

Abstract
Atherosclerosis represents a significant cause of morbidity and mortality in both the developed and developing countries. Animal models of atherosclerosis have served as valuable tools for providing insights on its aetiology, pathophysiology and complications. They can be used for invasive interrogation of physiological function and provide a platform for testing the efficacy and safety of different pharmacological therapies. Compared to studies using human subjects, animal models have the advantages of being easier to manage, with controllable diet and environmental risk factors. Moreover, pathophysiological changes can be induced either genetically or pharmacologically to study the harmful effects of these interventions. There is no single ideal animal model, as different systems are suitable for different research objectives. A good understanding of the similarities and differences to humans enables effective extrapolation of data for translational application. In this article, we will examine the different mouse models for the study and elucidation of the pathophysiological mechanisms underlying atherosclerosis. We also review recent advances in the field, such as the role of oxidative stress in promoting endoplasmic reticulum stress, mitochondrial dysfunction and mitochondrial DNA damage, which can result in vascular inflammation and atherosclerosis. Finally, novel therapeutic approaches to reduce vascular damage caused by chronic inflammation using microRNA and nano-medicine technology, are discussed.
Keywords: Mouse models, Atherosclerosis, ApoE, LDL receptor, Reactive oxygen species, ER stress, Mitochondrial dysfunction
Background
Atherosclerosis is responsible for acute myocardial infarction and cerebrovascular accidents, accounting for the majority of cardiovascular deaths. It is a chronic inflammatory disease characterized by the development of complex atherosclerotic plaques, leading to hardening and narrowing of the arterial lumen. Atherosclerotic plaque formation is initiated and sustained by a combination of endothelium dysfunction and chronic exposure to cardiovascular risk factors that promote vascular inflammation, such as hyperlipidemia, hypertension, smoking, male gender and diabetes.
Among these, the single most important risk factor is high plasma low density lipoprotein (LDL) level, of which a monogenetic cause is familial hypercholesterolemia [1]. Other risk factors are important in individuals with normal LDL levels. In the presence of a LDL level permissive of atherosclerotic plaque formation, these risk factors are important in explaining the development and progression of atherosclerotic lesions [2, 3]. Conversely, these are less important in individuals with very low LDL levels, who are unlikely to develop atherosclerosis irrespective of other risk factors [4]. With this in mind, cholesterol loading in the lesion-containing foam cells has been the focal point of intense research and has been studied extensively using different animal models. The extent of cholesterol loading is the result of lipoprotein uptake and lipid efflux in foam cells has been targeted therapeutically to reduce lesion size [5].
LDL accumulation
Excess LDL in plasma accumulate in the sub-endothelial space of arterial wall and undergoes oxidation to become oxidized LDL (oxLDL) [6]. This then triggers an inflammatory response and induces the expression of chemotactic protein such as vascular cell adhesion molecule-1 (VCAM-1), E-selectin and P-selectin in the endothelium [7, 8]. The expression of adhesion molecules attracts blood cells into the injured arterial wall, with monocytes being the most prominent cell type [9]. Upon migration into the intimal layer, monocytes differentiate into macrophages which avidly internalize oxLDL and become foam cells themselves [10].
Foam cells present antigens to immune cells, such as circulating monocytes and T-cells, whose activation contributes critically to plaque development [11]. They also secrete mediators to perpetuate the inflammatory process in the arterial wall and to stimulate migration of smooth muscle cells from the tunica media into sub-endothelial space. Mediated by platelet derived growth factor, smooth muscle cells exhibit abnormally high proliferation rates and secrete extracellular matrix proteins that contribute to fibrous cap formation [12, 13]. The fibrous cap protects the core of the plaque from circulating blood cells, especially platelets that are responsible for the thrombosis associated with plaque rupture. This maladaptive response of a non-resolving inflammation is the driving force of atherosclerotic plaque development. It is worth noting that the SMCs from different regions of the microvasculature have different developmental origins [14], which can contribute to site-specific atherosclerosis responses [15].
During plaque evolution, macrophages proliferate and undergo apoptosis and efferocytosis [16, 17]. Depending on the efficiency of efferocytosis, apoptotic cells may be removed, leading to reduction of lesion size [18], or they may accumulate and be subjected to secondary necrosis, producing a necrotic core characteristic of advanced plaques. The accumulation of apoptotic bodies will enhance the plaque instability by triggering inflammation [19, 20].
Whilst foam cells are the most abundant leukocytes within the atherosclerotic lesion, studies in mouse models have implicated other cell types, including neutrophils, T-and B-cell, and mast cells, in atherogenesis [21, 22]. These cells, though contribute little to the lesion mass, can influence atherosclerosis by secreting variety of proteins that regulate other cells or components within the plaque. Experiment studies using mice have demonstrated that among the subsets of T-cells, Th1 cells and natural killer T-cells are notably pro-atherogenic, whilst the role of Th2 and Th17 cells are less well understood [23–25].
Plaque rupture
Plaque rupture is responsible for the adverse consequences of ischemic events in acute myocardial infarction and cerebrovascular accidents and death [26]. At advanced stages of atherosclerosis, rupture of vulnerable plaques exposes their thrombogenic compounds, thereby leading to luminal thrombosis [27]. Macrophage-derived proteases, especially metalloproteases, can destabilize plaques, but the exact underlying mechanism of plaque vulnerability remained incompletely elucidated [28].
Animal models of atherosclerosis
The first key piece of evidence that atherosclerosis is inducible in laboratory animals was provided by Ignatowski in 1908, who demonstrated lesion formation in the aortic wall of rabbits that were fed a diet enriched in animal protein (mainly meat, milk, and egg yolk). Since then, various animal species, such as rabbits, mice, rats, guinea pigs, hamsters, birds, dogs and non-human primates, have been used for experimentation. Of these, rodents, swine and rabbits have provided crucial information for the pathophysiological mechanisms underlying the initiation and subsequent development of atherosclerotic plaques. In spite of the many differences between the animal models, all of these demonstrate the requirement of high blood cholesterol levels for plaque development. The observations of this remarkable characteristic supported the discovery of the essential role of cholesterol in atherosclerosis development. Animal models have been extensively used for the study of human cardiovascular diseases [29–40], and their use has led to opportunities for translational application [32, 41–45]. In this review, the different models for examining atherosclerosis, their own advantages and disadvantages (Table 1) and the molecular pathways involved (Table 2), will be discussed in turn.
Table 1.
Advantages and disadvantages of mouse models for studying atherosclerosis
| Advantages | Disadvantages |
|---|---|
| Ease of genetic manipulation | Toxic diet needed to induce atherosclerosis in wild-type mice |
| Low maintenance cost | Different lipid metabolism compared to humans: high HDL, no CETP |
| Short generation time means less time consuming for research projects | Differing cardiovascular anatomy and physiology with different predisposed site for the development atherosclerosis |
| Wide availability | Small body size limits frequent blood collection and increases difficulty of dissection of small arteries |
Table 2.
Molecular pathways involved in atherosclerosis
| Molecular mechanisms | Role in Atherosclerosis | References |
|---|---|---|
| Expression of vascular cell adhesion molecule-1 (VCAM-1), E-selectin and P-selectin | Inflammatory response induced by LDL oxidation | [7, 8] |
| Nuclear factor-kappa B (NF-κB) activation | Chemotaxis via regulation of chemokines, such as CCL2, CCL5, CCL8, and CXCL9 | [166, 169] |
| Release of platelet derived growth factor | Fibrous cap formation | [12, 13] |
| CHOP activation | Macrophage apoptosis via endoplasmic reticulum stress | [107, 108] |
| Pattern recognition receptor activation | Macrophage apoptosis via activation of CD36-TLR2 pathway | [111, 112] |
| Activation of mitochondrial, Ca2+-dependent pathways | Vascular smooth muscle cell apoptosis via calpain/mPTP/cytochrome C/caspase-3 and apoptosis-inducing factor | [140] |
| cytochrome c release and activation of caspase-9 and the effector caspases | Macrophage apoptosis induced by cholesterol loading | [141] |
| Toll-like receptor activation | Immune activation through recognition of mitochondrial DNA, which can act as damage-associated molecular patterns (DAMPs) | [148] |
| Upregulation of transient receptor potential cation (TRPC) channels | VSMC, migration, proliferation and apoptosis; neointimal proliferation | [158, 160–162] |
Mouse
With its small size and ease of genetic manipulation, the mouse is currently the most frequently used model in atherosclerosis research [46]. Development of atherosclerosis is influenced by a number of genes, which interact with each other and the environment to affect the disease phenotype [47]. Manipulation can take place for single genes, and indeed in a time-dependent manner, which permits the elucidation of the molecular mechanisms operating in different phase of plaque evolution, whilst the manipulation of gene expression in specific cell types has increased our understanding of the contributions of different immune cells to atherosclerosis.
Dietary models
The first mouse model was developed by Wissler and coworkers in 1960s [48]. They fed the mice with a diet of high fat 30%, high cholesterol 5% and cholic acid 2%. This diet successfully promoted hypercholesterolemia, and induced the formation of fatty streaks in different vascular regions [49]. However, the high fat diet induced a high level of acute inflammatory changes, but did not simulate the chronic low-grade inflammatory environment observed in human atherosclerosis [50]. Also, the pro-inflammatory diet as highly toxic, which led to weight loss and increased the susceptibility of the animals to infections.
A less toxic diet of 15% fat, 1.25% cholesterol and 0.5% cholic acid was introduced, resulting in high variability of diet-induced atherosclerosis in different strains of mice [51]. The most susceptible strain was C57BL/6, which developed mild hypercholesterolemia — around 200 mg/ml — and fatty streak lesions in the aortic root after 3–9 months of fat feeding [52]. However, the lesion was not comparable to that observed in humans, with small lesions found in the aorta, consisted almost exclusively of macrophages and did not progress to fibrous plaques. The difference in morphology of the plaque formation between mouse and human hampers data extrapolation. Nevertheless, from experiments conducted in dietary models, a number of genetic loci that increase the susceptibility to atherosclerosis has been identified. An example is Ath-1, which was mapped to the short arm of chromosome 1 [53].
Recent experiments in germ-free mice generated by feeding with an antibiotic cocktail have demonstrated dietary lipid phosphatidylcholine (lecithin) was identified by a metabolomics approach, and these were subsequently shown to be predictive of atherosclerosis in humans, highlighting the role of intestinal microbiome in regulating plasma lipoprotein homeostasis [54].
ApoE knockout and LDLR-deficient models
Given the toxicity of high fat diet and differences in lesion morphology, limitations of using wild-type mice for studying atherosclerosis were recognized. The development of genetically modified mice represented a breakthrough. Gene knockout and knock-in techniques, together with the ability to control the spatial and temporal patterns of gene expression, have proven useful for studying atherosclerosis. The first genetically modified mouse model developed was the Apolipoprotein E (ApoE) knockout model [55]. ApoE is a structural component of all lipoprotein except for LDL and is a critical ligand for the hepatic clearance of plasma lipoproteins mediated by LDLR and LDLR-related protein [56–58]. ApoE knockout mice exhibited significant hypercholesterolemia (400 mg/dl, which represents a five-fold increase in plasma cholesterol level compared to wild type mice) despite being on a low fat diet [59–61]. An advantage of using ApoE knockout mice is that the high toxic diet can be avoided. Moreover, it shares greater degrees of similarity in the atherosclerotic development to humans compared to wild-type models. There is dramatic shift in plasma lipoprotein profile, with the proatherogenic VLDL as the most abundant circulating lipoprotein, similar to type II hyperlipidemia in humans [62]. Spontaneous development of atherosclerotic lesions in several vascular beds, predominantly in the aortic root, aortic arch and different branch point along the aorta, are observed [63]. Another advantage is the rapid development of lesions compared to wild-type. When ApoE knockout mice are fed with a high fat diet [64, 65] (Western type diet, which contains 0.15% cholesterol, 21% fat derived from milk and without the use of cholic acid), they exhibit high plasma lipid level of over 1000 mg/dl and develop complex fibrous plaques in the aortic sinus after 10–14 weeks of diet.
However, ApoE knockout mice has its own drawbacks which limits the extrapolation of data derived from this model to humans. Firstly, the lipid metabolism is dissimilar, with the majority of the plasma cholesterol as VLDL but not LDL as observed in humans [66, 67]. Secondly, ApoE was found to provide extra athero-protective properties in addition to mediating lipoprotein clearance, by virtue of its anti-oxidative, anti-proliferative and anti-inflammatory actions [68]. Therefore, atherosclerosis in ApoE knockout model may be independent of plasma lipid levels. Conversely, low levels of ApoE expressed in adrenal cells can reduce the severity of atherosclerosis without affecting plasma lipid levels in ApoE-deficient mice [69].
To overcome the problems of the above model, LDLR deficient mouse model was developed. Compared to ApoE, LDLR has fewer functions and therefore the effects due to its absence are more easily attributed to lipoprotein homeostasis than other processes such as cell proliferation or inflammation [70]. LDLR deficiency impairs lipoprotein uptake and clearance, resulting in a greater preponderance of LDL as the cholesterol-carrying plasma lipoprotein. Compared to ApoE knockout mice, this model shows a milder plasma cholesterol increase of 250 mg/dl when fed with standard low fat diet and the elevated lipoprotein is mainly LDL [71]. Whilst on high fat diet, LDLR-deficient animals developed severe hypercholesterolemia of 900 mg/dl with accumulation of VLDL and LDL, as well as extensive atherosclerosis [72].
Compared with ApoE deficiency, the absence of functional LDLR in humans is more common and leads to familial hypercholesterolemia, greatly increasing the cardiovascular risk [73]. This model shares the characteristics observed in human familial hypercholesterolemia, including the lesions in aortic valves and the aortic root [74]. This model has been useful for examining the relationship between diabetes and atherosclerosis, which often co-exist [72]. It was found that LDLR deficient mice were more prone to obesity and insulin resistance than ApoE deficient mice.
However, the LDLR model is imperfect with its own shortcoming. Firstly, compared with the ApoE knockout mice, the LDLR deficient mouse model is more resistant to injury-induced neointimal formation [75]. Therefore, ApoE knockout mice remains the better alternative for investigating the molecular mechanisms underlying restenosis following angioplasty [76]. Secondly, limited lesions developed only in older LDLR deficient mice with standard diet [77, 78]. Consequently, high fat diets with different levels of cholesterol have been employed to induce more significant atherosclerotic changes [79]. With a variety of dietary cholesterol intake, this model cannot be standardized across models generated by different laboratories.
Advantages and disadvantages of mouse models
The advantages of using small sized animals over larger animals are lower costs to purchase, breed, feed and maintain. The use of antibodies and drugs for intervention becomes more affordable with its small size. Another advantage is its rapid growth rate and short generation time [80]: it takes 6 to 8 weeks for the mouse to reach sexual maturity and approximately an additional 3 weeks for gestation [81]. It is relatively easy to control multiple genes by interbreeding given the wide availability of inbred strains [82], or using genetic knockout techniques to elucidate the role of individual genes in atherosclerosis [83]. The ease of genetic manipulation comes in the well-defined genetic makeup and availability of inbred strains.
The major limitation of mouse models is their natural resistance to atherosclerosis for several reasons. Firstly, cholesterol metabolism and lipoprotein pattern are different from those of humans due to the absence of cholesteryl ester transfer protein (CETP) [84], a carrier that facilitates the transport of cholesteryl esters and triglycerides between different lipoproteins [85]. Thus, mice usually have a lower plasma cholesterol levels of 60–100 mg/dl compared to humans, with the high density lipoprotein being the major circulating lipoprotein [86]. This contrasts with the deleterious LDL, which is the predominant form found in plasma of humans [87]. Thus, genetically-modified mice have been used to induce hypercholesterolemia, although concerns have been raised because this may be non-physiological and the pathogenesis may differ from that of human atherosclerosis [88].
Although the comparatively smaller size of mice results in more convenient use for experimentation, it also limits many aspects of practical investigation. For example, the coronary arteries are too small for visualization. Moreover, blood collection can be difficult [89]. There is also considerable difference in the anatomy of the cardiovascular system between mice and humans. The arterial intima consists of only endothelium overlying the internal elastic lamina without smooth muscle cells or connective tissues found in humans [90]. Moreover, the tunica media is less thick and the vasa vasorum is absent in mice [91].
The morphology of atherosclerotic lesions observed in mice is different from that of humans, in that the thick fibrous cap is absent [92]. In mice, vasa vasorum is not needed since the thin layers of tunica intima and intima do not pose a significant barrier to oxygen diffusion [91]. The absence of the vasa vasorum explains the lack of ingrowth of neovessels into the base of the lesions. The neovessel is thought to be derived from the vasa vasorum, considered as an important entry path for immune cells and contributes to chronic inflammation and development of the necrotic core [93]. The predilection sites for atherosclerosis development in mouse models are the aortic sinus and innominate artery, whereas the coronary arteries are commonly affected in humans. Unstable plaque is rarely observed in mouse models, and it is therefore difficult to examine plaque rupture and overlying luminal thrombosis, which is the direct cause of acute ischemic events responsible for cardiovascular deaths, using these systems [94–96].
Recent advances
Oxidative stress, dysfunction of intracellular organelles and vascular inflammation
Recent research efforts have focused on elucidating the mechanisms by which dysfunction of two intracellular organelles, the endoplasmic reticulum (ER) and the mitochondrion of various cell types, promotes vascular inflammation and atherosclerosis.
ER stress
The ER is responsible for protein synthesis, proper folding, maturation and assembly prior to further processing by the Golgi apparatus [97]. The highly oxidative environment within the ER lumen facilitates the formation of tertiary and quaternary structures, aided by chaperone proteins and a high concentration of luminal Ca2+ to facilitate their interactions [98]. Abnormalities in these processes can lead to misfolding or unfolding of proteins, which then accumulate within the ER lumen. This would increase ER stress, thereby activating the unfolded protein response (UPR) [99]. The UPR is a normal adaptive and protective mechanism to reduce the rate of protein synthesis, increase folding ability of proteins and aid misfolded or unfolded protein to cellular degradation pathways [100].
Prolonged ER stress of different cell types involved in atherosclerosis, including macrophages, VSMCs and endothelial cells, has been observed in atherosclerosis [101–103]. In macrophages, LDL and cholesterol are transported from late endosomes to the ER, where esterification of cholesterol and formation of lipid droplets occur [104]. In macrophages of atherosclerotic plaques, the esterification process is greatly reduced, which is responsible for cholesterol accumulation in foam cells [105]. It has been shown that UPR activation of macrophages takes place during the different stages of atherosclerotic lesion development in ApoE knockout mice [106]. Prolonged ER stress leads to macrophage apoptosis associated with expression of the UPR sensor C/EBPα-homologous protein (CHOP). CHOP inactivation could reduce the rate of macrophage apoptosis and plaque necrosis, suggesting that targeting of the UPR has the potential to hinder the progression of atherosclerosis [107, 108]. In early plaques, macrophages are efficient in clearing cells which have undergone apoptosis [109]. However, in advanced plaques, they are unable to do so [110], resulting in the formation of necrotic core [111]. Other mechanisms contributing to apoptosis of macrophages require activation of pattern recognition receptors (PRRs) of the innate immune system by oxidized lipids [111, 112]. PRR activation induces apoptosis via the CD36-TLR2 pathway [112]. In this pathway, the enzyme NADPH oxidase plays a key role in mediating oxidative stress, activation of the double-stranded RNA-dependent protein kinase, which induces CHOP and apoptosis [113].
ER stress in endothelial cells can be activated by shear stress [114, 115] or oxLDL [116]. This results in increased oxidative stress and therefore increased oxidation of sarcoplasmic/endoplasmic reticulum Ca2+-dependent ATPase, leading to atherogenesis [116]. In VSMCs, a number of cellular stressors, such as cholesterol loading, can initiate ER stress [117, 118], thereby activating apoptosis [119]. This may then affect collagen synthesis adversely. Homocysteine, which is raised in hypertension and diabetes, can initiate UPR [117]. ER stress plays a role in cardio-metabolic disorders such as hypertension and diabetes, and partly explains why atherosclerosis is worsened in the presence of these co-morbid conditions [120–122]. The use of mouse models has discovered novel mechanisms by which traditional pharmacological agents act to exert their vascular protective effects through modifying ER stress response [122–124]. For example, recent experiments have demonstrated that metformin, a commonly used anti-diabetic drug, protected endothelial function in obese, diabetic mice by inhibiting ER stress [120].
Mitochondrial dysfunction
The mitochondria are the cellular powerhouses because of their ability to generate ATP by oxidative phosphorylation. They have other functions such as, redox status, reactive oxygen species (ROS) production and regulation of cellular apoptosis [125]. They are the only intracellular organelle apart from the nucleus to contain DNA. During ATP synthesis, electron is transferred from complex I to complex IV of the respiratory chain, which leads to pumping of protons from the mitochondrial matrix into the intermembrane space [126]. Much of the cellular ROS is derived from the mitochondria, mostly from complex I and complex III and enzymes including the alpha-ketoglutarate dehydrogenase complex and those participating in fatty acid beta-oxidation [127–130]. Production of oxygen free radicals is more efficient by reverse electron transfer dependent on succinate (through complex I to NAD+) than forward electron transfer with NADH [131]. This reverse electron transfer is an important mechanism of ROS production in many pathological conditions such as hypertension, playing an important role in the development of atherosclerosis [132]. Mitochondrial ROS can itself increase its own production in a positive feedback loop, which is termed ROS-induced ROS release (RIRR) [133, 134]. This is recognized to play a key role initiating mitochondrial depolarization [134].
Elevated ROS production can damage a number of macromolecules within the cells, such as proteins, lipids and mitochondrial DNA of different cell types, contributing to atherosclerosis development (Fig. 1). For example, 4-Hydroxynonenal (HNE), an end-product of membrane lipid peroxidation [135], increased ROS production, producing mitochondrial dysfunction and inducing VSMC apoptosis [136]. Reperfusion after ischaemia, which a consequence of atherosclerotic disease, can lead to opening of the mitochondrial permeability transition pore (MPTP) [137, 138]. MPTP opening transiently leads to depolarization of the mitochondrial membrane potential. However, prolonged opening is harmful, as it leads to swelling of the mitochondrial matrix, and rupture of the outer membrane, releasing pro-apoptotic factors to initiate apoptosis [139]. VSMC apoptosis induced by oxidize LDL occurs via two distinct mitochondrial, Ca2+-dependent pathways, calpain/mPTP/cytochrome C/caspase-3 and apoptosis-inducing factor [140]. Other cell types such as macrophages also demonstrate mitochondrial dysfunction. Thus, cholesterol loading in macrophages triggers cytochrome c release and activation of caspase-9 and the effector caspases, leading to macrophage apoptosis [141]. Moreover, oxLDL can increase the release of peroxyl radicals, inducing both mitochondrial depolarization, dysfunction and cell lysis in macrophages [142].
Fig. 1.
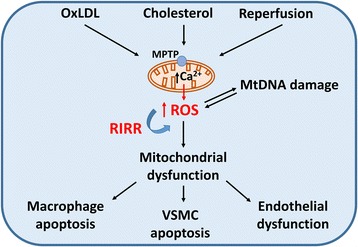
The role of mitochondrial dysfunction in promoting atherosclerosis. OxLDL: oxidized LDL. MtDNA: mitochondrial DNA. ROS: reactive oxygen species. RIRR: ROS-induced ROS release. VSMC: vascular smooth muscle cell
There has been a recent interest in the consequences of mitochondrial DNA damage in atherosclerosis. The relationship between mitochondrial DNA damage and oxidative stress has been studied in detail using genetically knockout mice with expressing a proofreading-deficient version of the mitochondrial DNA Polymerase G (POLG) [143, 144]. Mitochondrial DNA damage occurs early in atherogenesis [145]. Mice with double knockout of ApoE and POLG accumulate mitochondrial DNA damage, which promoted atherosclerosis and was associated with the formation of vulnerable plaques [146]. In ApoE-deficient recipients of mutator-mouse bone marrow, fibrous cap thinning and increased necrotic core, which are characteristic of vulnerable plaques, were observed [147]. It is thought that mitochondrial DNA can act as damage-associated molecular patterns (DAMPs), which are recognized by Toll-like receptors (TLRs). TLRs can activate the innate immune system [148].
Current treatment options and complications
The mainstay treatment options for occlusive vascular disease are stent insertion or bypass grafting. However, significant complications such as stent thrombosis, re-stenosis or vein graft failure can arise [149–151]. There is therefore a need to elucidate the molecular mechanisms underlying these harmful processes in order to devise ways to prevent them. In recent years, the role of oxidative stress, endoplasmic reticulum stress and mitochondrial dysfunction in promoting neointimal proliferation has been intensively studied [152]. VSMCs, the main component of the neointima, are capable of switching from a quiescent and contractile to a proliferative phenotype [153]. The latter is responsible for luminal occlusion. Increased ROS release causes neointimal hyperplasia by promoting VSMC migration and proliferation, as well as collagen deposition within the extracellular matrix [16, 154, 155]. Altered Ca2+ signalling plays a critical role in these processes [156, 157]. For example, L-type Ca2+ channels are downregulated and transient receptor potential cation (TRPC) channels are upregulated in VSMCs [156]. Of the TRPCs, the Ca2+-permeable TRPM2 channel was suggested to be a sensor of oxidative stress [158], and its activation by ROS leads to intracellular Ca2+ overload inflammation [159], VSMC, migration, proliferation and apoptosis [158, 160, 161]. Another TRPC isoform, TRPC1, has also been implicated in neointimal proliferation [162].
Future therapeutic approaches: microRNAs and nanoparticle delivery
The roles of microRNAs in atherosclerosis have been the focus of research in recent years [163]. The use of this model has led to novel therapeutic approaches. MicroRNAs are non-coding RNAs involved in post-transcriptional regulation of genes by RNA silencing [164]. Their generation is under tight control spatially and temporally. Recent work has demonstrated their regulation of flow-dependent vascular remodelling [165]. It appears that some microRNAs, such as miR-10a, miR-19a, and miR-23b are inducible by shear stress and play a protective role in atherosclerosis [166–168]. Other microRNAs, such as MiR-146a and miR-181b, have anti-inflammatory properties by inhibiting the translation of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) and importin α3. This results in inhibition of nuclear factor-kappa B (NF-κB) activity, which is critical in regulating the expression of several chemokines, such as CCL2, CCL5, CCL8, and CXCL9 [166, 169]. In a recent study, polyethylene glycol-polyethyleneimine nanoparticles were used as vectors for microRNA delivery targeting E-selectin of inflamed endothelium of ApoE-deficient mice, which ameliorated endothelial inflammation and atherosclerosis [170]. Phosphatidylserine-based nanoparticles delivering the natural dietary compound curcumin was able to target macrophages and reduce their pro-inflammatory action [171]. Polyethylene glycol-conjugated polyion complex (PIC) vesicles containing drugs were injected into carotid arteries of rats using a balloon catheter, resulting in sustained targeted delivery, providing a proof-of-concept that this approach can be used for treating atherosclerosis in the future [172].
Conclusions
There is no single ideal animal model for studying a particular clinical condition [173–175]. The general criteria for an appropriate animal model lies are the size, docility, ease of breeding and housing, known genetic profile, analogies with humans and the costs involved. Mice are amenable to genetic modification, allowing identification of genes contributing to the development of atherosclerosis. The understanding on the risk factors and natural history of atherosclerosis offer insights on disease prevention. Identification of the molecular events is important for developing therapeutic strategies to improve endothelial dysfunction, which could slow or even reverse disease progression in atherosclerosis [176, 177].
Acknowledgements
None.
Funding
YC is supported by a project grant from the ESRC for her doctoral studies at the University of Cambridge. GT was supported by the BBSRC Doctoral Training Award and Assistant Professorships from The Croucher Foundation of Hong Kong. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
YL Literature review; design of manuscript; drafted and critically revised the manuscript for important intellectual content; preparation of figure. HL Literature review; Drafted and critically revised the manuscript for important intellectual content. VL critically revised the manuscript for important intellectual content. YC critically revised the manuscript for important intellectual content. OT critically revised the manuscript for important intellectual content. BY critically revised the manuscript for important intellectual content. TL critically revised the manuscript for important intellectual content. WW critically revised the manuscript for important intellectual content. GT Literature review; drafted and critically revised the manuscript for important intellectual content; preparation of figure. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
N/A
Abbreviations
- ApoE
ApolipoproteinE
- CETP
Cholesteryl ester transfer protein
- CHOP
C/EBPα-homologous protein
- DAMP
Damage-associated molecular patterns
- ER
Endoplasmic reticulum
- LDL
Lipoprotein
- MPTP
Mitochondrial permeability transition pore
- NF-κB
Nuclear factor-kappa B
- oxLDL
Oxidized LDL
- POLG
Polymerase G
- PRR
Pattern recognition receptors
- RIRR
ROS-induced ROS release
- ROS
Reactive oxygen species
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- TRAF6
TNF-associated factor 6
- TRPC
Transient receptor potential cation channel
- VSMC
Vascular smooth muscle cell
Contributor Information
Yee Ting Lee, Email: andrealyt@gmail.com.
Hiu Yu Lin, Email: venicehy@hku.hk.
Yin Wah Fiona Chan, Email: ywfc2@cam.ac.uk.
Ka Hou Christien Li, Email: K.H.Li3@newcastle.ac.uk.
Olivia Tsz Ling To, Email: oliviato@cuhk.edu.hk.
Bryan P Yan, Email: bryan.yan@cuhk.edu.hk.
Tong Liu, Email: liutongdoc@126.com.
Guangping Li, Email: tjcardiol@126.com.
Wing Tak Wong, Email: jack_wong@cuhk.edu.hk.
Wendy Keung, Email: wkeung@hku.hk.
Gary Tse, Phone: +852 2632 3717, Email: tseg@cuhk.edu.hk.
References
- 1.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European atherosclerosis society. Eur Heart J. 2013;34:3478–3490a. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 4.Steinberg D, Glass CK, Witztum JL. Evidence mandating earlier and more aggressive treatment of hypercholesterolemia. Circulation. 2008;118:672–677. doi: 10.1161/CIRCULATIONAHA.107.753152. [DOI] [PubMed] [Google Scholar]
- 5.Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010;30:139–143. doi: 10.1161/ATVBAHA.108.179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parthasarathy S, Quinn MT, Steinberg D. Is oxidized low density lipoprotein involved in the recruitment and retention of monocyte/macrophages in the artery wall during the initiation of atherosclerosis? Basic Life Sci. 1988;49:375–380. doi: 10.1007/978-1-4684-5568-7_58. [DOI] [PubMed] [Google Scholar]
- 7.Braun M, Pietsch P, Felix SB, Baumann G. Modulation of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 on human coronary smooth muscle cells by cytokines. J Mol Cell Cardiol. 1995;27:2571–2579. doi: 10.1006/jmcc.1995.0044. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13:197–204. doi: 10.1161/01.ATV.13.2.197. [DOI] [PubMed] [Google Scholar]
- 9.Joris I, Stetz E, Majno G. Lymphocytes and monocytes in the aortic intima—an electron-microscopic study in the rat. Atherosclerosis. 1979;34:221–231. doi: 10.1016/S0021-9150(79)80003-9. [DOI] [PubMed] [Google Scholar]
- 10.Proudlock JW, Day AJ, Tume RK. Cholesterol-esterifying enzymes of foam cells isolated from atherosclerotic rabbit intima. Atherosclerosis. 1973;18:451–457. doi: 10.1016/0021-9150(73)90075-0. [DOI] [PubMed] [Google Scholar]
- 11.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. doi: 10.1161/01.ATV.6.2.131. [DOI] [PubMed] [Google Scholar]
- 12.Sano H, Sudo T, Yokode M, Murayama T, Kataoka H, Takakura N, Nishikawa S, Nishikawa SI, Kita T. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103:2955–2960. doi: 10.1161/01.CIR.103.24.2955. [DOI] [PubMed] [Google Scholar]
- 13.Kozaki K, Kaminski WE, Tang J, Hollenbach S, Lindahl P, Sullivan C, Yu JC, Abe K, Martin PJ, Ross R, et al. Blockade of platelet-derived growth factor or its receptors transiently delays but does not prevent fibrous cap formation in ApoE null mice. Am J Pathol. 2002;161:1395–1407. doi: 10.1016/S0002-9440(10)64415-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 15.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 16.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887–893. doi: 10.1161/ATVBAHA.111.224873. [DOI] [PubMed] [Google Scholar]
- 19.Kolodgie FD, Narula J, Haider N, Virmani R. Apoptosis in atherosclerosis. Does it contribute to plaque instability? Cardiol Clin. 2001;19:127–139. doi: 10.1016/S0733-8651(05)70199-5. [DOI] [PubMed] [Google Scholar]
- 20.Martinet W, Kockx MM. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol. 2001;12:535–541. doi: 10.1097/00041433-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 21.Baetta R, Corsini A. Role of polymorphonuclear neutrophils in atherosclerosis: current state and future perspectives. Atherosclerosis. 2010;210:1–13. doi: 10.1016/j.atherosclerosis.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 22.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 23.Lahoute C, Herbin O, Mallat Z, Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol. 2011;8:348–358. doi: 10.1038/nrcardio.2011.62. [DOI] [PubMed] [Google Scholar]
- 24.Getz GS, Vanderlaan PA, Reardon CA. Natural killer T cells in lipoprotein metabolism and atherosclerosis. Thromb Haemost. 2011;106:814–819. doi: 10.1160/TH11-05-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butcher M, Galkina E. Current views on the functions of interleukin-17A-producing cells in atherosclerosis. Thromb Haemost. 2011;106:787–795. doi: 10.1160/TH11-05-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YC, Huang AL, Kyaw TS, Bobik A, Peter K. Atherosclerotic plaque rupture: identifying the straw that breaks the Camel’s back. Arterioscler Thromb Vasc Biol. 2016;36:e63–72. doi: 10.1161/ATVBAHA.116.307993. [DOI] [PubMed] [Google Scholar]
- 27.Kalz J, ten Cate H, Spronk HM. Thrombin generation and atherosclerosis. J Thromb Thrombolysis. 2014;37:45–55. doi: 10.1007/s11239-013-1026-5. [DOI] [PubMed] [Google Scholar]
- 28.Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 29.Choy L, Yeo JM, Tse V, Chan SP, Tse G. Cardiac disease and arrhythmogenesis: mechanistic insights from mouse models. Int J Cardiol Heart Vasc. 2016;12:1–10. doi: 10.1016/j.ijcha.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Chen Z, Wang Y, Jiang J, Tse G, Xu W, Ge J, Sun B. Effects of granulocyte colony-stimulating factor on rabbit carotid and swine heart models of chronic obliterative arterial disease. Mol Med Rep. 2016. [DOI] [PMC free article] [PubMed]
- 31.Tse G, Lai ET, Tse V, Yeo JM. Molecular and electrophysiological mechanisms underlying cardiac arrhythmogenesis in diabetes mellitus. J Diabetes Res. 2016;2016:2848759. doi: 10.1155/2016/2848759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tse G. Both transmural dispersion of repolarization and of refractoriness are poor predictors of arrhythmogenicity: a role for iCEB (QT/QRS)? J Geriatr Cardiol. 2016;13(9):813–4. doi: 10.11909/j.issn.1671-5411.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Sun B, Tse G, Jiang J, Xu W. Reversibility of both sinus node dysfunction and reduced HCN4 mRNA expression level in an atrial tachycardia pacing model of tachycardia-bradycardia syndrome in rabbit hearts. Int J Clin Exp Pathol. 2016;9:8526–8531. [Google Scholar]
- 34.Chuang C-S, Ho S-C, Lin C-L, Lin M-C, Kao C-H. Risk of cerebrovascular events in pneumoconiosis patients: a population-based study, 1996–2011. Medicine. 2016;95:e2944. doi: 10.1097/MD.0000000000002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin HY, Lee YT, Chan YW, Tse G. Animal models for the study of primary and secondary hypertension in humans. Biom Rep. 2016;5:653–9. doi: 10.3892/br.2016.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo JM, Tse V, Kung J, Lin HY, Lee YT, Kwan J, Yan BP, Tse G. Isolated heart models for studying cardiac electrophysiology: a historical perspective and recent advances. J Basic Clin Physiol Pharmacol. 2016. in press. [DOI] [PubMed]
- 37.Fu H, Li G, Liu C, Li J, Cheng L, Yang W, Tse G, Zhao J, Liu T. Probucol prevents atrial ion channel remodeling in an alloxan-induced diabetes rabbit model. Oncotarget. 2016. ahead of print. [DOI] [PMC free article] [PubMed]
- 38.Tse G, Yeo JM, Tse V, Kwan SK, Sun B. Gap junction inhibition by heptanol increases ventricular arrhythmogenicity by decreasing conduction velocity without affecting repolarization properties or myocardial refractoriness in Langendorff-perfused mouse hearts. Mol Med Rep. 2016;14(5):4069–74. doi: 10.3892/mmr.2016.5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tse G, Liu T, Li KH, Laxton V, Chan YW, Keung W, Li RA, Yan BP. Electrophysiological mechanisms of Brugada syndrome: insights from pre-clinical and clinical studies. Front Physiol. 2016;7:467. doi: 10.3389/fphys.2016.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tse G, Lai ETH, Yeo JM, Tse V, Wong SH. Mechanisms of electrical activation and conduction in the gastrointestinal system: lessons from cardiac electrophysiology. Front Physiol. 2016;7:182. doi:10.3389/fphys.2016.00182. [DOI] [PMC free article] [PubMed]
- 41.Tse G. (Tpeak — Tend)/QRS and (Tpeak — Tend)/(QT x QRS): novel markers for predicting arrhythmic risk in the Brugada syndrome. Europace. 2016. ahead of print. [DOI] [PubMed]
- 42.Tse G, Yan BP. Traditional and novel electrocardiographic conduction and repolarization markers of sudden cardiac death. Europace. 2016. ahead of print. [DOI] [PubMed]
- 43.Tse G. Novel conduction-repolarization indices for the stratification of arrhythmic risk. J Geriatr Cardiol. 2016;9:811–812. doi: 10.11909/j.issn.1671-5411.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tse G, Wong ST, Tse V, Yeo JM. Variability in local action potential durations, dispersion of repolarization and wavelength restitution in aged wild-type and Scn5a+/− mouse hearts modelling human Brugada syndrome. J Geriatr Cardiol. 2016. in press. [DOI] [PMC free article] [PubMed]
- 45.Tse G, Wong ST, Tse V, Yeo JM. Determination of action potential wavelength restitution in Scn5a+/− mouse hearts modelling human Brugada syndrome. J Geriatr Cardiol. 2016. in press. [DOI] [PMC free article] [PubMed]
- 46.Mukhopadhyay R. Mouse models of atherosclerosis: explaining critical roles of lipid metabolism and inflammation. J Appl Genet. 2013;54:185–192. doi: 10.1007/s13353-013-0134-4. [DOI] [PubMed] [Google Scholar]
- 47.Simmons RD, Kumar S, Jo H. The role of endothelial mechanosensitive genes in atherosclerosis and omics approaches. Arch Biochem Biophys. 2016;591:111–131. doi: 10.1016/j.abb.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vesselinovitch D, Wissler RW, Doull J. Experimental production of atherosclerosis in mice. Atherosclerosis. 1968;8:483–495. doi: 10.1016/S0368-1319(68)80104-8. [DOI] [PubMed] [Google Scholar]
- 49.Thompson JS. Atheromata in an inbred strain of mice. J Atheroscler Res. 1969;10:113–122. doi: 10.1016/S0368-1319(69)80090-6. [DOI] [PubMed] [Google Scholar]
- 50.Okada Y, Yamaguchi K, Nakajima T, Nishikawa T, Jo M, Mitsumoto Y, Kimura H, Nishimura T, Tochiki N, Yasui K, et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013;33:301–311. doi: 10.1111/liv.12033. [DOI] [PubMed] [Google Scholar]
- 51.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 52.Schreyer SA, Wilson DL, LeBoeuf RC. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis. 1998;136:17–24. doi: 10.1016/S0021-9150(97)00165-2. [DOI] [PubMed] [Google Scholar]
- 53.Paigen B, Mitchell D, Reue K, Morrow A, Lusis AJ, LeBoeuf RC. Ath-1, a gene determining atherosclerosis susceptibility and high density lipoprotein levels in mice. Proc Natl Acad Sci U S A. 1987;84:3763–3767. doi: 10.1073/pnas.84.11.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- 56.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 57.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 58.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 59.Plump AS, Breslow JL. Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr. 1995;15:495–518. doi: 10.1146/annurev.nu.15.070195.002431. [DOI] [PubMed] [Google Scholar]
- 60.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1994;91:9607–9611. doi: 10.1073/pnas.91.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paszty C, Maeda N, Verstuyft J, Rubin EM. Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest. 1994;94:899–903. doi: 10.1172/JCI117412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghiselli G, Schaefer EJ, Gascon P, Breser HB., Jr Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science. 1981;214:1239–1241. doi: 10.1126/science.6795720. [DOI] [PubMed] [Google Scholar]
- 63.Smith JD, Breslow JL. The emergence of mouse models of atherosclerosis and their relevance to clinical research. J Intern Med. 1997;242:99–109. doi: 10.1046/j.1365-2796.1997.00197.x. [DOI] [PubMed] [Google Scholar]
- 64.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.ATV.14.1.133. [DOI] [PubMed] [Google Scholar]
- 65.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.ATV.14.1.141. [DOI] [PubMed] [Google Scholar]
- 66.Pendse AA, Arbones-Mainar JM, Johnson LA, Altenburg MK, Maeda N. Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J Lipid Res. 2009;50:S178–S182. doi: 10.1194/jlr.R800070-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Investig. 1999;103:1579–1586. doi: 10.1172/JCI6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50(Suppl):S156–161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klotz S, Hay I, Zhang G, Maurer M, Wang J, Burkhoff D. Development of heart failure in chronic hypertensive Dahl rats: focus on heart failure with preserved ejection fraction. Hypertension. 2006;47:901–911. doi: 10.1161/01.HYP.0000215579.81408.8e. [DOI] [PubMed] [Google Scholar]
- 70.Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merat S, Casanada F, Sutphin M, Palinski W, Reaven PD. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but Do Not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels Are achieved by a fructose-rich diet. Arterioscler Thromb Vasc Biol. 1999;19:1223–1230. doi: 10.1161/01.ATV.19.5.1223. [DOI] [PubMed] [Google Scholar]
- 73.Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133–170. doi: 10.1146/annurev.ge.24.120190.001025. [DOI] [PubMed] [Google Scholar]
- 74.VanderLaan PA, Reardon CA, Thisted RA, Getz GS. VLDL best predicts aortic root atherosclerosis in LDL receptor deficient mice. J Lipid Res. 2009;50:376–385. doi: 10.1194/jlr.M800284-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian J, Pei H, Sanders JM, Angle JF, Sarembock IJ, Matsumoto AH, Helm GA, Shi W. Hyperlipidemia is a major determinant of neointimal formation in LDL receptor-deficient mice. Biochem Biophys Res Commun. 2006;345:1004–1009. doi: 10.1016/j.bbrc.2006.04.180. [DOI] [PubMed] [Google Scholar]
- 76.Ali ZA, Alp NJ, Lupton H, Arnold N, Bannister T, Hu Y, Mussa S, Wheatcroft M, Greaves DR, Gunn J, Channon KM. Increased in-stent stenosis in ApoE knockout mice: insights from a novel mouse model of balloon angioplasty and stenting. Arterioscler Thromb Vasc Biol. 2007;27:833–840. doi: 10.1161/01.ATV.0000257135.39571.5b. [DOI] [PubMed] [Google Scholar]
- 77.Herijgers N, Van Eck M, Groot PHE, Hoogerbrugge PM, Van Berkel TJC. Effect of bone marrow transplantation on lipoprotein metabolism and atherosclerosis in LDL receptor-knockout mice. Arterioscler Thromb Vasc Biol. 1997;17:1995–2003. doi: 10.1161/01.ATV.17.10.1995. [DOI] [PubMed] [Google Scholar]
- 78.Merat S, Fruebis J, Sutphin M, Silvestre M, Reaven PD. Effect of aging on aortic expression of the vascular cell adhesion molecule-1 and atherosclerosis in murine models of atherosclerosis. J Gerontol A Biol Sci Med Sci. 2000;55:B85–94. doi: 10.1093/gerona/55.2.B85. [DOI] [PubMed] [Google Scholar]
- 79.Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y. Hyperlipidemia and atherosclerotic lesion development in ldlr-deficient mice on a long-term high-Fat diet. PLoS One. 2012;7:e35835. doi: 10.1371/journal.pone.0035835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. 2016;2016:170–176. doi: 10.1093/emph/eow014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray SA, Morgan JL, Kane C, Sharma Y, Heffner CS, Lake J, Donahue LR. Mouse gestation length is genetically determined. PLoS One. 2010;5:e12418. doi: 10.1371/journal.pone.0012418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21:434–440. doi: 10.1097/MOL.0b013e32833ded6a. [DOI] [PubMed] [Google Scholar]
- 83.Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RRS, Eskin E, Yan M, et al. Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLoS Genet. 2015;11:e1005711. doi: 10.1371/journal.pgen.1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vergeer M, Holleboom AG, Kastelein JJP, Kuivenhoven JA. The HDL hypothesis: does high-density lipoprotein protect from atherosclerosis? J Lipid Res. 2010;51:2058–2073. doi: 10.1194/jlr.R001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hayek T, Azrolan N, Verdery RB, Walsh A, Chajek-Shaul T, Agellon LB, Tall AR, Breslow JL. Hypertriglyceridemia and cholesteryl ester transfer protein interact to dramatically alter high density lipoprotein levels, particle sizes, and metabolism. Studies in transgenic mice. J Clin Invest. 1993;92:1143–1152. doi: 10.1172/JCI116683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bullock BC, Clarkson TB, Lehner ND, Lofland HB, Jr, St Clair RW. Atherosclerosis in cebus albifrons monkeys. [. clinical and pathologic studies. Exp Mol Pathol. 1969;10:39–62. doi: 10.1016/0014-4800(69)90047-1. [DOI] [PubMed] [Google Scholar]
- 87.Whitman SC. A Practical Approach to Using Mice in Atherosclerosis Research. Clin Biochem Rev. 2004;25:81–93. [PMC free article] [PubMed] [Google Scholar]
- 88.Singh V, Tiwari RL, Dikshit M, Barthwal MK. Models to study atherosclerosis: a mechanistic insight. Curr Vasc Pharmacol. 2009;7:75–109. doi: 10.2174/157016109787354097. [DOI] [PubMed] [Google Scholar]
- 89.Parasuraman S, Raveendran R, Kesavan R. Blood sample collection in small laboratory animals. J Pharmacol Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kostamaa H, Donovan J, Kasaoka S, Tobis J, Fitzpatrick L. Calcified plaque cross-sectional area in human arteries: correlation between intravascular ultrasound and undecalcified histology. Am Heart J. 1999;137:482–488. doi: 10.1016/S0002-8703(99)70496-5. [DOI] [PubMed] [Google Scholar]
- 91.Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovasc Res. 2007;75:649–658. doi: 10.1016/j.cardiores.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosenfeld ME, Averill MM, Bennett BJ, Schwartz SM. Progression and disruption of advanced atherosclerotic plaques in murine models. Curr Drug Targets. 2008;9:210–216. doi: 10.2174/138945008783755575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 94.Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788–792. doi: 10.1161/01.ATV.0000014587.66321.B4. [DOI] [PubMed] [Google Scholar]
- 95.Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, George S, Jackson C. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. 2005;111:1422–1430. doi: 10.1161/01.CIR.0000158435.98035.8D. [DOI] [PubMed] [Google Scholar]
- 96.Zhou J, Moller J, Danielsen CC, Bentzon J, Ravn HB, Austin RC, Falk E. Dietary supplementation with methionine and homocysteine promotes early atherosclerosis but not plaque rupture in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1470–1476. doi: 10.1161/hq0901.096582. [DOI] [PubMed] [Google Scholar]
- 97.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 98.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 99.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 100.Tsang KY, Chan D, Bateman JF, Cheah KS. In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci. 2010;123:2145–2154. doi: 10.1242/jcs.068833. [DOI] [PubMed] [Google Scholar]
- 101.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 102.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 105.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 106.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 107.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, Yano M, Iwawaki T, Kohno K, Araki K, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 109.Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73:470–480. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 110.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 111.Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type a scavenger receptor. J Cell Biol. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A. 2009;106:8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C, Wu M, Choi HC, Lyons TJ, Zou MH. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes. 2010;59:1386–1396. doi: 10.2337/db09-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou J, Werstuck GH, Lhotak S, de Koning AB, Sood SK, Hossain GS, Moller J, Ritskes-Hoitinga M, Falk E, Dayal S, et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- 118.Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis. 2009;207:123–130. doi: 10.1016/j.atherosclerosis.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 119.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY, Yao X, Wang N, Huang Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5’ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler Thromb Vasc Biol. 2014;34:830–836. doi: 10.1161/ATVBAHA.113.301938. [DOI] [PubMed] [Google Scholar]
- 121.Wong WT, Tian XY, Huang Y. Endothelial dysfunction in diabetes and hypertension: cross talk in RAS, BMP4, and ROS-dependent COX-2-derived prostanoids. J Cardiovasc Pharmacol. 2013;61:204–214. doi: 10.1097/FJC.0b013e31827fe46e. [DOI] [PubMed] [Google Scholar]
- 122.Zhang Y, Liu J, Luo JY, Tian XY, Cheang WS, Xu J, Lau CW, Wang L, Wong WT, Wong CM, et al. Upregulation of angiotensin (1–7)-mediated signaling preserves endothelial function through reducing oxidative stress in diabetes. Antioxid Redox Signal. 2015;23:880–892. doi: 10.1089/ars.2014.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, Jin L, Lian Q, Huang Y, Ding H, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation. 2015;131:1861–1871. doi: 10.1161/CIRCULATIONAHA.115.015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu A, Huang Y. A Tireless Giant in Vascular Research. J Cardiovasc Pharmacol. 2016;67(5):359–60. doi: 10.1097/FJC.0000000000000389. [DOI] [PubMed] [Google Scholar]
- 125.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 127.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 128.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 129.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Panov A, Dikalov S, Shalbuyeva N, Hemendinger R, Greenamyre JT, Rosenfeld J. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am J Physiol Cell Physiol. 2007;292:C708–718. doi: 10.1152/ajpcell.00202.2006. [DOI] [PubMed] [Google Scholar]
- 132.Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol. 2013;305:H1131–1140. doi: 10.1152/ajpheart.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. 2005;49:1044–1049. doi: 10.1002/mnfr.200500090. [DOI] [PubMed] [Google Scholar]
- 136.Lee JY, Jung GY, Heo HJ, Yun MR, Park JY, Bae SS, Hong KW, Lee WS, Kim CD. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicol Lett. 2006;166:212–221. doi: 10.1016/j.toxlet.2006.07.305. [DOI] [PubMed] [Google Scholar]
- 137.Di Lisa F, Canton M, Menabo R, Dodoni G, Bernardi P. Mitochondria and reperfusion injury. The role of permeability transition. Basic Res Cardiol. 2003;98:235–241. doi: 10.1007/s00395-003-0415-x. [DOI] [PubMed] [Google Scholar]
- 138.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 139.Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–258. doi: 10.1196/annals.1341.022. [DOI] [PubMed] [Google Scholar]
- 140.Vindis C, Elbaz M, Escargueil-Blanc I, Augé N, Heniquez A, Thiers J-C, Nègre-Salvayre A, Salvayre R. Two distinct calcium-dependent mitochondrial pathways Are involved in oxidized LDL-induced apoptosis. Arterioscler Thromb Vasc Biol. 2005;25:639–645. doi: 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- 141.Yao PM, Tabas I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J Biol Chem. 2001;276:42468–42476. doi: 10.1074/jbc.M101419200. [DOI] [PubMed] [Google Scholar]
- 142.Asmis R, Begley JG. Oxidized LDL promotes peroxide-mediated mitochondrial dysfunction and cell death in human macrophages: a caspase-3-independent pathway. Circ Res. 2003;92:e20–29. doi: 10.1161/01.RES.0000051886.43510.90. [DOI] [PubMed] [Google Scholar]
- 143.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 144.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 145.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, et al. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.CIR.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 146.Yu E, Baker L, Harrison J, Figg N, Mercer J, Calvert P, Vidal-Puig A, Murphy M, Bennett M. Mitochondrial DNA damage promotes atherosclerosis and is associated with vulnerable plaque. Lancet. 2013;381:S117. doi: 10.1016/S0140-6736(13)60557-7. [DOI] [Google Scholar]
- 147.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128:702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Levine GN, Chodos AP, Loscalzo J. Restenosis following coronary angioplasty: clinical presentations and therapeutic options. Clin Cardiol. 1995;18:693–703. doi: 10.1002/clc.4960181203. [DOI] [PubMed] [Google Scholar]
- 150.Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: pathogenesis, predisposition, and prevention. Circulation. 1998;97:916–931. doi: 10.1161/01.CIR.97.9.916. [DOI] [PubMed] [Google Scholar]
- 151.Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.RES.77.3.445. [DOI] [PubMed] [Google Scholar]
- 152.Tse G, Yan BP, Chan YW, Tian XY, Huang Y. Reactive oxygen species, endoplasmic reticulum stress and mitochondrial dysfunction: the link with cardiac arrhythmogenesis. Front Physiol. 2016;7:313. doi: 10.3389/fphys.2016.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thyberg J, Blomgren K, Hedin U, Dryjski M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: an electron-microscopic and stereological study. Cell Tissue Res. 1995;281:421–433. doi: 10.1007/BF00417860. [DOI] [PubMed] [Google Scholar]
- 154.Xu Q. Mouse models of arteriosclerosis: from arterial injuries to vascular grafts. Am J Pathol. 2004;165:1–10. doi: 10.1016/S0002-9440(10)63270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Napoli C, Lerman LO. Involvement of oxidation-sensitive mechanisms in the cardiovascular effects of hypercholesterolemia. Mayo Clin Proc. 2001;76:619–631. doi: 10.1016/S0025-6196(11)62413-0. [DOI] [PubMed] [Google Scholar]
- 156.House SJ. Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 2008;456:769–785. doi: 10.1007/s00424-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Munaron L, Antoniotti S, Fiorio Pla A, Lovisolo D. Blocking Ca2 + entry: a way to control cell proliferation. Curr Med Chem. 2004;11:1533–1543. doi: 10.2174/0929867043365008. [DOI] [PubMed] [Google Scholar]
- 158.Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol. 2006;209:31–41. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 159.Tano J-YK, Lee RH, Vazquez G. Macrophage function in atherosclerosis: potential roles of TRP channels. Channels. 2012;6:141–148. doi: 10.4161/chan.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ingueneau C, Huynh UD, Marcheix B, Athias A, Gambert P, Negre-Salvayre A, Salvayre R, Vindis C. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J Cell Mol Med. 2009;13:1620–1631. doi: 10.1111/j.1582-4934.2008.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ru X, Zheng C, Zhao Q, Lan H-Y, Huang Y, Wan S, Mori Y, Yao X. Transient receptor potential channel M2 contributes to neointimal hyperplasia in vascular walls. Biochim Biophys Acta (BBA) Mol Basis Dis. 2015;1852:1360–1371. doi: 10.1016/j.bbadis.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 162.Kumar B, Dreja K, Shah SS, Cheong A, Xu SZ, Sukumar P, Naylor J, Forte A, Cipollaro M, McHugh D, et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ Res. 2006;98:557–563. doi: 10.1161/01.RES.0000204724.29685.db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Feinberg MW, Moore KJ. MicroRNA regulation of atherosclerosis. Circ Res. 2016;118:703–720. doi: 10.1161/CIRCRESAHA.115.306300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 165.Neth P, Nazari-Jahantigh M, Schober A, Weber C. MicroRNAs in flow-dependent vascular remodelling. Cardiovasc Res. 2013;99:294–303. doi: 10.1093/cvr/cvt096. [DOI] [PubMed] [Google Scholar]
- 166.Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li YS, Chien S, Wang N. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A. 2010;107:3240–3244. doi: 10.1073/pnas.0914882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li YS, Chien S. Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci U S A. 2010;107:3234–3239. doi: 10.1073/pnas.0914825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci U S A. 2010;107:13450–13455. doi: 10.1073/pnas.1002120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen LJ, Chuang L, Huang YH, Zhou J, Lim SH, Lee CI, Lin WW, Lin TE, Wang WL, Chen L, et al. MicroRNA mediation of endothelial inflammatory response to smooth muscle cells and its inhibition by atheroprotective shear stress. Circ Res. 2015;116:1157–1169. doi: 10.1161/CIRCRESAHA.116.305987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ma S, Tian XY, Zhang Y, Mu C, Shen H, Bismuth J, Pownall HJ, Huang Y, Wong WT. E-selectin-targeting delivery of microRNAs by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci Rep. 2016;6:22910. doi: 10.1038/srep22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang J, Kang YX, Pan W, Lei W, Feng B, Wang XJ. Enhancement of Anti-Inflammatory Activity of Curcumin Using Phosphatidylserine-Containing Nanoparticles in Cultured Macrophages. Int J Mol Sci. 2016;17(5):E969. doi:10.3390/ijms17060969. [DOI] [PMC free article] [PubMed]
- 172.Taniguchi R, Miura Y, Koyama H, Chida T, Anraku Y, Kishimura A, Shigematsu K, Kataoka K, Watanabe T. Adequately-sized nanocarriers allow sustained targeted drug delivery to neointimal lesions in Rat arteries. Mol Pharm. 2016;13:2108–2116. doi: 10.1021/acs.molpharmaceut.6b00219. [DOI] [PubMed] [Google Scholar]
- 173.Tse G, Lai ET, Yeo JM, Yan BP. Electrophysiological mechanisms of Bayés syndrome: insights from clinical and mouse studies. Front Physiol. 2016;7:188. doi: 10.3389/fphys.2016.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tse G, Lai ET, Lee AP, Yan BP, Wong SH. Electrophysiological mechanisms of gastrointestinal arrhythmogenesis: lessons from the heart. Front Physiol. 2016;7:230. doi: 10.3389/fphys.2016.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tse G, Lai ET, Chan YW, Yeo JM, Yan BP. What is the arrhythmic substrate in viral myocarditis? insights from clinical and animal studies. Front Physiol. 2016;7:308. doi: 10.3389/fphys.2016.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chatzigeorgiou A, Halapas A, Kalafatakis K, Kamper E. The use of animal models in the study of diabetes mellitus. In Vivo. 2009;23:245–258. [PubMed] [Google Scholar]
- 177.Leong XF, Ng CY, Jaarin K. Animal models in cardiovascular research: hypertension and atherosclerosis. Biomed Res Int. 2015;2015:528757. doi: 10.1155/2015/528757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.