

Abstract
The t(8;21)(q22;q22) translocation, present in 10-15% of acute myeloid leukemia (AML) cases, generates the AML1/ETO fusion protein. To study the role of AML1/ETO in the pathogenesis of AML, we used the Ly6A locus that encodes the well characterized hematopoietic stem cell marker, Sca1, to target expression of AML1/ETO to the hematopoietic stem cell compartment in mice. Whereas germ-line expression of AML1/ETO from the AML1 promoter results in embryonic lethality, heterozygous Sca1+/AML1-ETO ires EGFP (abbreviated Sca+/AE) mutant mice are born in Mendelian ratios with no apparent abnormalities in growth or fertility. Hematopoietic cells from Sca+/AE mice have markedly extended survival in vitro and increasing myeloid clonogenic progenitor output over time. Sca+/AE mice develop a spontaneous myeloproliferative disorder with a latency of 6 months and a penetrance of 82% at 14 months. These results reinforce the notion that the phenotype of murine transgenic models of human leukemia is critically dependent on the cellular compartment targeted by the transgene. This model should provide a useful platform to analyze the effect of AML1/ETO on hematopoiesis and its potential cooperation with other mutations in the pathogenesis of leukemia.
The t(8;21)(q22;q22) translocation is one of the most commonly detected karyotypic abnormalities in acute myeloid leukemia (AML). This genetic alteration is found in up to 40% of de novo AML cases of the French-American-British M2 subtype, and in 12-15% of AML cases overall (1, 2). In many instances of AML, t(8;21) is the sole cytogenetic abnormality, suggesting that the translocation plays a key role in transformation. The t(8;21) translocation fuses sequences from the AML1 (RUNX1, CBFA2, and PEBPα) gene on chromosome 21 to the ETO (MTG8) gene on chromosome 8. This translocation results in production of the AML1/ETO protein, an in-frame fusion of the N terminus of AML1 and virtually the entire ETO protein. AML1 is the DNA-binding subunit of core-binding factor (CBF), a multimeric transcription factor complex that includes CBFβ and additional transcriptional coactivators. Mice lacking either Aml1 or Cbfβ fail to develop definitive intraembryonic hematopoiesis and die midgestation (3-6). Mice heterozygous for an AML1/ETO allele knocked into the Aml1 locus have an identical phenotype, providing genetic evidence that the AML1/ETO fusion protein acts as a dominant inhibitor of CBF activity (7, 8).
Although it is clear that CBF plays a critical role in hematopoietic development, several lines of evidence suggest that expression of its dominant inhibitor AML1/ETO is not sufficient to cause AML. AML1/ETO expression is frequently detectable in peripheral blood cells from t(8;21) AML patients for years into durable remission (9, 10). Conversely, clonotypic AML1/ETO sequences were identified in DNA retrospectively extracted from neonatal blood samples from 5 of 10 children with t(8;21) AML, preceding the development of AML by 5-10 years in these cases (11). Several strains of AML1/ETO-expressing transgenic mice have been generated (12-17). Although none of these mice develop leukemia spontaneously, two strains are highly susceptible to N-ethyl-N-nitrosourea (ENU)-induced leukemia (12, 13), providing evidence that AML1/ETO can cooperate with secondary mutations to cause AML.
Using the nonobese diabetic (NOD)/severe combined immunodeficient (scid) xenograft model, Bonnet and Dick (18) demonstrated that human AML originates in a primitive hematopoietic cell. These investigators showed that cells from patients with AML that are competent to adoptively transfer disease to NOD/scid recipients share a CD34+CD38- phenotype with normal hematopoietic stem cells (HSCs) (18). This concept was further supported by Weissman and colleagues (19), who showed that AML1/ETO mRNA is detectable in purified HSC from patients with t(8;21) AML. Taken together, these data suggest that the leukemia-initiating cell in t(8;21) AML (and probably all nonacute promyelocytic leukemia AML) has a primitive stem/progenitor phenotype. These observations led us to hypothesize that targeting AML1/ETO expression to HSC might be important for experimental models of this disease. We used the Sca1 (Ly6A) locus to target expression of AML1/ETO to the HSC compartment in mice. Our results confirm the paradigm that AML1/ETO expression is not sufficient to induce acute leukemia. In contrast to prior models, however, mice heterozygous for a mutant AML1/ETO allele at the Sca1 locus spontaneously develop a myeloproliferative disorder (MPD) at high penetrance.
Materials and Methods
Targeting Strategy. A targeting construct was modified from a vector containing 4.8 kb of isogenic genomic DNA that efficiently mediates homologous recombination at the Sca1 (Ly6A) locus (20). The 2.2-kb left targeting arm was reamplified with the following primers: 5′-GCTCTGGATCCCAAGACCACAGCGCATG and 5′-GGGTATACGCATCCTCAGAGAAGGG to introduce an AccI site at its 3′ end. A 2.3-kb AML1/ETO cDNA (kindly provided by S. Hiebert, Vanderbilt University, Nashville, TN) was amplified with the following primers: 5′-ATGCGTATACCCGTAGATGCCAGC and 5′-ATGCGGCCGCTAGCGAGGGGTTGTC to introduce AccI and NotI sites for subcloning. The AML1/ETO cDNA was released as a 2.3-kb AccI-NotI fragment and subcloned into the AccI site of the modified Sca1 left targeting arm and a NotI site in the plasmid polylinker. This cloning strategy introduces two silent polymorphisms in the AML1/ETO 5′ coding sequence and places the AML1/ETO ORF precisely after the Sca1 Kozak sequence. A modified ires EGFP SV40 polyA cassette (ires indicates internal ribosomal entry site) (BD Biosciences Clontech) was then subcloned between the AML1/ETO cDNA and a LoxP-flanked PGK-neo cassette (GenBank accession no. AF335420) followed by a 2.6-kb right targeting arm lacking the Sca1 translation initiation codon. The final 12.0-kb construct was linearized with XhoI and electroporated into RW-4 (129/SvJ) embryonic stem cells. Genotyping. Genomic DNA was extracted from peripheral blood leukocytes and amplified in a multiplex PCR (REDExtract-N-Amp Blood PCR kit, Sigma). The following primers were used: 5′-TGTGCAGCCCTTCTCTGAGG (Sca1 forward); 5′-ACTGAGCTCCACGTGTCCTT (Sca1 reverse); and 5′-CGTAGGCAGCACGGAGCAGA (AML1/ETO reverse). Amplification yielded a 330-bp amplicon from the WT allele and a 250-bp amplicon from the mutant allele.
RT-PCR. Total RNA was prepared from red cell-lysed peripheral blood leukocytes (High Pure, Roche, Indianapolis). Yield was quantified by using the Ribogreen reagent (Molecular Probes). Random-primed cDNA was synthesized from 10 ng of RNA with AMV reverse transcriptase (Promega). Twenty cycles of amplification were performed by using the following primers in AML1/ETO: 5′-GGCTGGCAATGATGAAAACT and 5′-CGCCATTCAAGGCTGTAGGAG. A second round of PCR was performed under identical conditions by using the first round product (diluted 1:20) as a template and the following nested primers in AML1/ETO: 5′-CACCTACCACAGAGCCATC and 5′-GTTGTCGGTGTAAATGAA.
Western Blot Analysis. Bone marrow cells were harvested from mice at 6-10 weeks of age. Lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (0.1% SDS/1.0% polyoxyethylene nonyl phenol/0.5% sodium deoxycholate/1 mM EDTA in PBS) supplemented with 1 mM PMSF and a protease inhibitor mixture (Roche). Nitrocellulose membranes were probed with polyclonal anti-hETO antisera (kindly provided by P. Erickson, University of Colorado, Denver, CO). The membranes were then stripped and reprobed with a monoclonal antibody against β-actin (Santa Cruz Biotechnology).
Flow Cytometric Analysis. Cells were stained with directly conjugated antibodies to Gr1, B220, CD3, CD4, or CD8 (BD Pharmingen). Double-negative thymocytes were enumerated by gating out cells positive for CD3, CD4, CD8, or B220 and analyzing for expression of CD44 and CD25. Twenty-five thousand scatter-gated events were acquired on a FACScan (BD Biosciences) and analyzed with flowjo 4.5.1 software (Treestar, Ashland, OR).
Hematologic Analysis. Complete blood counts with automated five-part differentials were determined with a Mascot Hemavet cell counter (CDC Technologies, Oxford, CT). For progenitor assays, 1.25 × 104 bone marrow cells or 1.25 × 105 splenocytes were plated in duplicate 1.25-ml methylcellulose cultures supplemented with recombinant mouse IL-3 (10 ng/ml), IL-6 (10 ng/ml), stem cell factor (SCF; 50 ng/ml), and erythropoietin (3 units/ml) (M3434, StemCell Technologies, Vancouver). Myeloid progenitors (CFU-G, CFU-GM, CFU-M) were enumerated after 8 days of culture. The plates were then stained with benzidine, and burst-forming units-erythroid (BFU-E) were counted, as described (21). Reticulocyte analysis was performed as described, with minor modifications (22). Whole blood was diluted 1:1,000 in fluorescence-activated cell sorter (FACS) buffer (PBS with 0.2% BSA, 0.01% NaN3) and stained with acridine orange (0.1 mcg/ml, Molecular Probes). Ten thousand scatter-gated red cells were acquired on a FACScan and analyzed with cellquest software (BD Biosciences). Hemolysis was induced in control mice with a single s.c. injection of phenylhydrazine (60 mg/kg, Sigma). Peripheral blood and bone marrow smears were stained with May-Grünwald/Giemsa.
Tissue Culture. Splenocytes were cultured in RPMI medium 1640 supplemented with 10% FCS and IL-3 (10 ng/ml, R & D Systems) with or without stem cell factor (100 ng/ml, R & D Systems). A density of 5 × 105 nonadherent cells per ml was maintained with twice weekly media changes. Trypan blue-excluding cells were counted at each media change. Kasumi-1 cells (kindly provided by L. Healy, Institute of Cancer Research, London) were grown in RPMI medium 1640/20% FCS.
Statistical Analysis. Cumulative probability of survival was estimated by the Kaplan-Meier method. Differences in survival were estimated by using the log-rank test. All other differences were assessed by using a two-tailed t test.
Results
Generation of Sca+/AE Mice. Several lines of evidence suggest that the leukemia-initiating cell in t(8;21) AML has a primitive stem/progenitor phenotype (18, 19, 23). We therefore chose to target the AML1/ETO cDNA to the Sca1 (Ly6A) locus, because we demonstrated previously that this strategy yields high-level transgene expression in the hematopoietic stem/progenitor compartment in adult mice (20). We engineered a targeting construct consisting of a human AML1/ETO cDNA upstream of an ires EGFP cassette (Fig. 6A, which is published as supporting information on the PNAS web site). Using an external probe from the 3′-flanking region of the Sca1 gene for Southern blot analysis, we identified six correctly targeted embryonic stem cell clones from three independent electroporations (Fig. 6B). The PGK-neo cassette was not excised because previous experiments in our laboratory demonstrated that retention of this selectable marker had no discernible effect on expression of the targeted Sca1 allele or on the tightly linked Ly6 genes Ly6G (Gr1), Ly6M, or Sca2 (20, 24). Chimeric founders were created by using three of these clones. Two of the lines transmitted the mutant allele through the germ line. Both of these were used to establish colonies of mice in a C57BL/6J × 129/SvJ background.
Initial strain characterization and tumor watches were performed at the F1 generation. The two independently derived Sca1+/AML1-ETO ires EGFP lines (abbreviated hereafter as Sca+/AE) were housed in separate specific pathogen-free barrier facilities and had identical phenotypes in all experiments. One line (line 8) was backcrossed to C57BL/6J (at least six generations).
Sca+/AE Mice Are Viable and Express the Transgene. In contrast to AML1+/AML1-ETO mice, Sca+/AE mice are viable. Heterozygous mutant mice are born in the predicted Mendelian ratios at the F1 generation and in subsequent backcrosses (Fig. 1A). Mutant Sca+/AE mice are healthy and fertile and display no overt developmental defects.
Fig. 1.

Transgene expression. (A) Genotypes of a representative F1 litter are shown. Genomic DNA extracted from peripheral blood leukocytes was amplified in a multiplex PCR containing a shared forward primer and specific reverse primers to detect the WT or targeted (AE) alleles. WT and heterozygous Sca+/AE mice are born in Mendelian ratios. (B) AML1/ETO mRNA expression is detectable by RT-PCR in peripheral blood leukocytes from F1 heterozygous mice. The positive lanes correlate with the genotypes shown above in A. (C) Western blot analysis of bone marrow extracts from the indicated mice demonstrates expression of AML1/ETO protein in Sca+/AE mice (2.0 × 105 cell equivalents per lane) and in the positive control Kasumi-1 cell line (4.0 × 104 cell equivalents per lane).
Hematopoiesis was normal at baseline in heterozygous Sca+/AE mice. At age 5-8 weeks, blood counts, bone marrow differentials, and cellularity were largely within normal limits (Table 1). There was a modest increase in the total WBC count and a decrease in the frequency of myeloid progenitors in bone marrow from heterozygous mice (P < 0.05). This finding is consistent with reports that AML1/ETO expression can affect colony formation and self-renewal capacity without impairing terminal differentiation of myeloid cells (25-27).
Table 1. Hematologic analysis in young Sca+/AE mice.
| Genotype | WBC, 103/μl | ANC, 103/μl | Hb, g/dl | Plt, 103/μl | CFU-GM/105 BMC | BFU-E/105 BMC | CFU-GEMM/105 BMC |
|---|---|---|---|---|---|---|---|
| Sca+/+ (n = 6) | 8.6 ± 0.4 | 1,947 ± 234 | 14.4 ± 0.7 | 705 ± 46 | 156 ± 14 | 11.8 ± 1.8 | 7.2 ± 1.5 |
| Sca+/AE (n = 6) | 11.2 ± 1.1* | 2,497 ± 428 | 13.9 ± 0.7 | 673 ± 38 | 113 ± 9.5* | 15.7 ± 2.9 | 7.3 ± 0.7 |
Results are displayed as mean values ± SEM. ANC, absolute neutrophil count; Plt, platelets; BMC, bone marrow cells; BFU-E, burst-forming units-erythroid.
, P < 0.05.
AML1/ETO mRNA expression was readily detectable in peripheral blood leukocytes from heterozygous animals (Fig. 1B). Western blot analysis of bone marrow extracts prepared from young heterozygous mutant mice demonstrated full-length AML1/ETO protein in total bone marrow at ≈5% of the level in the positive control Kasumi-1 cell line (Fig. 1C).
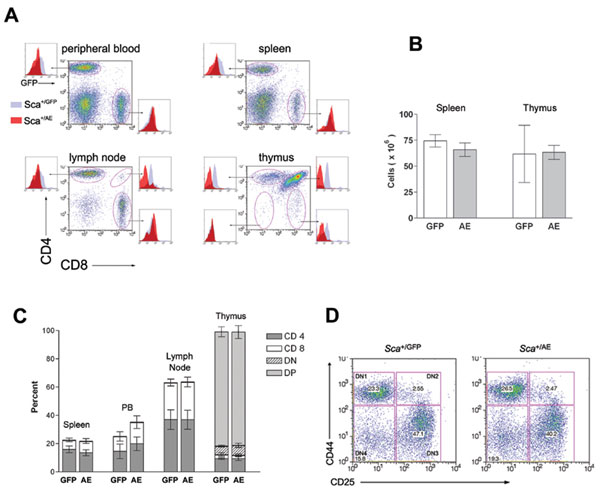
Incorporation of a bicistronic cassette in our targeting vector allowed us to use GFP activity as a surrogate for AML1/ETO expression in flow cytometric assays. Sca+/GFP mice, previously generated in our laboratory (20), served as a control for these experiments because the transgene was integrated by means of homologous recombination in precisely the same position in exon II of the Sca1 gene in both Sca+/GFP and Sca+/AE mice. GFP was highly expressed in peripheral blood and bone marrow cells from Sca+/AE mice. Two-color analysis revealed that myeloid (Gr1+) cells expressed the transgene in a pattern that was indistinguishable from control Sca+/GFP mice, as predicted (Fig. 2A).
Fig. 2.

Flow cytometric analysis. (A) Flow cytometric analysis was performed by using cells from mice heterozygous at the Sca1 locus for a null, GFP, or AML1/ETO ires GFP allele. Most of the myeloid (Gr1+) cells express GFP in the bone marrow and peripheral blood of Sca+/GFP and Sca+/AE mice. (B) In contrast to identically targeted Sca+/GFP mice, Sca+/AE mice demonstrate low level transgene expression in B220+ cells. (C) T cells from Sca+/AE mice express significantly lower transgene levels compared with cells from Sca+/GFP mice.
Analysis of transgene expression in the lymphoid compartment revealed unexpected differences between the strains. GFP expression was significantly lower in thymocytes and some peripheral T cells from Sca+/AE compared with Sca+/GFP mice (Fig. 2C). Because Runx factors are important for T cell development (28-30), the possibility was raised that AML1/ETO expression in Sca+/AE mice is deleterious to T cells, leading to impairment of differentiation, apoptosis, or silencing of the transgene. Expression of GFP was indistinguishable in peripheral blood, splenic, or mesenteric lymph node CD8+ T cells from Sca+/GFP and Sca+/AE mice (Fig. 7A, which is published as supporting information on the PNAS web site). In contrast, GFP expression was significantly lower in CD4+ T cells in these tissues and in immature thymic double positive (DP) and single positive (SP) CD4 and CD8 cells in Sca+/AE compared with Sca+/GFP mice (Fig. 7A). There was no significant change in the cellularity of thymus or spleen in Sca+/AE mice (Fig. 7B), nor were there significant differences in the size of T cell subsets in lymphoid organs from these mice (Fig. 7C). Furthermore, there was no evidence of a differentiation block in immature double negative (DN) thymocytes (Fig. 7D). Taken together, these results suggest that the unexpected down-regulation of transgene expression in T cells from Sca+/AE mice could be a compensatory response that allows cells to escape the alternative fates of maturation arrest or apoptosis.
The level of GFP expression in B cells from Sca+/AE mice diverged even further from control Sca+/GFP cells. Fully differentiated B cells are present in normal numbers in spleen, peripheral blood, and bone marrow, but these cells expressed GFP at levels barely detectable above background (Fig. 2B). Because Sca1 protein expression is not proportionately reduced in these cells (not shown), these results suggest that expression of the AML1/ETO mutant allele is selectively down-regulated in thymocytes, peripheral CD4+ T cells, and B cells.
Cells from Sca+/AE Mice Have Increased Viability. We wished to determine whether AML1/ETO expression was sufficient to alter growth of primary cells in culture. Previous reports (8, 12) have shown that bone marrow cells expressing AML1/ETO have extensive serial replating capacity in methylcellulose cultures. Splenocytes from Sca+/AE and control Sca+/GFP mice were cultured in cytokine-supplemented media (see Materials and Methods). Cells from all control mice (n = 4) died within 4 weeks. In contrast, splenocytes from Sca+/AE mice (n = 7 of 8) survived for up to 8 months in cultures containing IL-3 alone (Fig. 3) but remained growth factor-dependent during this period. Stem cell factor provided no additive effect to cultures containing IL-3 and was not sufficient to sustain long-term growth of Sca+/AE cells (data not shown). Although the survival of Sca+/AE cells was markedly extended, their rate of proliferation was extremely low, resulting in little to no net expansion of the cells in culture. Morphologic examination of the cultured cells revealed a mixture of mast cells and immature myeloid cells (Fig. 8, which is published as supporting information on the PNAS web site) with a concordant immunophenotype (CD3-B220-Gr1loCD11b-Kit+Sca+). Aliquots taken from all cultures at 8 months had persistent AML1/ETO mRNA expression by quantitative RT-PCR (not shown).
Fig. 3.

In vitro analysis. Splenocytes from Sca+/AE or WT littermate mice were cultured in RPMI medium 1640/10% FCS with IL-3 (10 ng/ml). Transgenic cells remain viable for significantly longer than WT cells. Representative data from one of three experiments are shown.
Sca+/AE Mice Develop an MPD. Although hematopoiesis appeared normal in young (between 5 and 8 weeks of age) Sca+/AE mice, more than half of the mice developed an increase in peripheral blood neutrophils and reticulocytes, bone marrow myeloid hyperplasia, splenomegaly, and extramedullary hematopoiesis (Table 2). The peripheral blood, bone marrow, and spleen of these mice were marked by an expansion of predominantly mature myeloid cells (CD11b+Gr1+) without evidence of maturation block (Fig. 4 and data not shown). Extramedullary hematopoiesis significantly disrupted the follicular architecture in the spleen (Fig. 4). The frequency of hematopoietic progenitors was increased in the spleens of affected mice (364 ± 45 vs. 198 ± 2 CFU-GM/106 splenocytes). The disorder was not lethal in most cases, although one animal in a cohort of 23 was moribund at the time of killing, presumably due to severe anemia (Hb = 4.1 g/dl). To objectively score for this phenotype, we adopted an operational definition that identifies animals >2 SDs above the mean of control values for both (i) peripheral blood absolute neutrophil count (>2,000/mcL), and (ii) spleen weight (≥0.25 g). The penetrance of this syndrome was 52% at 1 year and 82% at 14 months (Fig. 5). No clonal cytogenetic abnormalities were detected in splenocytes from Sca+/AE mice (n = 2) with severe extramedullary hematopoiesis. This syndrome conforms to the definition of MPD put forth recently in the “Bethesda proposal” for classification of murine myeloid neoplasms (31). Of note, lethality, monoclonality, and transplantability are not required to meet this standard of MPD (31).
Table 2. Hematologic analysis in aged Sca+/AE mice.
| Differential
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | WBC, 103/μl | Hb, g/dl | Plt, 103/μl | ANC, 103/μl | Early/mid | Late | Erythroid | Lymphoid | M/E | Retics,* % | Spleen weight,† g |
| Sca+/+ (n = 12) | 4.26 ± 0.51 | 13.9 ± 0.62 | 895 ± 71 | 1,101 ± 107 | 5.6 ± 0.4 | 60.1 ± 2.7 | 32.6 ± 2.3 | 1.8 ± 0.2 | 2.2:1 | 3.1 ± 0.2 | 0.15 ± 0.02 |
| Sca+/AE (n = 12) | 9.99 ± 1.68‡ | 13.0 ± 0.56 | 1,069 ± 119 | 4,702 ± 1,000‡ | 6.0 ± 0.7 | 74.4 ± 2.0§ | 18.5 ± 2.2§ | 1.0 ± 0.1 | 5.1:1§ | 6.5 ± 2.0 | 0.27 ± 0.04¶ |
Peripheral blood counts were obtained from consecutive, unselected pairs of littermate mice (age 9-12 months). Bone marrow differential counts and myeloid/erythroid (M/E) ratios were determined from manual 200-cell counts of May-Grünwald/Giemsa-stained bone marrow slides. Results are displayed as mean values ± SEM. Early/mid, myeloblast-myelocyte; late, metamyelocyte-neutrophil. Plt, platelets; ANC, absolute neutrophil count.
Sca+/+, n = 9; Sca+/AE, n = 10.
Sca+/+, n = 23; Sca+/AE, n = 18.
P < 0.005.
P < 0.001.
P < 0.05.
Fig. 4.

Morphologic analysis of MPD. Peripheral blood and bone marrow slides show evidence of myeloid hyperplasia with loss of erythroid and lymphoid precursors in the bone marrow and polychromasia in peripheral blood of Sca+/AE mice compared with WT littermate mice. Increased extramedullary hematopoiesis with disruption of follicular architecture is evident in the spleens of Sca+/AE mice.
Fig. 5.

Survival analysis. Kaplan-Meier plot demonstrates high penetrance and long latency of a nonlethal MPD in Sca+/AE mice compared with WT littermates (P < 0.0001).
To determine whether this MPD is transplantable, fresh or cryopreserved splenocytes from affected Sca+/AE mice were transferred by i.p. or i.v. injection into unirradiated immunocompetent congenic (n = 36) or immunodeficient NOD/scid (n = 14) recipients. All recipients remained healthy and survived for up to 6 months, although donor GFP+ cells were not detectable in peripheral blood or spleen. Sublethally irradiated (300 cGy) nude/NOD/scid (n = 3) or β2-microglobulin/NOD/scid (n = 10) mice engrafted with donor Sca+/AE cells (80-95% and 50-100% chimeric in bone marrow and spleen, respectively) but did not develop neutrophilia or splenomegaly within 6 months. Although these experiments suggest that the MPD is not transplantable, the limited lifespan and predisposition to lymphoma that characterize these immunodeficient strains preclude surveillance for diseases of long latency.
The reticulocytosis noted in Sca+/AE mice does not seem to be a response to hemolysis in these mice because lethal irradiation (1,000 cGy) induced no change in their hemoglobin over 72 h, whereas control mice irradiated after a single dose of phenylhydrazine that induces brisk hemolysis developed significant anemia and died on day 3 after irradiation (data not shown). This result suggests that the reticulocytosis in Sca+/AE mice is more likely a consequence of extramedullary hematopoiesis.
Unexpectedly, Sca+/AE mice also developed skin pathology. Skin biopsies of the ears of Sca+/AE mice revealed epidermal hyperplasia in all animals as early as 4 weeks of age (data not shown). At later time points, hyperkeratosis and ulceration were evident histopathologically in the majority of specimens. These lesions were most extensive in the ears, often bilateral, and progressed to squamous cell carcinoma in 3 of 23 mice by 1 year of age (T.S.F. and T.A.G., unpublished results).
Discussion
In this study, we targeted expression of AML1/ETO using the well characterized mouse HSC marker Sca1, encoded by the Ly6A locus. Heterozygous mutant Sca+/AE mice are viable and express the AML1/ETO transgene in all hematopoietic tissues. Although Sca+/AE mice did not develop acute leukemia, >80% of the animals developed a spontaneous MPD by 14 months of age. In addition, splenocytes from Sca+/AE mice have markedly extended survival in vitro and a progressive rise in myeloid clonogenic progenitors throughout life. Interestingly, nearly 15% of the Sca+/AE mice also developed squamous cell carcinomas of the skin, indicating that AML1/ETO can contribute to transformation of nonhematopoietic cells.
Because of the dominant negative effect of AML1/ETO on AML1 function, this targeting strategy was deemed “high risk” for an embryonic lethal phenotype, similar to targeted integration of AML1/ETO into the AML1 locus (7, 8). We anticipated that Sca+/AE mice would be viable because (i) they are WT at the AML1 locus (rather than haploinsufficient, as in the case of AML1+/AML1-ETO mice) and (ii) Sca1 and AML1 expression are only partially overlapping in the midgestation embryo (32). Indeed, the Sca+/AE mice are viable and express the transgene broadly in hematopoietic cells.
Using flow cytometric analysis of GFP as a surrogate for AML1/ETO expression, we found that the transgene is at the limit of detection in B lymphocytes from bone marrow, spleen, and peripheral blood. This finding is in sharp contrast to identically targeted Sca+/GFP mice, in which nearly all B220+ cells coexpress GFP. Similarly, immature thymocytes and CD4+ T cells in spleen, lymph nodes, and peripheral blood of Sca+/AE mice have significantly reduced transgene expression compared with Sca+/GFP mice. This finding corresponds closely to the requirement for Runx factors in lymphocyte development. Runx1 is expressed in B cells (33) and is required for generation of B220+ cells after bone marrow transplantation (30). Similarly, both Runx1 and Runx3 are critical for T cell development (29, 30). We hypothesize that in vivo selection for lymphocytes with low (or absent) AML1/ETO expression in the Sca+/AE mice may be a compensatory response to the “toxicity” of dominant negative inhibition of Runx function in B and CD4+ T lymphocytes. In contrast, Sca+/AE granulocytes tolerate high-level transgene expression in concordance with the lack of a requirement for Runx factors in terminal myeloid differentiation. This finding is consistent with the tendency of human t(8;21) AML blasts to retain their capacity for terminal myeloid differentiation despite continued AML1/ETO mRNA expression.
In contrast to other AML1/ETO models, the Sca+/AE mice develop a spontaneous MPD at high penetrance. Conditional activation of AML1/ETO from the AML1 promoter induced lymphomas in 2 of 15 mice with a latency of 1 year (12). Human CD34+ peripheral blood cells retrovirally transduced with AML1/ETO retain self-renewal and multilineage differentiation capacity in culture for >7 months but do not cause disease in NOD/scid recipients (34). Retroviral transduction of murine bone marrow cells with AML1/ETO followed by transplantation into WT recipients performed by several laboratories has led to varying degrees of altered hematopoiesis, including HSC expansion (14), or impaired B cell (17) or myeloid differentiation (35). Transgenic mice expressing AML1/ETO, either from the MRP8 promoter (13) or by using a tetracycline-inducible system (15), have no detectable in vivo hematopoietic phenotype. Finally, transient expression of an AML1/ETO transgene in zebrafish embryos caused vascular abnormalities and dysplastic myeloid and erythroid differentiation (36). Taken together, these results support the notion that AML1/ETO expression is not sufficient to induce leukemia but suggest that differences in the cellular compartment expressing AML1/ETO or differences in the transgene dose may be critical determinants for the capacity of AML1/ETO to alter hematopoiesis. Recent experiments with other fusion genes associated with human leukemia (e.g., PML/RARα and AML1/MDS1/EVI1) support the surprising conclusion that low levels of transgene expression may be optimal for high disease penetrance (37, 38).
Young (6- to 8-week-old) Sca+/AE mice uniformly develop epidermal hyperplasia. Within 1 year, these lesions progress to squamous cell carcinomas in ≈15% of cases. AML1/ETO may initiate this process by means of direct or indirect mechanisms. The bulge region of the hair follicle contains a population of self-renewing, multipotent stem cells that may be the target of transformation in squamous carcinomas (39). We recently demonstrated that Sca-1 is expressed in mammary gland epithelial progenitor cells (40), raising the possibility that the Sca1 promoter may also direct expression of AML1/ETO to skin progenitor cells in Sca+/AE mice. AML1/ETO may substitute for classical initiators of epithelial carcinogenesis (e.g., mutant H-ras), but the incomplete penetrance and long latency of these tumors suggest that additional factors are required for the complete phenotype. A chronic inflammatory response to the premalignant proliferative skin lesions may provide the stimulus for tumor progression (41). Alternatively, AML1/ETO may initiate squamous carcinomas through a non-cell intrinsic mechanism. AML1/ETO expression in hematopoietic cells trafficking through the skin may induce keratinocyte proliferation by means of paracrine factors. There is increasing evidence that hematopoietic cells can participate in epithelial transformation (42).
The high penetrance of skin pathology in Sca+/AE mice raises the possibility that the hematologic abnormalities in these mice are secondary. It is unlikely that the MPD is entirely a reactive process for the following reasons: (i) hematopoietic cells from Sca+/AE mice have a cell-autonomous increase in survival in vitro, (ii) some Sca+/AE mice develop MPD without dermatitis, and (iii) the dermatitis is not severe in most cases. Nevertheless, it remains a formal possibility that the MPD is at least in part a reactive process. This hypothesis will be tested using reciprocal bone marrow transplantation experiments.
In summary, targeted expression of the AML1/ETO fusion protein associated with t(8;21) acute myeloid leukemia using the Ly6A (Sca1) locus induces a highly penetrant, long latency MPD. Importantly, these mice do not spontaneously develop AML. This finding suggests that additional secondary genetic alterations must be present, in combination with AML1/ETO, in order for AML to develop. Based on existing clinical data, hemizygous loss of AML1 or gain of activated receptor tyrosine kinase alleles (e.g., c-kit and Flt3) are leading candidates. The Sca+/AE mice should be a useful platform for modeling the ability of AML1/ETO to cooperate with these and other specific secondary mutations in the pathogenesis of AML.
Supplementary Material
Acknowledgments
We thank Dr. Chih-Lin Hsieh (University of Southern California) for cytogenetic analyses. This work was supported by National Institutes of Health Grant K08 HL03872 and by American Cancer Society Grant IRG 58-010-42 (to T.A.G.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AML, acute myeloid leukemia; Sca+/AE, Sca1+/AML1-ETO ires EGFP; CBF, core-binding factor; MPD, myeloproliferative disorder; NOD, nonobese diabetic; scid, severe combined immunodeficient; HSC, hematopoietic stem cell.
References
- 1.Downing, J. R. (1999) Br. J. Haematol. 106, 296-308. [DOI] [PubMed] [Google Scholar]
- 2.Nucifora, G. & Rowley, J. D. (1995) Blood 86, 1-14. [PubMed] [Google Scholar]
- 3.Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G. & Downing, J. R. (1996) Cell 84, 321-330. [DOI] [PubMed] [Google Scholar]
- 4.Wang, Q., Stacy, T., Binder, M., Marin-Padilla, M., Sharpe, A. H. & Speck, N. A. (1996) Proc. Natl. Acad. Sci. USA 93, 3444-3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang, Q., Stacy, T., Miller, J. D., Lewis, A. F., Gu, T. L., Huang, X., Bushweller, J. H., Bories, J. C., Alt, F. W., Ryan, G., et al. (1996) Cell 87, 697-708. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki, K., Yagi, H., Bronson, R. T., Tominaga, K., Matsunashi, T., Deguchi, K., Tani, Y., Kishimoto, T. & Komori, T. (1996) Proc. Natl. Acad. Sci. USA 93, 12359-12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yergeau, D. A., Hetherington, C. J., Wang, Q., Zhang, P., Sharpe, A. H., Binder, M., Marin-Padilla, M., Tenen, D. G., Speck, N. A. & Zhang, D. E. (1997) Nat. Genet. 15, 303-306. [DOI] [PubMed] [Google Scholar]
- 8.Okuda, T., Cai, Z., Yang, S., Lenny, N., Lyu, C. J., van Deursen, J. M., Harada, H. & Downing, J. R. (1998) Blood 91, 3134-3143. [PubMed] [Google Scholar]
- 9.Kusec, R., Laczika, K., Knobl, P., Friedl, J., Greinix, H., Kahls, P., Linkesch, W., Schwarzinger, I., Mitterbauer, G., Purtscher, B., et al. (1994) Leukemia 8, 735-739. [PubMed] [Google Scholar]
- 10.Nucifora, G., Larson, R. A. & Rowley, J. D. (1993) Blood 82, 712-715. [PubMed] [Google Scholar]
- 11.Wiemels, J. L., Xiao, Z., Buffler, P. A., Maia, A. T., Ma, X., Dicks, B. M., Smith, M. T., Zhang, L., Feusner, J., Wiencke, J., et al. (2002) Blood 99, 3801-3805. [DOI] [PubMed] [Google Scholar]
- 12.Higuchi, M., O'Brien, D., Kumaravelu, P., Lenny, N., Yeoh, E. J. & Downing, J. R. (2002) Cancer Cell 1, 63-74. [DOI] [PubMed] [Google Scholar]
- 13.Yuan, Y., Zhou, L., Miyamoto, T., Iwasaki, H., Harakawa, N., Hetherington, C. J., Burel, S. A., Lagasse, E., Weissman, I. L., Akashi, K. & Zhang, D. E. (2001) Proc. Natl. Acad. Sci. USA 98, 10398-10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Guzman, C. G., Warren, A. J., Zhang, Z., Gartland, L., Erickson, P., Drabkin, H., Hiebert, S. W. & Klug, C. A. (2002) Mol. Cell. Biol. 22, 5506-5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhoades, K. L., Hetherington, C. J., Harakawa, N., Yergeau, D. A., Zhou, L., Liu, L. Q., Little, M. T., Tenen, D. G. & Zhang, D. E. (2000) Blood 96, 2108-2115. [PubMed] [Google Scholar]
- 16.Buchholz, F., Refaeli, Y., Trumpp, A. & Bishop, J. M. (2000) EMBO Rep. 1, 133-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwieger, M., Lohler, J., Friel, J., Scheller, M., Horak, I. & Stocking, C. (2002) J. Exp. Med. 196, 1227-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonnet, D. & Dick, J. E. (1997) Nat. Med. 3, 730-737. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto, T., Weissman, I. L. & Akashi, K. (2000) Proc. Natl. Acad. Sci. USA 97, 7521-7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanson, P., Mathews, V., Marrus, S. H. & Graubert, T. A. (2003) Exp. Hematol. 31, 159-167. [DOI] [PubMed] [Google Scholar]
- 21.Cooper, M. C., Levy, J., Cantor, L. N., Marks, P. A. & Rifkind, R. A. (1974) Proc. Natl. Acad. Sci. USA 71, 1677-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graubert, T. A., Hug, B. A., Wesselschmidt, R., Hsieh, C. L., Ryan, T. M., Townes, T. M. & Ley, T. J. (1998) Nucleic Acids Res. 26, 2849-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Passegue, E., Jamieson, C. H., Ailles, L. E. & Weissman, I. L. (2003) Proc. Natl. Acad. Sci. USA 100, Suppl. 1, 11842-11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson, J. M., Johnson, M. H., Zimonjic, D. B. & Graubert, T. A. (2000) Blood 95, 3125-3132. [PubMed] [Google Scholar]
- 25.Ahn, M. Y., Huang, G., Bae, S. C., Wee, H. J., Kim, W. Y. & Ito, Y. (1998) Proc. Natl. Acad. Sci. USA 95, 1812-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westendorf, J. J., Yamamoto, C. M., Lenny, N., Downing, J. R., Selsted, M. E. & Hiebert, S. W. (1998) Mol. Cell. Biol. 18, 322-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulloy, J. C., Cammenga, J., MacKenzie, K. L., Berguido, F. J., Moore, M. A. & Nimer, S. D. (2002) Blood 99, 15-23. [DOI] [PubMed] [Google Scholar]
- 28.Taniuchi, I., Osato, M., Egawa, T., Sunshine, M. J., Bae, S. C., Komori, T., Ito, Y. & Littman, D. R. (2002) Cell 111, 621-633. [DOI] [PubMed] [Google Scholar]
- 29.Woolf, E., Xiao, C., Fainaru, O., Lotem, J., Rosen, D., Negreanu, V., Bernstein, Y., Goldenberg, D., Brenner, O., Berke, G., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 7731-7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ichikawa, M., Asai, T., Saito, T., Yamamoto, G., Seo, S., Yamazaki, I., Yamagata, T., Mitani, K., Chiba, S., Hirai, H., et al. (2004) Nat. Med. 10, 299-304. [DOI] [PubMed] [Google Scholar]
- 31.Kogan, S. C., Ward, J. M., Anver, M. R., Berman, J. J., Brayton, C., Cardiff, R. D., Carter, J. S., de Coronado, S., Downing, J. R., Fredrickson, T. N., et al. (2002) Blood 100, 238-245. [DOI] [PubMed] [Google Scholar]
- 32.de Bruijn, M. F., Ma, X., Robin, C., Ottersbach, K., Sanchez, M. J. & Dzierzak, E. (2002) Immunity 16, 673-683. [DOI] [PubMed] [Google Scholar]
- 33.North, T. E., Stacy, T., Matheny, C. J., Speck, N. A. & de Bruijn, M. F. (2004) Stem Cells 22, 158-168. [DOI] [PubMed] [Google Scholar]
- 34.Mulloy, J. C., Cammenga, J., Berguido, F. J., Wu, K., Zhou, P., Comenzo, R. L., Jhanwar, S., Moore, M. A. & Nimer, S. D. (2003) Blood 102, 4369-4376. [DOI] [PubMed] [Google Scholar]
- 35.Grisolano, J. L., O'Neal, J., Cain, J. & Tomasson, M. H. (2003) Proc. Natl. Acad. Sci. USA 100, 9506-9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalev-Zylinska, M. L., Horsfield, J. A., Flores, M. V., Postlethwait, J. H., Vitas, M. R., Baas, A. M., Crosier, P. S. & Crosier, K. E. (2002) Development (Cambridge, U.K.) 129, 2015-2030. [DOI] [PubMed] [Google Scholar]
- 37.Westervelt, P., Lane, A. A., Pollock, J. L., Oldfather, K., Holt, M. S., Zimonjic, D. B., Popescu, N. C., DiPersio, J. F. & Ley, T. J. (2003) Blood 102, 1857-1865. [DOI] [PubMed] [Google Scholar]
- 38.Ren, R. (2004) Curr. Opin. Hematol. 11, 25-34. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Losada, J. & Balmain, A. (2003) Nat. Rev. Cancer 3, 434-443. [DOI] [PubMed] [Google Scholar]
- 40.Welm, B. E., Tepera, S. B., Venezia, T., Graubert, T. A., Rosen, J. M. & Goodell, M. A. (2002) Dev. Biol. 245, 42-56. [DOI] [PubMed] [Google Scholar]
- 41.Coussens, L. M. & Werb, Z. (2002) Nature 420, 860-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coussens, L. M., Tinkle, C. L., Hanahan, D. & Werb, Z. (2000) Cell 103, 481-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}