

Abstract
Background
Activated hepatic stellate cell (HSC) is the main fibrogenic cell type in the injured liver. miRNA plays an important role in activation and proliferation of HSC.
Methods
Our previous study examined the expression profiles of microRNAs in quiescent and activated HSC. Real-time PCR and western blot were used to detect the expression of Collagen type I (Col 1) and Alpha-Smooth Muscle Actin (α-SMA). CCK-8 and Edu assay was used to measure the proliferation rate of HSC. Luciferase reporter gene assay was used to tested the binding between miR-338-3p and Cyclin-dependent kinase 4 (CDK4).
Results
We found overexpression of miR-338-3p could inhibit Col 1 and α-SMA, two major HSC activation markers, whereas miR-338-3p inhibitor could promote them. Besides, miR-338-3p overexpression could suppress the growth rate of HSC. Further, we found that CDK4, a pleiotropic signaling protein, was a direct target gene of miR-338-3p. Moreover, we found that overexpression of CDK4 could block the effects of miR-338-3p.
Conclusions
We found miR-338-3p is an anti-fibrotic miRNA which inhibits cell activation and proliferation. Our findings suggest that miR-338-3p/CDK4 signaling pathway participates in the regulation of HSC activation and growth and may act as a novel target for further anti-fibrotic therapy.
Keywords: Liver fibrosis, miR-338, CDK4
Background
Liver fibrosis is a common consequence of most chronic liver diseases [1]. Liver fibrosis received more attention until the hepatic stellate cell (HSC) was identified as the main ECM-producing cells in the injured liver [2]. Under the normal physiologic condition, HSCs reside in the space of disse and store a large amount of vitamin A. After suffering from liver injury, HSCs will be activated, and then proliferate, eventually transdifferentiate into myofibroblast-like cells [3].
microRNAs (miRNAs), short (~22 nt) conceding RNA molecules, can directly regulate gene expression by binding to the 3’UTR region of target mRNA to participate in lots of regulation of physiological process and diseases [4–8]. Recently, researchers focused on the role of miRNA in liver fibrosis pathophysiology to determine their regulatory effects on proliferation, differentiation of HSC [9–11]. Several abnormally expressed miRNAs were found and identified between quiescent and activated HSCs by using miRNA array or RT-PCR [12–19]. Previous studies have reported that miRNAs were critically involved in the activation of HSCs. Among them, miR-29b precursor allowed activated HSCs to switch to a more quiescent state [10]. Similarly, overexpression of miR-27a/b could lead HSCs to a quiescent phenotype [20]. microRNA-338 (miR-338), a newly identified miRNA, played a crucial role in a variety of carcinomas. Aberrant expression of miR-338 was closely related to cell proliferation, invasion, early detection and clinic pathologic variables in liver cancer, colorectal cancer, gastric cancer and neuroblastoma [21–25]. In previous research, our miRNA microarray data have found altered expression of miR-338-3p during culture activation of HSC [14]. However, little is known about the role of miR-338-3p in liver fibrosis.
Cyclin-dependent kinase 4 (CDK4) is found to be involved in cell cycle regulation. Activation of cyclin D—CDK4 promotes the cell cycle progression through G1/S transition [26]. Inhibition of CDK4 shows promising efficacy on advanced breast cancer [27]. In liver tissue and hepatoma cells, CDK4/6 inhibition is a potent mediator of cytostasis [28]. However, whether the CDK4 participates in the fibrogenic process and regulates HSC activation and proliferation remains largely unknown. In this study, RT-PCR data suggested that miR-338-3p expression in fully activated HSCs were significantly decreased compared with that in quiescent HSCs. Transforming growth factor (TGF-β) is deemed to be the most potent fibrogenic cytokine. The results showed that there was a negative relationship between TGF-β and miR-338-3p. Therefore, we speculated that miR-338-3p was closely associated with HSCs function. Then, we found overexpression of miR-338-3p could suppress HSCs activation and proliferation while inhibition of miR-338-3p could promote HSCs activation and proliferation. Based on the Bioinformatics prediction, we found that CDK4 was a potential target gene of miR-338-3p. Further luciferase reporter assay and RT-PCR confirmed their complementary binding. Moreover, our results indicated that overexpression of CDK4 could partially block miR-338-3p-inhibited cell activation and proliferation.
Methods
Primary rat HSCs, cell lines and culture
The isolation method of primary rat HSCs was according to the previous literature [29]. Primary Rat HSCs, HSC-T6 and HEK293T (human embryonic kidney cell line) were kindly gifted from Dr. Gao (Tongji University, Shanghai, China). The primary cells and cell lines were cultured in DMEM (Dulbecco’s modified Eagle’s medium, Thermo, Waltham, MA, USA) containing 10% FBS (Fetal bovine serum, FBS, Gibco, Grand Island, NY, USA) at 37 °C in a humidified atmosphere of 5% CO2.
Plasmid construction
Wild-type 3’UTR containing predicted miR-338-3p binding sites were amplified from HSC-T6 genomic DNA and inserted into the PGL3 luciferase reporter vector. Mutant 3’UTR was generated using the Quick Change Lighting Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). The CDK4 expression vector was obtained by cloning the CDK4-coding sequence into the pcDNA.
RT-PCR analysis
Total RNA was extracted from cultured HSCs using Trizol Reagent (Takara, Dalian, China). The primer sequences used for mRNA detection in this study were listed as follows: GAPDH (PF: CAGTGCCAGCCTCGTCTCAT, PR: AGGGGCCATCCACAGTCTTC); ColI (PF: ATCCTGCCGATGTCGCTAT, PR: CCACAAGCGTGCTGTAGGT); α-SMA (PF: CCGAGATCTCACCGACTACC, PR: TCCAGAGCGACATAGCACAG); CDK4 (PF: GAAGAAGAAGCGGAGGAAGAGG, PR: TTAGGTTAGTGCGGGAATGAAT).
CCK-8 assay and Edu assay
Cell proliferation was performed using CCK-8 assay (Dojindo, Japan) and Edu (Ribibio, Guangzhou, China) assay. For CCK-8, HSC-T6 was transfected with the miR-338 precursor, miR-338 inhibitor (Ribibio, Guangzhou, China) or pcDNA-CDK4 in 96 well culture plates. Proliferation rates were tested at 24, 48 and 72 h after transfection. The EdU assay was conducted according to the protocol of Ribibio Edu Kit.
Luciferase assay
Luciferase assay was performed with the Dual Luciferase Reporter Assay System (Promega, Madison, WI). Transfection was carried out in 48 well plates using Fugen (Roche). There were two groups. One was co-transfected with 200 ng wild-type-CDK4-3’UTR, 20 nm miR-338 precursor, and 20 ng Renilla. Another was co-transfected with 200mutant CDK4 3’UTR without binding site of miR-338-3p, 20 nm miR-338 precursor, and 20 ng Renilla. 48 h later, Firefly and Renilla luciferase activities were tested.
Western blotting
Cells were lysed in SDS sample buffer. Antibodies against GAPDH (Biogot, 1:5000 dilution, Nanjing, China), Col1 (Abcam, 1:1500 dilution, Cambridge, MA, USA), α-sma (Sigma, 1:1000 dilution, Shanghai, China) and CDK4 (Biogot, 1:3000 dilution, Nanjing, China) were used in this study. Signals were visualized with ImageQuant LAS 4000 (GE Healthcare Life Sciences).
Statistical analysis
The statistical analysis in our study was performed by using SPSS 22.0. Data were given as mean ± SEM. Two tailed t-test was used to determine between two groups. Statistical significance level was set at p < 0.05.
Results
miR-338-3p was downregulated in fully activated HSCs
Based on the microarray data, multitude ectopic miRNAs were detected in the HSCs. In our previous study, we isolated rat primary HSCs and extracted total RNA to perform miRNA microarray assay. Our attention was focused on miR-338-3p, a new underlying member of liver fibrosis. The microarray data showed that the expression of miR-338-3p was sharply reduced by 90% at day 7 (partially activated HSCs) [14]. To validate this finding, RT-PCR was carried out to measure the endogenous miR-338-3p expression in a quiescent state and an activated state. As primary HSCs were gradually activated during culture, we assessed miR-338-3p expression at day 2, day 7 and day 14 after isolation. Our data indicated that endogenous miR-338-3p expression was obviously reduced at day 7 and day 14 (Fig. 1a). Meanwhile, collagen type I (Col1) and α-sma (α-smooth muscle actin, α-SMA), two key biomarkers of HSCs activation, was gradually increased (Fig. 1b, c). In addition, we found that treatment with transforming growth factor (TGF-β, 2 ng/ml) in quiescent HSCs (day 2), the expression of miR-338-3p was reduced rapidly (Fig. 1d). When cells were fully activated (day 14), we treated them with SB431542, a potent and specific inhibitor of TGF-β and detected the level of miR-338-3p. The results suggested that the expression of miR-338-3p was increased compared to that in control group (Fig. 1e).
Fig. 1.
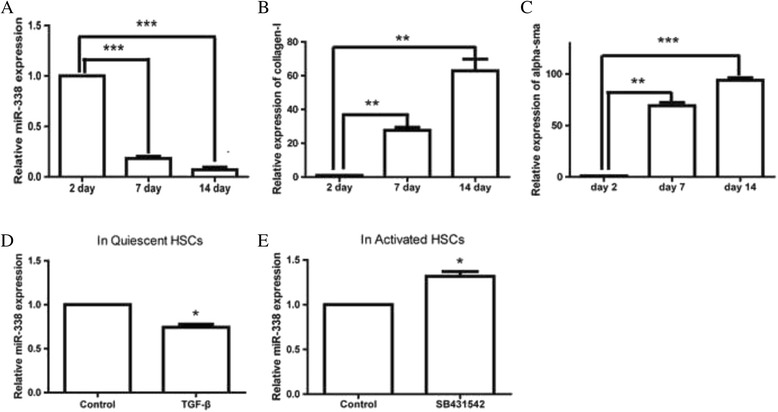
Expression of miR-338-3p is reduced in HSCs during culture activation. a miR-338-3p expression in the HSCs during culture activation. Data shown are means ± SD (n = 3), ***P < 0.001. b mRNA level of Col1 in the HSCs during cell culture. Data shown are means ± SD (n = 3), **P < 0.01. c mRNA level of α-sma in the HSCs during cell culture. Data shown are means ± SD (n = 3), **P < 0.01, ***P < 0.001. d miR-338-3p expression of quiescent HSCs was reduced upon TGF-β treatment. Data shown are means ± SD (n = 3), *P < 0.05. e miR-338-3p expression of activated HSCs was increased upon SB431542 treatment. Data shown are means ± SD (n = 3), *P < 0.05
Overexpression of miR-338-3p suppressed HSC activation while inhibition of it promoted HSC activation
As we discovered, miR-338-3p was significantly decreased during the process of activation, we transfected miR-338 precursor to repair its loss at the early stage of primary HSCs. On day 7, cells transfected with miR-338 precursor showed a more original shape with less peripheral protrusions. However, the control group cells showed a more irregular shape with more peripheral protrusions (Fig. 2a). As results showed in Figs. 1b, c and 2a, it seemed that there was a negative relationship between miR-338 expression and HSCs activation. We assumed that the expression of miR-338-3p was involved in this activation process. To confirm our hypothesis, we alter the miR-338-3p expression in primary HSC by transfecting miR-338 precursor. Interestingly, the expression of Col 1 and α-sma was slightly decreased due to the upregulation of miR-338-3p (Fig. 2b). Due to the low efficiency of transfection in primary cells, HSC-T6 cell line was further used to conduct the following studies. To replicate the results, HSC-T6 cell line was transfected with miR-338 precursor or negative control. 48 h later, cells were collected and transfection efficiency was confirmed by RT-PCR (Fig. 2c). Then the expression of Col1 and α-sma in two groups was measured using RT-PCR. As expected, the data showed miR-338-3p inhibited HSCs activation. The expression of Col1 and α-sma was respectively reduced as a result of miR-338-3p overexpression (Fig. 2d, e). Furthermore, inhibition of miR-338-3p could upregulate Col1 and α-sma which confirm their association on the other direction (Fig. 2f, g). Their expression was also confirmed at the protein level (Fig. 2h).
Fig. 2.
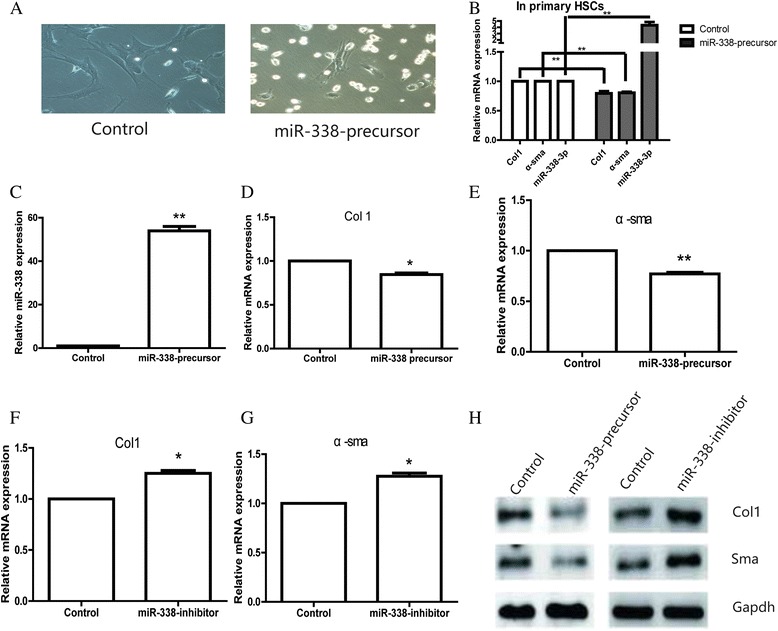
Overexpression of miR-338-3p could inhibit cell activation, whereas inhibition of miR-338 could promote cell activation. a The cell morphology of HSCs after transfecting with miR-338 precursor or negative control. b The expression of miR-338, Col1, and α-SMA in primary HSCs was tested after miR-338-precursor transfection. c miR-338-3p transfection efficiency was confirmed by qRT-PCR. Data shown are means ± SD (n = 3), **P < 0.01. d mRNA level of Col1 in the HSCs transfected with miR-338 precursor or negative control. Data shown are means ± SD (n = 3), *P < 0.05. e mRNA level of α-sma in the HSCs transfected with miR-338 precursor or negative control. Data shown are means ± SD (n = 3), **P < 0.01. f mRNA level of Col1 in the HSCs transfected with miR-338-3p inhibitor or negative control. Data shown are means ± SD (n = 3), *P < 0.05. g mRNA level of α-sma in the HSCs transfected with miR-338-3p inhibitor or negative control. Data shown are means ± SD (n = 3), *P < 0.05. h Protein level of Col1 and sma by western blot
Overexpression of miR-338 suppressed HSC-T6 proliferation
Next, we examined the impacts of miR-338 on HSCs proliferation. HSC-T6 were transfected with miR-338 precursor, inhibitor or corresponding negative control. In the CCK-8 assay, the cell growth curves suggested that overexpression of miR-338 significantly restrained HSC-T6 proliferation in a time-dependent manner (Fig. 3a.). Moreover, cells transfected with miR-338 inhibitor showed a higher proliferative ability (Fig. 3b). Edu incorporation assay also demonstrated that miR-338 precursor reduced the proliferation of HSC-T6 (Fig. 3c-d). Besides proliferation, we also assessed the role of miR-338-3p in HSCs migration. As Fig. 3e, f showed, miR-338-3p has no effects on cell migration.
Fig. 3.
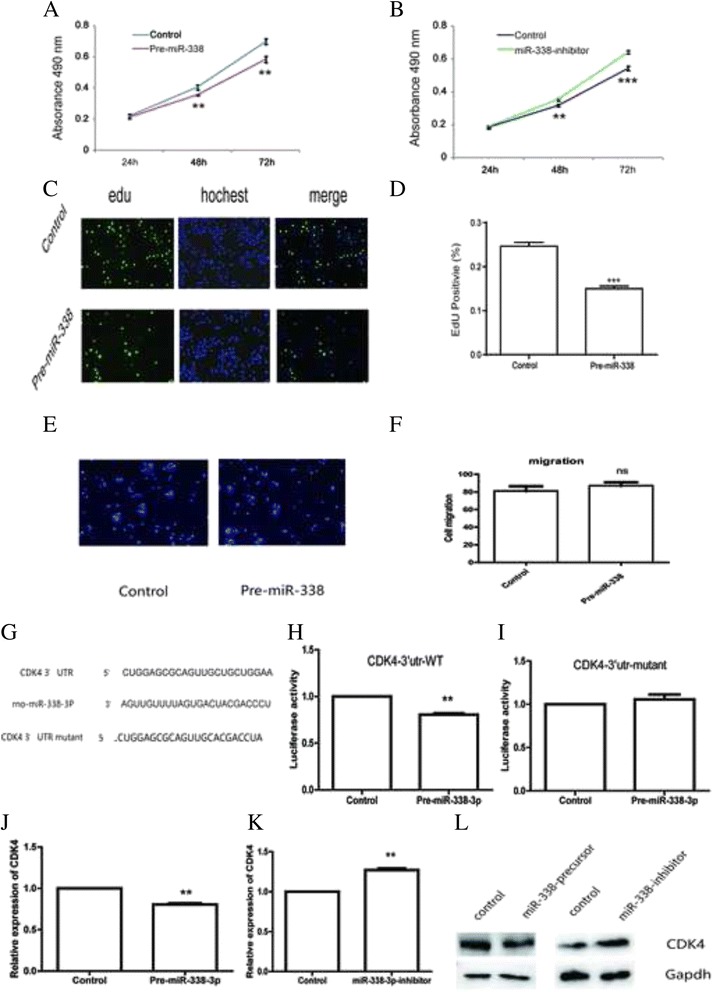
miR-338-3p regulates cell proliferation and CDK4. a The proliferation analysis of HSC-T6 transfected with miR-338 precursor or negative control. Data shown are means ± SD (n = 3). **P < 0.01 versus the corresponding control. b The proliferation analysis of HSC-T6 transfected with miR-338 inhibitor or negative control. Data shown are means ± SD (n = 3). **P < 0.01 versus the corresponding control. c Micrograph of HSC-T6 transfected with miR-338 precursor. d Edu incorporation assay demonstrated that miR-338 precursor reduced the proliferation of HSC-T6. Data shown are means ± SD (n = 3). ***P < 0.001. e, f HSCs migration was measured by transwell assay. g The predicted sequence of binding region between miR-338-3p and CDK4. h Luciferase activity of CDK4 3’UTR WT reporter vector co-transfected with miR-338-3p. Data shown are means ± SD (n = 3). **P < 0.01. i Luciferase activity of CDK4 3’UTR mutant reporter vector co-transfected with miR-338-3p. Data shown are means ± SD (n = 3). j mRNA level of CDK4 in the HSCs transfected with Pre-miR-338-3p or negative control. Data shown are means ± SD (n = 3). **P < 0.01. k mRNA level of CDK4 in the HSCs transfected with miR-338-inhibitor or negative control. Data shown are means ± SD (n = 3). **P < 0.01. l The expression of CDK4 was restrained by miR-338-precursor, whereas recovered by miR-338 inhibitor
miR-338 repressed CDK4 expression by directly binding to its 3’UTR region
Based on the prediction of Bioinformatics (TargetScan, PicTar), some genes are predicted to be the targets of miR-338-3p. Among them, we hypothesized CDK4, an oncogene in liver cancer, might be a putative target gene of miR-338-3p in liver fibrosis. The predicted sequence of interaction was showed in Fig. 3g. To test this prediction, 3’UTR with miR-338-3p binding sites were cloned into the PGL3 luciferase reporter vector. A mutant 3’UTR of CDK4 with anti-sense mutation in the predicted sites was also constructed. The reporter construct was co-transfected into HEK293T with Renilla plasmid and miR-338 precursor or negative control. The wild-type CDK43’UTR luciferase activity was suppressed due to miR-338-3p overexpression. By contrast, the activity of mutant-3’UTR-CDK4 remained relatively unaffected (Fig. 3h, i). Additionally, CDK4 expression was decreased in the miR-338 precursor group while increased in the miR-338 inhibitor group (Fig. 3j, k). miR-338-3p could also regulate CDK4 at protein level (Fig. 4l). Taken together, these data strongly suggested that miR-338-3p repressed CDK4 expression by directly binding to its 3’UTR region.
Fig. 4.
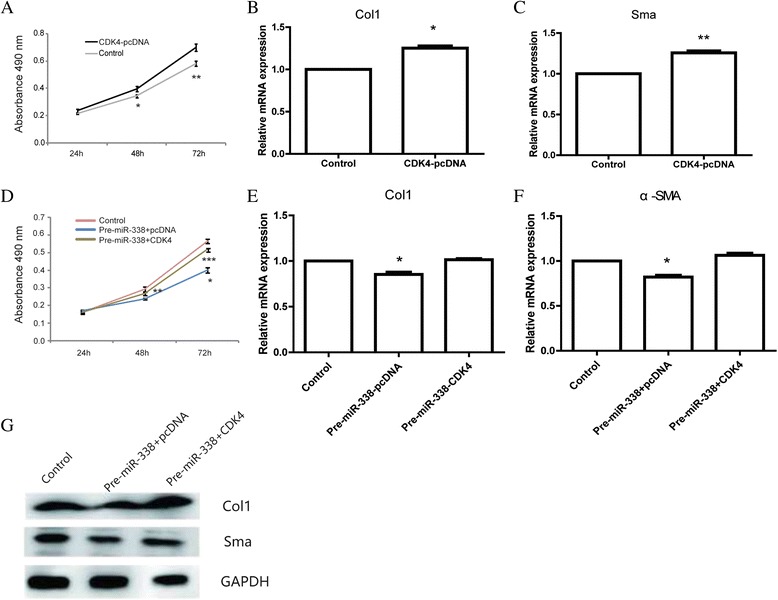
Overexpression of CDK4 could partially rescue the effects of miR-338-3p upon HSCs. a The proliferation analysis of HSC-T6 cells transfected with CDK4 vector or empty control. Data shown are means ± SD (n = 3). *P < 0.05, **P < 0.01 versus the corresponding control. b mRNA level of Col1 in the HSCs transfected with CDK4 vector or empty control. Data shown are means ± SD (n = 3), *P < 0.05. c mRNA level of α-sma in the HSCs transfected with CDK4 vector or empty control. Data shown are means ± SD (n = 3), **P < 0.01. d The proliferation analysis of HSC-T6 cells co-transfected with miR-338 precursor and CDK4 plasmid. Data shown are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. e mRNA level of Col1 in the HSC-T6 cells co-transfected with miR-338 precursor and CDK4 plasmid. Data shown are means ± SD (n = 3), *P < 0.05. f mRNA level of α-sma in the HSC-T6 cells co-transfected with miR-338 precursor and CDK4 plasmid. Data shown are means ± SD (n = 3), **P < 0.01. g Protein level of Col1, α-sma in the HSC-T6 cells co-transfected with miR-338 precursor and CDK4 plasmid
CDK4 rescued miR-338-inhibition of activation and proliferation of HSCs
Since miR-338 regulated cell growth, cell migration and cell invasion in liver cancer and colorectal carcinoma by targeting CDK4 [21, 30], the role of CDK4 in liver fibrosis remained unclear.
We conduct CCK-8 assay to determine the proliferation of HSC-T6 transfected with CDK4 plasmid or control empty vector. The results showed that cell transfected with CDK4 plasmid displayed a higher proliferative capacity compared with the control group (Fig. 4a). Besides, we found cells transfected with CDK4 could promote cell activation with upregulation of Col1 and α-Sma (Fig. 4b, c). We co-transfected HSC-T6 cells with miR-338 precursor and CDK4 plasmid to investigate whether CDK4 would rescue the inhibition effect of miR-338 on the cell activation and proliferation or not. The results suggested that overexpression of CDK4 partially block the repression effect of miR-338 on the activation and proliferation of HSC-T6. The growth curves of three groups (Control, Pre-miR-338/pcDNA, Pre-miR-338/CDK4) were shown in Fig. 4d. The expression of activation associated markers, Col1 andα-Sma, was showed in Fig. 4e-f.
Discussion
Liver fibrosis is a scarring response to liver damage. It’s a common pathological process for most of the liver disorder. A small number of patients go on to progress cirrhosis and/or hepatocellular carcinoma. Fortunately, liver fibrosis can be reversed if the inflammation was controlled [31].
Aberrant expression of miRNAs have been involved in liver fibrosis and regarded as a potential treatment strategy. Intervening miRNAs expression could assist activated HSCs to return to a quiescent phenotype. miRNA microarray and RT-PCR was carried out to determine abnormally expressed miRNAs during HSCs activation. Our results suggested that miR-338-3p was significantly downregulated in this process. miR-338 is located on chromosome 17q25.3 with a length of 22 nt and produces two mature forms, miR-338-3p and miR-338-5p. miR-338 was first reported in neurodegeneration and gradually studied in various disease [32]. In hepatocellular carcinoma, miR-338 downregulation was associated with tumor size, TNM stage, vascular invasion and in trahepatic metastasis [21, 22]. In colorectal carcinoma, miR-338 expression was significantly increased in both blood and tissue samples. It might appear to be a potential biomarker for early detection in colorectal carcinoma [23]. In gastric carcinoma, miR-338 was epigenetically silenced and its reduction was related to pathological variables. Overexpression of miR-338 could suppress cell proliferation, migration, invasion and tumorigenicity [24]. Moreover, combined with other six miRNAs, miR-338 could be used to predict gastric cancer prognosis [33]. Despite in cancer, miR-338 was also involved in idiopathic pulmonary fibrosis [34].
This is the first study to identify the biological function of miR-338-3p in liver fibrosis. Our results demonstrated that miR-338 precursor transfection suppressed the activation and proliferation of HSC-T6, whereas inhibition of miR-338-3p promoted cell activation and proliferation.
To understand the underlying mechanism of miR-338-mediated inhibition of proliferation, we identified CDK4, a member of the cyclin-dependent kinase family, as a candidate target gene. CDK4 usually work with Cyclin D to regulate the cell cycle in G1/S stage. Aberrant activation of CDK4 was closely associated with various kinds of carcinomas. CDK4 expression is significantly upregulated in lung cancer tissues and function as an important element for cell proliferation [35, 36]. In breast cancer, inhibition of CDK4 can induce G1 arrest [37]. These observations suggest that inhibition of CDK4 might be beneficial for cancer treatment. An increasing body of clinical trials targeting CDK4 has been launched. However, the role of CDK4 in liver fibrosis remains largely unknown. In this study, Luciferase reporter assay showed that there was a combination of miR-338 and CDK4. Hence, we deduced that miR-338-3p inhibited HSCs’ activation and proliferation likely through silencing CDK4. Our data indicated that restoring CDK4 expression could partially rescue miR-338-inhibited cell activation and proliferation.
Conclusions
In conclusion, our study identified a new anti-fibrosis miRNA that may play an important role in the development of liver fibrosis.
Acknowledgment
We thank Tongji University School of Medicine for technical help.
Funding
This work was supported by grants from National Natural Science Foundation of China [grants 81402756, 81101644, 11472300, 11272342]; Science and Technology Research Project from Guangxi Education Department (YB2014067). The funding body approved the study design and the methods that were used for data acquisition and analysis, but had no involvement with the interpretation of data or with the composition of the manuscript.
Availability of data and materials
All the data supporting the findings of the study is contained within the manuscript.
Authors’ contributions
Conceived and designed the experiments: BD, JH, HG, CC Performed the experiments: JH, BD, TZ, XL. Analyzed the data: JH, SL, CW, YZ. Wrote the paper: JH, BD, LZ, WL. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was carried out in strict accordance with the International Council for Laboratory Animal Science. All animal experiments were conducted in accordance with protocols approved by the Experimental Animal Ethical Committee of Tongji University.
ᅟ
- CDK4
Cyclin-dependent kinase 4
- Col 1
Collagen type I
- HSC
Hepatic stellate cell
- miR-338
microRNA-338
- α-SMA
Alpha-Smooth Muscle Actin
Contributor Information
Bensong Duan, Email: 9biosong_duan@tongji.edu.cn.
Jiangfeng Hu, Email: doctorhjf@foxmail.com.
Tongyangzi Zhang, Email: smile_yangzi@163.com.
Xu Luo, Email: luoxuha@qq.com.
Yi Zhou, Email: elian_zhou@163.com.
Shun Liu, Email: leeshun2010@163.com.
Liang Zhu, Email: czzhuliang@126.com.
Cheng Wu, Email: wucheng19799@126.com.
Wenxiang Liu, Email: LWX3987897@sina.com.
Chao Chen, Email: 15900611429@163.com.
Hengjun Gao, Email: hengjun_gao@tongji.edu.cn.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabele E, Brenner DA, Rippe RA. Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci. 2003;8:d69–77. doi: 10.2741/887. [DOI] [PubMed] [Google Scholar]
- 3.Marra F. Hepatic stellate cells and the regulation of liver inflammation. J Hepatol. 1999;31:1120–30. doi: 10.1016/S0168-8278(99)80327-4. [DOI] [PubMed] [Google Scholar]
- 4.Liu Q, Wang G, Chen Y, Li G, Yang D, Kang J. A miR-590/Acvr2a/Rad51b axis regulates DNA damage repair during mESC proliferation. Stem Cell Rep. 2014;3:1103–17. doi: 10.1016/j.stemcr.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan IK, Wang X, Asmann YW, Haga H, Patel T. Circulating extracellular RNA markers of liver regeneration. PLoS One. 2016;11:e0155888. doi: 10.1371/journal.pone.0155888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeliger C, Balmayor ER, van Griensven M. miRNAs related to skeletal diseases. Stem Cells Dev. 2016;25(17):1261-1281. [DOI] [PubMed]
- 7.Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–10. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–74. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 9.Yu F, Guo Y, Chen B, Dong P, Zheng J. MicroRNA-17-5p activates hepatic stellate cells through targeting of Smad7. Lab Investig. 2015;95:781–9. doi: 10.1038/labinvest.2015.58. [DOI] [PubMed] [Google Scholar]
- 10.Sekiya Y, Ogawa T, Yoshizato K, Ikeda K, Kawada N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem Biophys Res Commun. 2011;412:74–9. doi: 10.1016/j.bbrc.2011.07.041. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Tang N, Wu K, Dai W, Ye C, Shi J, Zhang J, Ning B, Zeng X, Lin Y. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS One. 2014;9:e108005. doi: 10.1371/journal.pone.0108005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–18. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 13.Kitano M, Bloomston P.M. Hepatic Stellate Cells and microRNAs in Pathogenesis of Liver Fibrosis. J Clin Med. 2016;5(3):38. doi:10.3390/jcm5030038. [DOI] [PMC free article] [PubMed]
- 14.Chen C, Wu CQ, Zhang ZQ, Yao DK, Zhu L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res. 2011;317:1714–25. doi: 10.1016/j.yexcr.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, Thannickal VJ, Cardoso WV, Lu J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:287–94. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo CJ, Pan Q, Cheng T, Jiang B, Chen GY, Li DG. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 2009;276:5163–76. doi: 10.1111/j.1742-4658.2009.07213.x. [DOI] [PubMed] [Google Scholar]
- 17.Li WQ, Chen C, Xu MD, Guo J, Li YM, Xia QM, Liu HM, He J, Yu HY, Zhu L. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J. 2011;278:1522–32. doi: 10.1111/j.1742-4658.2011.08075.x. [DOI] [PubMed] [Google Scholar]
- 18.Hu J, Chen C, Liu Q, Liu B, Song C, Zhu S, Wu C, Liu S, Yu H, Yao D, et al. The role of the miR-31/FIH1 pathway in TGF-beta-induced liver fibrosis. Clin Sci (Lond) 2015;129:305–17. doi: 10.1042/CS20140012. [DOI] [PubMed] [Google Scholar]
- 19.Wu K, Ye C, Lin L, Chu Y, Ji M, Dai W, Zeng X, Lin Y. Inhibiting miR-21 attenuates experimental hepatic fibrosis by suppressing both ERK1 pathway in HSC and hepatocyte EMT. Clin Sci. 2016;130(16):1469-1480. [DOI] [PubMed]
- 20.Ji J, Zhang J, Huang G, Qian J, Wang X, Mei S. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 2009;583:759–66. doi: 10.1016/j.febslet.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 21.Huang XH, Chen JS, Wang Q, Chen XL, Wen L, Chen LZ, Bi J, Zhang LJ, Su Q, Zeng WT. miR-338-3p suppresses invasion of liver cancer cell by targeting smoothened. J Pathol. 2011;225:463–72. doi: 10.1002/path.2877. [DOI] [PubMed] [Google Scholar]
- 22.Huang XH, Wang Q, Chen JS, Fu XH, Chen XL, Chen LZ, Li W, Bi J, Zhang LJ, Fu Q, et al. Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: miR-338 is downregulated. Hepatol Res. 2009;39:786–94. doi: 10.1111/j.1872-034X.2009.00502.x. [DOI] [PubMed] [Google Scholar]
- 23.Yong FL, Law CW, Wang CW. Potentiality of a triple microRNA classifier: miR-193a-3p, miR-23a and miR-338-5p for early detection of colorectal cancer. BMC Cancer. 2013;13:280. doi: 10.1186/1471-2407-13-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P, Chen X, Su L, Li C, Zhi Q, Yu B, Sheng H, Wang J, Feng R, Cai Q, et al. Epigenetic silencing of miR-338-3p contributes to tumorigenicity in gastric cancer by targeting SSX2IP. PLoS One. 2013;8:e66782. doi: 10.1371/journal.pone.0066782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Pan M, Han L, Lu H, Hao X, Dong Q. miR-338-3p suppresses neuroblastoma proliferation, invasion and migration through targeting PREX2a. FEBS Lett. 2013;587:3729–37. doi: 10.1016/j.febslet.2013.09.044. [DOI] [PubMed] [Google Scholar]
- 26.Jia X, Liu B, Shi X, Ye M, Zhang F, Liu H. Roles of the ERK, JNK/AP-1/cyclin D1-CDK4 pathway in silica-induced cell cycle changes in human embryo lung fibroblast cells. Cell Biol Int. 2011;35:697–704. doi: 10.1042/CBI20100298. [DOI] [PubMed] [Google Scholar]
- 27.O’Sullivan CC. CDK4/6 inhibitors for the treatment of advanced hormone receptor positive breast cancer and beyond: 2016 update. Expert Opin Pharmacother. 2016;17:1657-1667. [DOI] [PubMed]
- 28.Rivadeneira DB, Mayhew CN, Thangavel C, Sotillo E, Reed CA, Grana X, Knudsen ES. Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology. 2010;138:1920–30. doi: 10.1053/j.gastro.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riccalton-Banks L, Bhandari R, Fry J, Shakesheff KM. A simple method for the simultaneous isolation of stellate cells and hepatocytes from rat liver tissue. Mol Cell Biochem. 2003;248:97–102. doi: 10.1023/A:1024184826728. [DOI] [PubMed] [Google Scholar]
- 30.Sun K, Deng HJ, Lei ST, Dong JQ, Li GX. miRNA-338-3p suppresses cell growth of human colorectal carcinoma by targeting smoothened. World J Gastroenterol. 2013;19:2197–207. doi: 10.3748/wjg.v19.i14.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Tamayo R. Cirrhosis of the liver: a reversible disease? Pathol Annu. 1979;14(Pt 2):183–213. [PubMed] [Google Scholar]
- 32.Saba R, Goodman CD, Huzarewich RL, Robertson C, Booth SA. A miRNA signature of prion induced neurodegeneration. PLoS One. 2008;3:e3652. doi: 10.1371/journal.pone.0003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Zhang Y, Zhang Y, Ding J, Wu K, Fan D. Survival prediction of gastric cancer by a seven-microRNA signature. Gut. 2010;59:579–85. doi: 10.1136/gut.2008.175497. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Liu X, Chen S, Wu J, Ye X, Xu L, Chen H, Zhang D, Tan R, Wang Y. Tectorigenin inhibits the in vitro proliferation and enhances miR-338* expression of pulmonary fibroblasts in rats with idiopathic pulmonary fibrosis. J Ethnopharmacol. 2010;131:165–73. doi: 10.1016/j.jep.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 35.Lingfei K, Pingzhang Y, Zhengguo L, Jianhua G, Yaowu Z. A study on p16, pRb, cdk4 and cyclinD1 expression in non-small cell lung cancers. Cancer Lett. 1998;130:93–101. doi: 10.1016/S0304-3835(98)00115-3. [DOI] [PubMed] [Google Scholar]
- 36.Wu A, Wu B, Guo J, Luo W, Wu D, Yang H, Zhen Y, Yu X, Wang H, Zhou Y, et al. Elevated expression of CDK4 in lung cancer. J Transl Med. 2011;9:38. doi: 10.1186/1479-5876-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56:2973–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data supporting the findings of the study is contained within the manuscript.