12–14] is the primary database where data from multi-assay experiments is shared publicly. Other reference databases are dbSNP [15, 16], a deposit for short genetic variations and Database of Genomic Variants archive (DGVa), for longer structural variants [17, 18].
All of these data resources are accessible through standard Bioconductor classes (eSet and SummarizedExperiment). There are numerous packages used to retrieve and transform data from public repositories. For instance, GEOquery [19] that obtains GEO data as an eSet object. Such packages aim to facilitate downstream analyses for Bioconductor’s packages. However, Bioconductor lacks a standard class to efficiently manage different datasets obtained from the same individuals.
Several R/Bioconductor packages have implemented methods to integrate and visualize biological data: PMA [20–22], mixOmics [23–25], made4 [26, 27], RGCCA [28, 29], omicade4 [30, 31], CNAmet [32, 33], RTopper [34, 35], iClusterPlus [36, 37] and STATegRa [38] among others. Each of these packages implements a different strategy to face the integration analysis. They typically use their own data structure, which is usually a list of matrices. The use of such structure makes it difficult to perform usual operations such as subsetting data across data sets and selecting samples (e.g. complete cases are usually required in all integration analysis). The specificity of the data structures to each method further hinders the user’s disposition to perform different integration analyses on one study. Therefore, a standard structure to manage the different datasets of the same individuals will promote the use of current and future integration methods, allowing the implementation of general methods for management and processing.
In this article, we present MultiDataSet, a new R class based on Bioconductor standards developed to encapsulate multiple datasets. MultiDataSet deals with the usual difficulties of managing multiple and non-complete datasets while offering a simple way of subsetting features and selecting individuals. We describe the internal structure of MultiDataSet and illustrate its use in three examples (Additional files 1, 2 and 3) which cover three common situations in integration analyses.
Design and implementation
MultiDataSet is a S4 class of R implemented under Bioconductor guidelines [39]. Its structure is an extension of the abstract eSet class. MultiDataSet is therefore a data-storage class that comprises datasets of different omic data (assay data), feature data and phenotypic data. Despite its general form, MultiDataSet maintains the specific characteristics of the datasets (e.g. it preserves matrices of calls and probabilities of a SnpSet).
Internal structure of MultiDataSet
MultiDataSet comprises five fields that are R standard lists. Their names match other Bioconductor classes: assayData that contains the measurement values; phenoData that stores the description of the samples; featureData and rowRanges that have the description of the features; and return_method that allows recovering the original dataset. Relation between fields is shown in Fig. 1. In each dataset, samples are shared between assayData and phenoData, and features between assayData, featureData and rowRanges. We have programmed a function to recover the original datasets. The class is designed such that the different data is coordinated. A particular feature of MultiDataSet is the storing of datasets from different experiments that may not share the full set of samples between them.
Fig. 1.
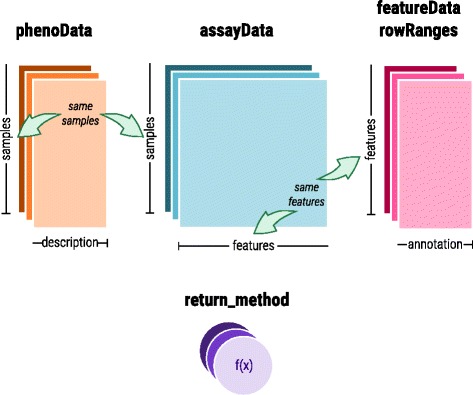
This schema shows how the information is stored in the five attributes of a MultiDataSet and how the different parts are linked. phenoData and assayData share the dimension corresponding to samples. featureData, rowRanges and assayData share the dimension corresponding to features. All the attributes are linked through the data-sets name
Six accesors are available to retrieve information from each MultiDataSet’s fields: assayData, pData, fData, rowRanges, rowRangesElements and sampleNames. The first four retrieve the content of assayData (a list of environments), phenoData (a list of AnnotatedDataFrames), featureData (a list of AnnotatedDataFrames) and rowRanges (a list of GenomicRanges with NAs for the datasets with features without genomic coordinates). rowRangesElements returns the names of datasets with a genomic coordinates in a GenomicRanges. The accessor sampleNames returns a named list with the samples names of each data set.
Adding datasets to MultiDataSet
Following Bioconductor guidelines, MultiDataSet objects are created empty through its constructor. Once the object is created, datasets can be added with add_eset and add_rse. The first function adds an object of class eSet while the second adds a SummarizedExperiment object and its extensions. The two functions have the same arguments: the MultiDataSet object, the dataset to be added, a tag for the type of dataset (i.e. methylation, expression…) and a name for each dataset. MultiDataSet thus allows the storage of multiple dataset of the same type, under different names. For features with genomic coordinates, a GenomicRanges object is created from the dataset’s featureData. In order to maintain the consistency across all datasets, the names of the samples are given by those in the phenotype dataset (a column called “id” is requested). If not present, object’s sampleNames are used.
MultiDataSet package incorporates three specific functions to include specific omic data sets: ExpressionSet (Biobase package), MethylationSet (MultiDataSet package) and SnpSet (Biobase package). These specific functions call general functions to add the data after performing extensive or specific checks (e.g. checking the class of the set or checking fData’s columns). As a result, only datasets with defined features can be introduced to MultiDataSet through a specific function.
Specific functions should always be used by users to ensure that the sets are properly added to MultiDataSet. The two basic functions add_eset and add_rse are intended to be used only by developers to develop new specific functions. The hierarchy between the specific and basic functions is shown in Fig. 2.
Fig. 2.
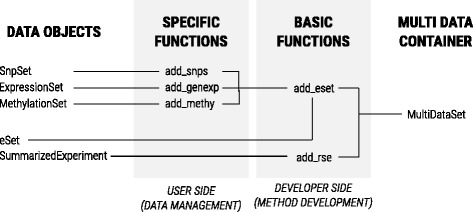
This figure represents the organization of the specific and basic functions to add datasets to a MultiDataSet object. The basic functions, that receive generic eSet and SummarizedExperiment objects, directly interact with the MultiDataSet objects and developers should use them to extend the functionality of MultiDataSet. Specific functions receive more specific datasets and interact with the MultiDataSet object through basic functions. They check the structure of the dataset and users should use them
Subsetting MultiDataSet
We have implemented two methods to perform subsetting. The operator ‘[’ can be used to select individuals, datasets and/or features. In the case of having tables with different samples, subsetting is performed by considering the union of samples from the different tables. For instance, let us assume that table 1 contains individuals A and B. Table 2 has individuals A, B and C, and Table 3 is having individuals A, C. Let us also assume that we are interested in getting information from tables 1, 2 and 3 for individuals A and C. Our subsetting method will return a MultiDataSet object containing individuals A for table 1 and individuals A and C for tables 2 and 3. We think that this procedure is better than returning a MultiDataSet object only having individual A (i.e. intersection) for the three tables. Therefore, subsetting by individuals may not return complete cases. Notice that the package has another function (commonSamples) that can be applied to this object to get complete cases if necessary.
When subsetting by datasets, if only one is selected, the original dataset is returned (aka. SnpSet, MethylationSet…). GenomicRanges object can be used as an argument to select the features present in a given genomic range. In this process, sets with no genomic coordinates (e.g. metabolomic data) are discarded. We extended the subsetting function of R (subset) to select specific features within a dataset, such as features associated to a gene or filtering individuals given a phenotype.
Results
We illustrate three common situations where MultiDataSet is useful. The examples are used to show package strengths and advantages. The first example shows how to use MultiDataSet to perform two integration analyses with already existing tools (R packages omicade4 and iClusterPlus). Second example shows how to develop a function to integrate gene expression and methylation data. Third example illustrates how to incorporate a new type of dataset that is not available in MultiDataSet to store proteome data including limit of detection.
Using MultiDataSet with third party R packages
In the first example, we show how to perform an integration analysis with three datasets using two approaches: 1) multivariate co-inertia analysis and 2) clustering of multiples tables. These methods are implemented in the R packages omicade4 and iClusterPlus (see code in Additional file 1). For both analyses, we integrate three datasets stored in a single MultiDataSet object. We use two gene expression datasets and one methylation dataset. The analysis includes three steps. First, the different datasets are added to a MultiDataSet through specific functions. Second, a MultiDataSet method is used to easily select those samples present in all the datasets. Finally, a wrapper is used to pass the MultiDataSet object to mcia function (from omicade4). This process is also done for the clustering analysis of iClusterPlus. Both wrappers, for mcia and iClusterPlus, are included in MultiDataSet package. We also provide the code of the wrappers to encourage developers to create their own.
Integration of methylation and expression using MultiDataSet
In the second example, we show how to develop a function to implement a new method to integrate two types of data. We describe the different parts of correlationMethExprs, a function from MEAL package [40] that performs an integrative analysis of methylation and expression data. The function has four main steps: 1) checking of data, 2) preparation of data structures, 3) implementation of the integration algorithm and 4) formatting of the results. Checking the correct format of original datasets is performed whenever the sets are added to the MultiDataSet, so an integration function only needs to check the global structure of the MultiDataSet. MultiDataSet’s methods facilitate the preparation of data structures and the recovery of original data in Bioconductor classes for the use of any Bioconductor’s methods. In addition, we illustrate common steps of an integration analysis that are easily performed thanks to commonSamples joint to the list conversion (as.list).
Adding a new type of data to MultiDataSet objects
In the third example, we show how to create a new specific function to deal with a type of data that is not natively supported in MultiDataSet package (see Additional file 3). We also cover how to implement checks of the object’s structure inside the function to ensure MultiDataSet’s consistency. This is illustrated by extending an eSet object to include proteins’ data. Detectors used in multiplex protein quantification work inside the dynamic range of protein concentrations. However, real protein concentrations may be outside these ranges so values outside the limit of detection (LOD) are generated. To address this issue, we introduce a new function to load protein data and to modify values out of the LOD. Then, we implement ProteomeSet, an eSet extension, to include the original data, the transformed data and the description of the limit of detection of each protein. Finally, we develop a new specific function to add ProteomeSets and to check that it contains all the components required with the right names. MultiDataSet can thus be extended to potentially contain any data type.
Discussion
The reduction of costs of microarray technologies and next generation sequencing has promoted the expansion of projects that collect different type of biological data on individuals. Consequently, it is useful to have a standard procedure to combine the information from different sources for further processing and analysis. We have developed MultiDataSet as a package that helps to storage different datasets, including common Bioconductor’s classes. MultiDataSet can also be extended to store novel data types.
MultiDataSet is implemented so it runs extensive checks on the datasets being integrated. It checks for the consistency within and across datasets. In addition, regular operations like selecting complete cases or subsetting are included in the package. MultiDataSet design facilitates data validation, as each kind of omic data is introduced using a specific function. These specific functions include the dataset’s type in the objects’ name, thereby standardizing MultiDataSet objects. Integration functions thus require fewer checks because many are done in the previous steps (see Additional files 1, 2 and 3). In addition, the checking can be modified by re-implementing a specific function, without altering the structure of the package.
MultiDataSet is designed to manage multiple datasets while preserving their original characteristics. Thus, the assay data, the phenotypic data and the feature data are stored independently for each dataset. In the case of the phenotypic data, we prefer this approach by several reasons. In omic datasets, the phenotypic data also stores technical information. For instance, if we were interested in encapsulating two MethylationSets, we will have technical variables with the same name but different information. Storing this information in one pheno data will lead to rename both columns to keep the information. In studies with longitudinal data, some phenotypic variables of the individuals change in the datasets. As an example, if we had two ExpressionSets, one obtained at age 4 and the other at age 18, the variable age will have very different values in each dataset. Finally, we decided to keep the pheno data separately to allow the user recovering the original datasets. Consequently, users can create their MultiDataSet with different datasets, perform some subsetting operations (e.g. selecting complete cases) and then recover the objects with the original classes that can be used to perform downstream analysis.
MultiDataSet performs common subsetting operations as well as managing experiments with multiple omic sources. It contains functions to select the individuals with information in all datasets or those features of each dataset that are inside a region of the genome. Moreover, MultiDataSet can also perform other complex subsetting operations, such as selecting subjects with specific phenotypes or selecting features belonging to a gene, which can be of great help in candidate gene studies. In addition, MultiDataSet easily recovers the original dataset for the use with native Bioconductor functions.
A Bioconductor group interested on combinable experiments has started a repository (https://github.com/vjcitn/MultiAssayExperiment). This group aims to use R/Bioconductor to define interfaces allowing efficient selection and combination of high-dimensional assays. However, it seems that the project focuses on TCGA. The package, called MultiAssayExperiment, shares similar capabilities for managing data, specially related to subsetting. Nevertheless, addition of sets is not standardized, lacking some of the advantages that we mentioned in the previous sections. Overall, we think that MultiDataSet is a strong candidate to become a standard class in integration of multiple omic data sets since it is already available in Bioconductor (https://bioconductor.org/packages/release/bioc/html/MultiDataSet.html), while MultiAssayExperiment is still under development. Another disadvantage is that MultiAssayExperiment is being developed by a large number of contributors making package improvements slow and tedious.
One of the limitations of MultiDataSet is that the memory management is not optimized as its uses straightforward extensions of Bioconductor’s classes. As datasets grow in size, further developments on the storage need to be devised. We have used MultiDataSet to encapsulate two MethylationSets having 450 K CpGs, on SnpSet with 800,000 SNPS and an ExpressionSet having 70,000 probes without problems. The other limitation is that, nowadays, MultiDataSet can only be used with three integration pipelines: by mcia (omicade4) and iClusterPlus through a wrapper and as input object for MEAL package.
Currently, we have two lines of work to increase the usage of our package. On one hand, we are contacting integration methods developers to allow using MultiDataSet objects as input in their pipelines. On the other hand, we are developing a new function to download data from GEO and include them in a MultiDataSet object.
Conclusion
MultiDataSet is a new R class designed to manage data from different omics experiment in the same individuals. It includes methods to perform typical operations, such as subsetting or selecting common samples, making integrative analysis more accessible to users with a low expertise in bioinformatics. In addition, its implementation eases standardization of multiple omic sets, which will also ease the development of new integration functions. Altogether, MultiDataSet is a suitable standard for data-storage in integration analysis done under R and Bioconductor framework.
Availability and requirements
Project name: MultiDataSet
Project home page: https://bioconductor.org/packages/release/bioc/html/MultiDataSet.html
Operating Systems: Windows, UNIX and MAC;
Programming Language: R
Other requirements: none
License: MIT.
Acknowledgements
The authors want to thank Alejandro Cáceres for carefully reading the last version of the manuscript.
Funding
This work has been partly funded by the Spanish Ministry of Economy and Competitiveness (MTM2015-68140-R). CH-F was supported by a grant from European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no308333 – the HELIX project. CR-A was supported by a FI fellowship from Catalan Government (#016FI_B 00272). The funding body had no role in the design of the study, the collection, analysis, and interpretation of data or in writing the manuscript.
Availability of data and materials
MultiDataSet is freely available in Bioconductor since version 3.3 (https://bioconductor.org/packages/release/bioc/html/MultiDataSet.html). Synthetic datasets used in Additional files 1 and 3 have been simulated for illustrative purposes. They can be obtained upon request. Data used in Additional file 2 was obtained from MEALData package which is available at Bioconductor: https://bioconductor.org/packages/release/data/experiment/html/MEALData.html.
Authors’ contributions
CH-F conceived the original idea of MultiDataSet. CH-F and CR-A developed the R package. CR-A and AB-G contributed improving the R packages. CH-F, CR-A and JRG wrote the manuscript. JRG supervised the project. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- DGVs
Database of genomic variants archive
- GEO
Gene expression omnibus
- ICGC
The International Cancer Genome Consortium
- LOD
Limit of detection
- SNP
Single nucleotide polymorphism
- TCGA
The cancer genome atlas
Additional files
“Using MultiDataSet with third party R packages”. This file illustrates how to perform an integration analysis using multivariate co-inertia analysis (omicade4) and clustering of multiples tables (iClusterPlus). (ZIP 38 kb)
“Integration of methylation and expression using MultiDataSet”. This file is a tutorial of how to develop a function to implement a new method using MultiDataSet capabilities. (HTML 78 kb)
“Adding a new type of data to MultiDataSet objects”. This file exemplifies how to create a method to incorporate data from S4 classes to a MultiDataSet object. (HTML 66 kb)
Contributor Information
Carles Hernandez-Ferrer, Email: carles.hernandez@isglobal.org.
Carlos Ruiz-Arenas, Email: carlos.ruiz@isglobal.org.
Alba Beltran-Gomila, Email: alba.beltran@isglobal.org.
Juan R. González, Email: juanr.gonzalez@isglobal.org
References
- 1.Pineda S, Gomez-Rubio PA, et al. Framework for the integration of genomics, epigenomics and transcriptomics in complex diseases. Hum Hered. 2015 doi: 10.1159/000381184. [DOI] [PubMed] [Google Scholar]
- 2.Suravajhala P, Kogelman L, Kadarmideen H. Multi-omic data integration and analysis using systems genomics approaches: methods and applications in animal production, health and welfare. Genet Sel Evol. 2016 doi: 10.1186/s12711-016-0217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentleman R, Carey V, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004 doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huber W, Carey V, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015 doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kannan L, Ramos M, et al. Public data and open source tools for multi-assay genomic investigation of disease. Brief Bioinform. 2015 doi: 10.1093/bib/bbv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Cancer Genome Atlas Research Network The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013 doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Cancer Genome Atlas. http://cancergenome.nih.gov/; Web Page (last visit: 2016/05/09).
- 8.The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015 doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.IGSR: The International Genome Sample Resource (Providing ongoing support for the 1000 Genomes Project data). http://www.1000genomes.org/; Web Page (last visit: 2016/05/09).
- 10.The International Cancer Genome Consortium International network of cancer genome projects. Nature. 2010 [Google Scholar]
- 11.International Cancer Genome Consortium. https://icgc.org/; Web Page (last visit: 2016/05/09).
- 12.Edgar R, Domrachev M, Lash A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002 doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett T, Wilhite S, et al. NCBI GEO: archive for functional genomics data sets – update. Nucleic Acids Res. 2013 doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gene Expression Omnibus. http://www.ncbi.nlm.nih.gov/geo/; Web Page (last visit: 2016/05/09).
- 15.Sherry S, Ward M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001 doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.dbSNP short genetic variations. http://www.ncbi.nlm.nih.gov/SNP/; Web Page (last visit: 2016/05/09).
- 17.MacDonald J, Ziman R, et al. The database of genomic variants: a curated collection of structural variation in the human genome. Nucl Acids Res. 2013 doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Database of Genomic Variants archive. http://www.ebi.ac.uk/dgva; Web Page (last visit: 2016/05/09).
- 19.Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007 doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 20.Witten D, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009 doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witten D, Tibshirani R. Extensions of sparse canonical correlation analysis, with applications to genomic data. Stat Appl Genet Mol Biol. 2009 doi: 10.2202/1544-6115.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witten W. PMA R package. CRAN; https://cran.r-project.org/web/packages/PMA/index.html.
- 23.Lé Cao K-A, González I, Déjean S. intergrOmics: an R package to unravel relationships between two omics data sets. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González I, Lé Cao K-A, Déjean S. mixOmics: Omics Data Integration Project. 2011; URL: http://www.mixomics.org; Web Page (last visit: 2016/05/09).
- 25.Lé Cao K-A, Gonzalez I, et al. mixOmics R package. CRAN; https://cran.r-project.org/web/packages/mixOmics/index.html.
- 26.Culhane A, Thioulouse J. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005 doi: 10.1093/bioinformatics/bti394. [DOI] [PubMed] [Google Scholar]
- 27.Culhane A. made4 R package. Bioconductor 3.3; https://www.bioconductor.org/packages/release/bioc/html/made4.html
- 28.Tenenhaus A, Tenenhaus M. Regularized generalized canonical correlation analysis for multiblock or multigroup data analysis. Eur J Oper Res. 2014 [Google Scholar]
- 29.Tenenhaus A, Guillemot V. RGCCA R package. CRAN; https://cran.r-project.org/web/packages/RGCCA/index.html
- 30.Meng C, Kuster B, et al. A multivariate approach to the integration of multi-omics datasets. BMC Bioinformatics. 2014 doi: 10.1186/1471-2105-15-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng C. omicade4 R package. Bioconductor 3.3; https://www.bioconductor.org/packages/release/bioc/html/omicade4.html.
- 32.Louhimo R, Hautaniemi S. CNAmet: an R package for integrating copy number, methylation and expression data. Bioinformatics. 2011 doi: 10.1093/bioinformatics/btr019. [DOI] [PubMed] [Google Scholar]
- 33.CNAmet R package. Systems Biology Laboratory, University of Helsinki; http://csbi.ltdk.helsinki.fi/CNAmet/.
- 34.Tyekucheva S, Marchionni L, et al. Integrating diverse genomic data using gene sets. Genome Biol. 2011 doi: 10.1186/gb-2011-12-10-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchionni L, Tyekucheva S. RTopper R package. Bioconductor 3.3; https://bioconductor.org/packages/release/bioc/html/RTopper.html.
- 36.Shen R, Wang S, Mo Q. Sparse integrative clustering of multiple omics data sets. The Annals of Applied Statistics. 2013 doi: 10.1214/12-AOAS578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mo Q, Shen R. iClusterPlus R package. Bioconductor 3.3; https://www.bioconductor.org/packages/release/bioc/html/iClusterPlus.html.
- 38.Gomez-Cabrero D, Sebastián-León P, Ball G. STATegRa R package. Bioconductor 3.3; http://bioconductor.org/packages/release/bioc/html/STATegRa.html.
- 39.Bioconductor. Package Guidelines. URL: http://bioconductor.org/developers/package-guidelines/; Web Page (last visit: 2016/05/27).
- 40.Ruiz-Arenas C, Hernandez-Ferrer C, González JR. MEAL R package. Bioconductor 3.3. https://bioconductor.org/packages/release/bioc/html/MEAL.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
MultiDataSet is freely available in Bioconductor since version 3.3 (https://bioconductor.org/packages/release/bioc/html/MultiDataSet.html). Synthetic datasets used in Additional files 1 and 3 have been simulated for illustrative purposes. They can be obtained upon request. Data used in Additional file 2 was obtained from MEALData package which is available at Bioconductor: https://bioconductor.org/packages/release/data/experiment/html/MEALData.html.