

Abstract
Macrophages play essential roles in the response to injury and infection and contribute to the development and/or homeostasis of the various tissues they reside in. Conversely, macrophages also influence the pathogenesis of metabolic, neurodegenerative, and neoplastic diseases. Mechanisms that contribute to the phenotypic diversity of macrophages in health and disease remain poorly understood. Here we review the recent application of genome-wide approaches to characterize the transcriptomes and epigenetic landscapes of tissue-resident macrophages. These studies are beginning to provide insights into how distinct tissue environments are interpreted by transcriptional regulatory elements to drive specialized programs of gene expression.
Diverse macrophage phenotypes in health and disease
Macrophages are among the most phenotypically diverse cell types of mammalian organisms (1, 2). They inhabit all or nearly all tissues under healthy conditions where they play important roles as sentinels of infection and injury. These functions are enabled by the expression of a multitude of cell surface and internal receptors that recognize microbial associated molecular patterns (MAMPs) and/or damage-associated molecular patterns (DAMPS), exemplified by toll-like receptors (TLRs) (3). Engagement of these receptors by microbial components, such as bacterial lipopolysaccharide (LPS), initiates signaling cascades that lead to the activation of latent transcription factors, including NF-κB, interferon regulatory factors (IRFs), and members of the AP-1 family (4, 5). These factors, in turn, function to activate hundreds of genes that play key roles in the orchestration of the innate immune response and that influence the development of adaptive immunity (6, 7). In addition to this sentinel function, macrophages are professional phagocytes, serving to clear bacteria, apoptotic cells, and a diverse range of host-derived and environmental debris, thereby contributing to an additional layer of immunity and tissue homeostasis (2).
While the sentinel and phagocytic functions comprise central and shared macrophage characteristics, the various populations of tissue-resident macrophages also exhibit a striking range of phenotypic diversity (1). Consider, for example, the diverse morphologies and functions of microglia, Kupffer cells, alveolar macrophages, Langerhans cells, peritoneal macrophages, and splenic red pulp macrophages. Each of these cells retain phagocytic and sentinel functions, but have also acquired distinct patterns of gene expression that are linked to their tissue-specific functional roles, e.g., synaptic pruning in the case of microglia (8), clearance of surfactant by alveolar macrophages (9), and removal of senescent red blood cells by splenic macrophages (10).
Although macrophages normally play adaptive roles in immunity, tissue repair and homeostasis, they are also implicated in a broad spectrum of human diseases (2). For example, macrophages contribute to all phases of the development of atherosclerosis, from formation of the initial fatty streaks to the rupture of complex lesions that result in myocardial infarction (11). Adipose tissue macrophages and Kupffer cells are implicated in metabolic diseases that include insulin resistance and non-alcoholic steatohepatitis (12–14). Microglia have been linked to numerous neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (15). As an example, genome wide association studies provide strong evidence that genomic variants in or near genes expressed in microglia, such as TREM2, are associated with increased risk of Alzheimer’s disease (16). In cancer, macrophages have been shown to play complex roles in tumor initiation, growth, metastasis, and immune evasion (17, 18).
These observations raise a number of questions, including: 1) How are distinct macrophage identities achieved? 2) To what extent does their developmental origin specify their functional properties in relation to tissue-specific environmental signals? 3) If tissue environment is important, what are the signals driving macrophage specialization? 4) What are the mechanisms that lead to pathogenic roles of macrophages in diseases such as atherosclerosis, metabolic disease, neurodegenerative diseases, and cancer? 5) Do the phenotypes of resident macrophages change in response to a primary disease process and/or are pathogenic activities the result of infiltration by monocyte-derived macrophages? 6) Is it possible to alter macrophage phenotypes for therapeutic purposes?
Genome-wide approaches to define macrophage identity and function
Among the most widely used and successful approaches to define cellular identity is the use of antibodies to mark specific cell surface or internal proteins and to quantify these proteins by flow cytometry. This approach can be considered to define what a cell is ‘wearing’. In much the same way that a uniform enables one to deduce roles of a person dressed as a policemen or fireman, specific combinations of markers can be used define cell types with different functional properties, e.g., macrophages, T cells, and B cells. An alternative approach to define cell identity made possible by the development of chromatin immunoprecipitation linked to massively parallel DNA sequencing (ChIP-Seq), is to define the total repertoire of transcriptional regulatory elements in that cell, i.e., its enhancers and promoters (19). These elements are selected from the genome in a cell-specific fashion and provide the cell with its transcriptional identity and regulatory potential. Knowledge of such elements not only enables an understanding of the program of gene expression observed in that cell, but in principle also enables predictions of how the cell will respond to new internal and external signals. Thus, in contrast to flow cytometry, genomic approaches can be considered to provide insights into what the cell is ‘thinking’. From this perspective, answers to some of the questions posed above may be attained by systematic evaluation of the enhancer and promoter elements of diverse tissue-resident macrophage populations under normal and disease conditions. In the following sections, we briefly review general principles of enhancer selection and function and recent findings relevant to this process in various macrophage model systems and in tissue-resident macrophages.
Enhancers and super-enhancers regulate cellular identity and function
Gene regulation at the level of transcription is achieved through the coordinated functions of enhancers and promoters. Promoters activated by RNA polymerase II represent the sites of initiation of mRNAs and long non-coding RNAs. While promoters are often key points of regulation by signal-dependent transcription factors, they are often not sufficient to direct the highly divergent levels of transcription of primary transcripts observed across cell types or to fully capture cell-specific responses to developmental and homeostatic signals (20, 21). These functions are frequently mediated by enhancers, which are defined as DNA sequences that ‘enhance’ transcription from their target promoters (22). Enhancers are generally thought to act by looping to their target promoters and increasing the recruitment of RNA Polymerase II and/or its transition from a paused to elongating form (23). Thus, a major effort in molecular and developmental biology is to define the mechanisms by which different cell types select their specific complement of enhancers, and how they, in turn, act to regulate target genes.
General features of enhancers are indicated in Figure 1. Eukaryotic DNA is packaged in the nucleus through its association with histone octamers to form nucleosomes, the basic unit of chromatin (23). Each nucleosome consists of approximately 147 nucleotides of DNA wrapped around a heterodimeric octamer core of 2 copies each of histone proteins H2A, H2B, H3, and H4. Similar to promoters, enhancers are characterized by nucleosome-free regions that are occupied by sequence-specific transcription factors. These factors in turn recruit various co-acitvator proteins that facilitate nucleosome remodeling, histone tail modifications, and recruitment of core transcription factors, including RNA polymerase II (24, 25). Enhancers are defined by a distinct array of chromatin modifications as compared to promoters. Whereas promoters are characterized by a high H3K4me3/low H3K4me1 ratio, enhancers show a contrary makeup of low H3K4me3/high H3K4me1 (26). However, active enhancers and promoters are both associated with histone tail acetylation, for example of histone H3 lysine 27 (27). The use of ChIP-Seq for these histone modifications and global DNase I hypersensitivity assays to define open regions of chromatin in the genomes of many different cell types and tissues revealed the presence of hundreds of thousands of these putative enhancer elements in the human genome (28). Each cell type selects a subset of ~20 – 30 thousand of this vast repertoire of potential regulatory elements, which are presumed to play essential roles in establishing cell specific gene expression.
Figure 1.
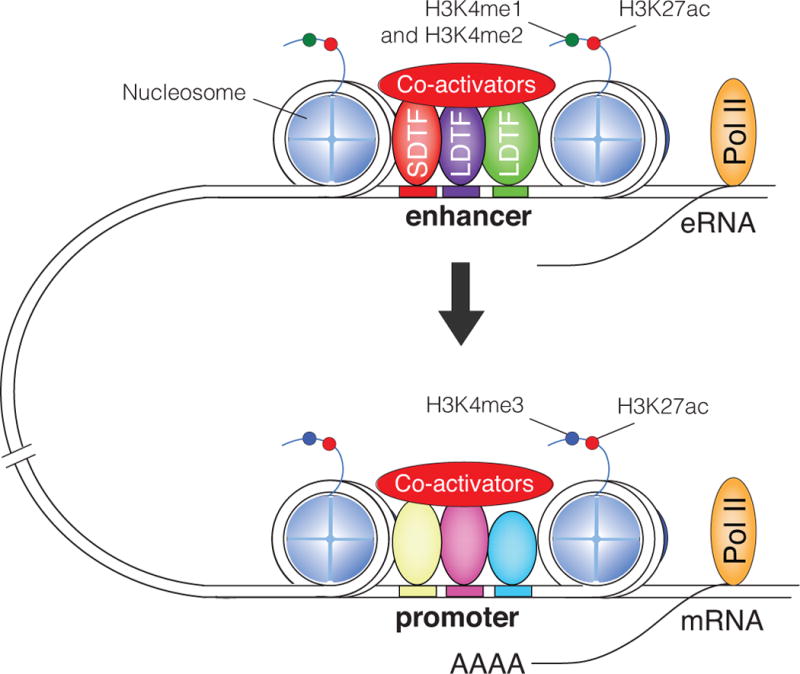
General organization of enhancers and promoters. DNA is packaged into nucleosomes that are displaced by sequence-specific transcription factors and co-activators. Promoters are primarily occupied by broadly expressed transcription factors, whereas enhancers are enriched for the binding of lineage-determining factors (LDTFs). Signal-dependent transcription factors (SDTFs) can bind to enhancers or promoters (here shown only at the enhancer). Promoters are distinguished by high levels of H3K4me3 compared to H3K4me1 and H3K4me2. Enhancers are characterized by high levels H3K4me1 relative to H3K4me3. Active enhancers and promoters are associated with transcriptional co-activators and acetylated histones, such as H3K27ac. Active enhancers are frequently associated with RNA polymerase II that generate enhancer RNAs (eRNAs).
An important caveat to these global approaches is that while genomic regions that have been genetically established to function as enhancers almost always exhibit features shown in Figure 1, genomic regions that have these characteristics are not necessarily active enhancers (29). To define enhancers, combinations of marks improve predictive power. For example, regions of the genome exhibiting open chromatin, transcription factor binding and H3K4me1, but not H3K27ac, are often considered ‘poised’, while the further addition of H3K27ac designates ‘active’ enhancer regions (27). However, the most rigorous test for enhancer function is to delete or otherwise mutate key elements of the DNA sequence of a putative enhancer and demonstrate reduced expression of the target promoter. While advances in genomic engineering greatly facilitate these types of experiments (30), the vast number of enhancer-like regions in the genome makes even these approaches insufficient for global analysis and improved methods for prediction of enhancer function are needed.
In addition to being occupied by sequence specific transcription factors and transcriptional co-regulators, a subset of enhancers recruit core transcription factors and RNA polymerase II and generate non-coding RNAs referred to as eRNAs (31, 32) (Figure 1). These are typically in the range of 200 to 1000 nucleotides long and are thus classified as long noncoding RNAs. The majority of these RNAs are capped at the 5′ end, but are not spliced or polyadenylated and are rapidly degraded in an exosome-dependent manner (32). The gain or loss of eRNA expression in response to signals, such as TLR4 ligation or ligands for nuclear receptors, is highly correlated with gain or loss in nearby gene expression (31). Thus, eRNA production may be an additional feature of enhancers that is predictive of enhancer function. The functional importance of enhancer transcription has not been fully established. Studies of enhancers that are newly selected in response to TLR4 ligation suggested that the process of enhancer transcription itself is important by serving to recruit histone methyltransferases that write the histone H3 lysine 4 mono- and dimethylation marks that are characteristic of enhancers (33). This was proposed to be due to recruitment of the Mll2 and Mll4 histone methyltransferases to the C-terminal domain of RNA polymerase II.
In addition, several lines of evidence suggest that at least some eRNAs contribute to enhancer function. In macrophages, the Rev-erb nuclear receptors were shown to repress gene expression by repressing enhancer transcription. This activity could be reproduced by using antisense oligonucleotides to reduce the expression of specific eRNAs in the nucleus (34). These and other studies suggest that if active, eRNAs primarily function in a local manner to regulate the expression of the particular target of the enhancer that they are generated from (34, 35). Several mechanisms have been suggested to account for eRNAs function, including promoting enhancer/promoter looping (35), release of the negative elongation factor NELF (36), and by trapping transcription factors such as YY1 (37). Disruption of the integrator complex, which is necessary for the 3′-end processing of non-polyadenylated, RNAPII-dependent, uridylate-rich, small nuclear RNA genes, which includes enhancers, results in a reduction of eRNA production and subsequently abrogates enhancer-promoter looping (38). Conversely, WDR82 and SET1 function to control termination of eRNAs, such that their loss of function results in unusually long eRNAs (39).
Examination of the distribution of genomic features associated with enhancers in various cell types, such as components of the Mediator complex and histone acetylation, revealed that they were clustered at very high density at a few hundred regions in each cell type. Such regions have been referred to as super-enhancers or stretch enhancers (40–42). Generally, super-enhancers are composed of clusters of ordinary enhancers that together extend more than an order of magnitude larger than individual ordinary enhancers (kilobases vs. hundreds of bases)(40, 41). Interestingly, the separate enhancers within super-enhancer regions have been shown to not work in an additive manner. Instead, there is a much more complex interplay where some regions are more important for activation and some regions negatively affect enhancer activity and may play a role in regulating enhancer strength (43). Furthermore, the eRNA transcription of the separate regions within super-enhancers is uniformly regulated rather than individually regulated (44).
Super-enhancer regions have been seen to overlap with and are thought to activate the transcriptional activators which are vital in the establishment of cell fate and identity (41). In macrophages, super-enhancers have been reported to overlap genes that are essential for macrophage development and function, including Spi1 (encoding PU.1), Cebpa, members of the Irf family, Csf1r, Fcgr2b, and Ctsb (45) (Figure 2). Similarly, in mouse embryonic stem cells (mESCs), genes required for pluripotency such as Oct4, Sox2, and Nanog are associated with super-enhancers (41). As well as having high levels of Mediator, super-enhancers are much more reliant on Mediator than regular enhancers. This is evidenced by knockdown experiments in mESCs against Med12, an integral component of the Mediator complex. Loss of Med12 resulted in a disproportionate down regulation of super-enhancer adjacent genes such as Oct4, Sox2, and Nanog (41). Further, a loss of Mediator in mouse embryonic stem cells (mESCs) affected pluripotency (46). To date, little work has been done to establish the role of super-enhancers in macrophages. However, data in other cell types suggest that super-enhancers play a disproportionately large part in controlling the expression of genes that establish cell fate, making them an ideal target for future studies of macrophage enhancer biology.
Figure 2.
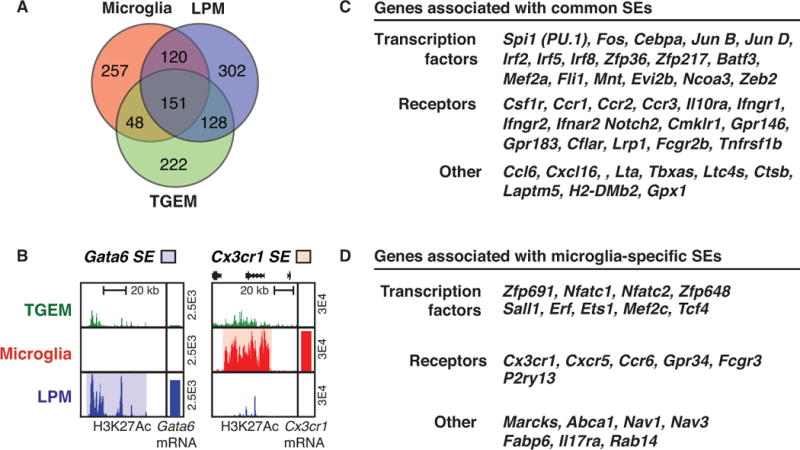
Super-enhancers in macrophages. A. Venn diagram of shared and subset-specific super-enhancers in thioglycollate-elicited macrophages (TGEMs), large peritoneal macrophages (LPMs), and microglia. B. Examples of subset-specific super-enhancers near the Gata6 and Cx3cr1 genes in TGEMs, LPMs and microglia. C. Partial listing of the 151 genes associated with super-enhancers found in all three macrophage subsets. D. Partial listing of genes associated with the 257 super-enhancers selectively found in microglia.
Selection and activation of macrophage-specific enhancers
The development of multicellular organisms involves hierarchically organized progenitor cells that ultimately give rise to terminally differentiated cell types with specialized functions. Cell fates are specified by lineage-determining transcription factors (LDTFs) whose expression is often not limited to a single cell type (47). Most transcription factors recognize short DNA motifs of ~8–12 base pairs in length, and considerable degeneracy can be tolerated for sequence specific binding in vivo. As a consequence, there are potentially tens of millions of binding sites for most transcription factors within the mammalian genome. Initial use of chromatin precipitation linked to massively parallel DNA sequencing (ChIP-Seq) to study the genome-wide binding patterns of transcription factors in a variety of species and cell types demonstrated that only a small fraction of these sites are actually occupied. Furthermore, different factors in the same cell type were often found to co-localize (48, 49) and the same factor in different cell types or at different stages of development exhibited different genome-wide binding patterns (50–52). However, mechanisms accounting for these binding patterns were unknown.
PU.1 is a LDTF required for the normal development of macrophages and B cells (53, 54), where it drives divergent programs of gene expression in each cell type. An initial application of ChIP-Sequencing approaches to investigate the genome wide binding patterns of the Ets domain transcription factor PU.1 in thioglycollate-elicited macrophages and splenic B cells provided evidence for a collaborative/hierarchical model (55) (Figure 3). Here, relatively simple combinations of lineage determining transcription factors (LDTFs) were suggested to play dominant roles in the selection of a large fraction of each cell type’s enhancers. While the binding of PU.1 to promoter regions was found to be similar in the two cell types, binding to regions of the genome distal to promoters, which include enhancers, was highly cell type specific. The recognition motif for PU.1 was identical at these sites in both cell types, indicating that the motif itself did not contribute to cell specific binding patterns. In contrast, genomic regions within 100 base pairs of PU.1 binding sites in macrophages were highly enriched for motifs for other macrophage LDTFs, particularly C/EBP and AP-1 motifs. Alternatively, genomic regions within 100 base pairs of PU.1 binding sites in B cells were highly enriched for motifs for B cell lineage determining factors, including Oct, E2A, EBF, and κB motifs. Gain of function experiments indicated that at regions of the genome exhibiting closely spaced binding sites for PU.1 and C/EBP, binding of C/EBP was dependent on PU.1. Conversely, loss of function experiments in B cells indicated that regions of the genome exhibiting closely spaced binding sites for PU.1 and E2A, binding of PU.1 was dependent on E2A (55). These observations suggested a ‘collaborative’ model of enhancer selection, in which PU.1 and alternative lineage determining factors such as C/EBPs and E2A are alone unable to bind to specific regions of the genome, but do so at genomic locations containing the appropriate combination of recognition motifs when they are co-expressed at high levels.
Figure 3.
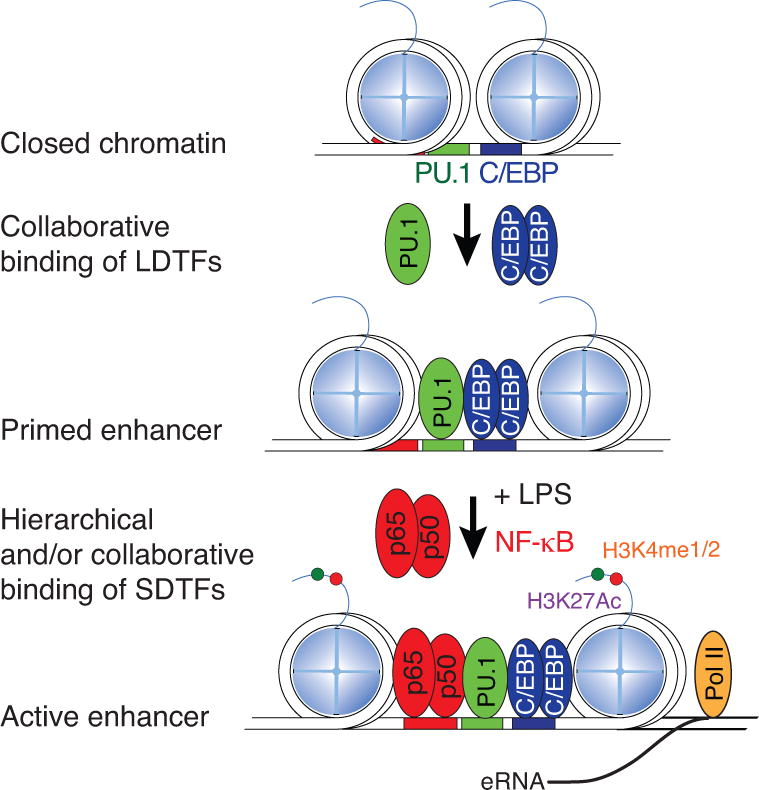
A collaborative/hierarchical model for selection and activation of macrophages enhancers. Macrophage LDTFs, exemplified by PU.1 and C/EBPs, collaborate with each other to bind to genomic regions containing closely spaced PU.1 and C/EBP recognition motifs to establish a primed enhancer. Signal-dependent activation of NFκB (here shown as p50 and p65) leads to its binding to primed enhancers and enhancer activation, resulting in histone acetylation and production of eRNAs.
Many regions of the genome that are bound by PU.1 and C/EBPs in macrophages exhibit H3K4me1, but not H3K27ac, consistent with features of poised enhancers. Notably, the majority of binding of signal-dependent transcription factors (SDTFs) in response to activating ligands in thioglycollate-elicited macrophages was found to occur at pre-existing poised or active enhancer-like regions. Initially observed for the nuclear receptors LXRα and LXRβ (55), this pattern was subsequently established for NF-κB (33), the glucocorticoid receptor (56), and Rev-erbs (34). These results are of interest because they provide a potential explanation for how broadly expressed signal dependent transcription factors can exert cell-specific effects on gene expression. In essence, these findings suggest that SDTFs are ‘instructed’ as to where to localize in each cell type based on the open regions of chromatin established by that cell type’s LDTFs and, consequently, how the cell will respond to signals based on the previous binding of LDTFs. For this class of interactions, the binding of LDTFs and SDTFs is ‘hierarchical’, in that, SDTF binding is proposed to be dependent on prior binding of LDTFs, whereas the binding of LDTFs is independent of the SDTFs (Figure 3). This relationship was established by gain and loss of function experiments for PU.1 and LXRs in the thioglycollate-elicited macrophage model system: at sites of co-localization LXR binding depended on the pre-existing binding of PU.1, but PU.1 binding was not dependent on LXRs (55).
The natural genetic variation existing between inbred strains of mice has been leveraged to provide a genetic test of the collaborative/hierarchical model of LDTF and SDTF binding in macrophages (57). These studies focused on regions of the genome at which PU.1 and C/EBP bound to their respective motifs in close proximity and were subsequently occupied by the p65 component of NF-κB. At these locations, selective mutations in the binding sites for PU.1 not only resulted in loss of PU.1 binding, but also the loss of C/EBP and the signal induced binding of p65. Similarly, selective mutations in binding sites for C/EBPs resulted in loss of C/EBP, PU.1 and p65. In contrast, mutations in NF-κB binding sites abolished binding of p65, but rarely affected the pre-existing binding of PU.1 and C/EBP (57). These findings supported both the collaborative model of LDTF binding and the hierarchical relationship of SDTFs and LDTFs. Furthermore, mutation of sites that abolished PU.1 and C/EBP binding also generally resulted in a loss of other features of enhancers, such as histone methylation and acetylation, indicating that these post translational modifications are dependent on the binding of the LDTFs.
De novo or latent enhancers
Although most of the genome-wide binding of p65 occurs at pre-existing enhancer like regions as described above, two independent studies demonstrated that a subset of binding events occurs at regions of the genome that do not have features of enhancers in resting macrophages but acquire them after TLR4 ligation (33, 58). Macrophage activation by the TLR4 agonist Kdo2 lipid A (KLA) resulted in the selection of a few thousand enhancers, called ‘de novo’ or ‘latent’ enhancers. These enhancers did not possess detectable levels of enhancer associated chromatin modifications, nor LDTF binding before KLA treatment. As well, unlike the vast majority of enhancers found after KLA stimulation, where LDTFs primed the environment and recruited SDTFs to further activate enhancer transcription, these enhancers required collaboration with LDTFs and SDTFs for the initial selection of the enhancer and subsequent indicative chromatin marks. A requirement for NF-κB was demonstrated through inhibition of NF-κB via an IKK inhibitor, where down-regulation of NF-κB prevented the establishment of these de novo enhancers (33). These data suggest that there are state-dependent enhancer landscapes that are only activated via external signals acting through SDTFs. Notably, histone modifications associated with latent enhancers were shown to persist following removal of the initiating signal. Genes associated with these regions exhibited more rapid and robust responses when cells were re-stimulated, suggesting that such modifications may be associated with an epigenetic ‘memory’ (58). These findings support the broader concept of a state of ‘trained immunity’ in which the epigenetic state of innate immune cells can be modulated to influence subsequent responses to a challenge (59).
Evidence for tissue environment as a determinant of macrophage phenotype
Transcriptional profiling of diverse macrophage populations has revealed striking differences in patterns of mRNA expression according to tissue bed (60). For example, a comparison between microglia and large peritoneal macrophages revealed nearly 1000 mRNAs that are more than 16-fold differentially expressed in each direction (45). While the functional roles of most differentially regulated genes remain poorly understood, these observations suggest a molecular basis for the phenotypic diversity of these cells. At the other end of the spectrum, at least two distinct populations of macrophages can be isolated from the mouse peritoneal cavity, referred to as large and small peritoneal macrophages and further distinguished by relatively high levels of MHC class II expression in the small peritoneal macrophages (61). In contrast to the substantial differences in gene expression observed between large peritoneal macrophages and microglia, only ~100 genes show >than 16-fold higher expression in comparing large and small peritoneal macrophages. Most of these are more highly expressed in the small peritoneal macrophage population and are enriched for functional annotations related to antigen presentation (45). Regardless of whether these differences are due to different developmental origins, different times of residence within the peritoneal cavity, or other mechanisms, these experiments indicate that macrophages can exhibit distinct phenotypes within a common tissue environment.
However, there is also emerging evidence that tissue environment is a strong determinant of macrophage phenotype, and that these phenotypes exhibit remarkable plasticity. One line of evidence is based on bone marrow transplantation, in which macrophages derived from adult hematopoietic stem cells replace tissue macrophages of embryonic origin. For example, lung and peritoneal macrophages from transplanted adult bone marrow acquire gene expression signatures similar to those of embryonically-derived macrophages (62). Second, peritoneal macrophages adoptively transferred to the lung adopt a lung macrophage-like pattern of gene expression (62). Third, transfer of peritoneal macrophages and microglia to tissue culture environments led to significant changes in the expression of hundreds of genes. Significantly, genes exhibiting down-regulation in peritoneal macrophages were enriched for those that made them most different from microglia, and vice-versa (45). Collectively, these experiments indicate that environmental factors are significant drivers of distinct macrophage phenotypes.
Enhancer landscapes of tissue-resident macrophages
Improvements in the sensitivity of genomic assays have enabled the recent analyses of enhancer landscapes in various tissue-resident macrophage populations (45, 62, 63). These studies documented the presence of tens of thousands of enhancer like regions in each population, the majority of which are shared. Putting these regions into the context of the collaborative/hierarchical model of enhancer selection and function, a large subset exhibit features of both ‘priming’ (presence of only H3K4 methylation) and ‘activation’ (presence of H3K4 methylation and H3K27 acetylation). For example, multiple enhancer-like regions exhibiting high levels of both H3K4me2 and H3K27ac reside in the vicinity of the Spi1 gene encoding PU.1 in all macrophage populations examined. This is consistent with the requirement of PU.1 in all macrophage subsets. In contrast, the Rarb gene, encoding the retinoic acid receptor β, exhibits H3K4 methylation in both microglia and peritoneal macrophages, but the H3K27ac mark of activation is only observed in the peritoneal macrophage population (45). This observation of the enhancer region being poised in microglia but active in peritoneal macrophages is consistent with the recent finding that the development of peritoneal macrophages is under the control of locally produced retinoic acid and longstanding evidence that the Rarb gene is itself retinoic acid inducible (64). These findings suggest that the enhancers driving Rarb expression are selected in both microglia and peritoneal macrophages by a common set of macrophage lineage determining factors, but that these enhancers only become active in the peritoneal cavity due to the selective presence of sufficient concentrations of retinoic acid in that tissue environment.
In addition to shared enhancers, each macrophage population also exhibits enhancer-like regions, corresponding to ~15–20% of the total, that are either specific to that cell type or are restricted to a subset of the various macrophage populations examined (45, 62, 63). The existence of such subset specific enhancers thus suggests that there are additional, context specific, transcription factor interactions necessary for their selection. Motif enrichment analysis of subset-specific enhancers returns binding sites for PU.1 as among the most highly enriched motifs. Furthermore, ChIP-Seq experiments confirmed binding of PU.1 to both common and subset specific enhancer-like regions, consistent with a requirement of PU.1 for the development of nearly all tissue-resident macrophages (45). However, within subset-specific enhancers, different transcription factor recognition motifs were identified in the vicinity of PU.1 binding. For example, a motif for Gata6, established to be essential for development of peritoneal macrophages, is significantly enriched near PU.1 in enhancers that are specific for peritoneal macrophages (45).
Using H3K27ac, super-enhancers were defined in large peritoneal macrophages, microglia and thioglycollate-elicited macrophages (45). This analysis revealed a core set of ~150 super-enhancers that were present in all three subsets (Figure 2). Genes associated with common super-enhancers included genes encoding transcription factors and receptors essential for development and survival of all macrophages, such as Spi1, which encodes PU.1 and Csf1R, which encodes the receptor for M-CSF. Intriguingly, each macrophage-subset exhibited ~200 super-enhancers that were specific for that subset and were highly correlated with subset-specific gene expression. Genes associated with super-enhancers specific to large peritoneal macrophages included the Rarb and Gata6 genes, were required for specification of the large peritoneal macrophage phenotype. Conversely, super-enhancers observed selectively in microglia were associated with numerous receptors and cell surface proteins associated with functions in the brain, including Cx3cr1, Nav1, and Bin1 (45). Notably, very little is known with respect to functions of many of the genes associated with super-enhancers in each macrophage subset. Given the strong enrichment of genes with essential roles in regulation of macrophage development and function in regions of the genome marked by super-enhancers, this designation may be a useful means for prioritization of analysis of genes with unknown functions. Further, the establishment of unique super-enhancers during differentiation may play a vital role in biasing the cell specific responses to extracellular differentiation signals (41, 65–67). Not surprisingly, super-enhancers have been shown to play an important role in determining cell function, acting as a fast switch to aid in cell-state transitions. For example, treatment of endothelial cells with TNF-α was found to result in drastic changes to cell super-enhancer selection through the activation of NF-κB, inducing a pro-inflammatory gene expression program (65).
In keeping with the dramatic changes in gene expression observed following transfer of peritoneal macrophages from an in vivo to an in vitro tissue culture environment, a corresponding change was observed in the enhancer landscape of these cells as measured by H3K4me2 and H3K27ac (45). Nearly half of the enhancer-like regions defined immediately after recovery from the peritoneal cavity exhibited a >50% reduction in signal for one or both of these marks by day 7 of tissue culture. This was observed at both regular enhancers and super-enhancers. Lost enhancers were generally associated with genes exhibiting reduced expression in vitro, providing correlative evidence for a functional relationship (45). These data support the concept that enhancers and super-enhancers are transcriptional modules that integrate multiple signals to regulate responsiveness of target genes (43).
Extending the collaborative/hierarchical model
While supported by gain and loss of function experiments and the effects of natural genetic variation, the collaborative/hierarchical model depicted in Figure 3 is vastly oversimplified. For example, the model does not account for the functions of the vast majority of transcription factors that are expressed in macrophages. Furthermore, since it is derived from responses of tissue culture macrophages to selective signals, its relevance to the mechanisms leading to selection and function of enhancers in tissue-resident macrophages is unclear. We next consider several different approaches to define transcription factors required for macrophage development, and place findings derived from these methods into the context of enhancer selection and function.
Gene deletion studies
One of the most powerful approaches to delineating gene function is through targeted generation of null alleles, either systemically or conditionally. Here, we briefly survey a representative subset of studies examining the consequences of loss of function of specific transcription factors on macrophage development and tissue-specific phenotypes. As noted previously, PU.1 is a key lineage-determining transcription factor for macrophages, neutrophils and B cells such that its deletion results in neonatal death and a general lack of these cell types (53, 54). Indeed, blocks in differentiation resulting from loss of PU.1 result in leukemic transformation of myeloid cells (68, 69). Genomic studies of PU.1 binding during distinct stages of B cell differentiation indicated progressive remodeling of its genomic locations in concert with sequential expression of additional lineage determining transcription factors for B cell development (55).
Specifications of branch points in cellular differentiation are achieved in part through expression of additional LDTFs. For example, IRF8 has been shown to be necessary for DC differentiation in knockout mouse models. In the absence of IRF8, myeloid progenitors undergo a DC to neutrophil reprogramming indicating a requirement for IRF8 in DC commitment (70, 71). In B cells, loss of function studies indicate that IRF8 and IRF4 compete to establish the differentiation of B cells to plasma cells (72). Whereas, expression of IRF4 promotes differentiation of plasma cells, expression of IRF8 inhibits this differentiation (72). Ultimately, this fate decision is determined by the relative expression of these two LDTFs. LDTFs can also function to inhibit each other to specify differentiation, as seen in the case of IRF8 inhibiting C/EBP chromatin association through a direct inhibitory interaction. The result of this interaction is a block in the differentiation into neutrophils from mononuclear phagocyte progenitors (73). Studies in macrophages characterizing the genome wide binding patterns of IRF8 in resting and LPS-stimulated macrophages indicate that it contributes to the selection of both basal enhancer elements as well as latent enhancers (74).
Several transcription factors have been identified that are required for tissue-specific macrophage subsets. The PU.1 related family member, Spi-C, was recently shown to be necessary for red pulp macrophage development. The Spi-C knockout mouse has a cell autonomous defect in splenic iron homeostasis, which led to the discovery of a specific lack of red pulp macrophages (75). One of the most well characterized examples of a subset specific LDTF is c-Fos. Mice lacking c-Fos were shown to have severe growth retardation and osteopetrosis resulting from a loss of osteoclasts (76–79). In the lung, differentiation of fetal monocytes into alveolar macrophages requires the expression of PPARγ. Knockout of PPARγ results in diminished lipid catabolism-associated gene expression and enhanced cholesterol esterification (80). Nr4a1-deficient mice result in an absence of Ly6C− monocytes, which are patrolling monocytes (81). Nr4a1-deficient mice exhibiti an increase in atherosclerosis in hypercholesterolemic LDL receptor knockout mice, which suggests that Ly6c− monocytes are necessary for controlling the inflammatory phenotype that leads to atherosclerotic plaque development (81). In the brain, TGFβ induced SMAD activity is necessary for maintenance of the microglia phenotype (82).
While these gene deletion experiments provide strong evidence for functional roles of the corresponding transcription factors, they do not establish the mechanisms by which they exert their effects on macrophage development and function. Of the factors discussed above, only PU.1 and IRF8 have been studied thus far at a genome-wide level in macrophages. The subset-selective activities of cFos in osteoclasts or SpiC in splenic macrophages are as yet not understood. Given the biologicial insights derived from these studies, it will be of considerable interest to evaluate the genome-wide binding and functions of thiese and other transcription factors required for the acquisition of tissue specific phenotypes. A further limitation of gene deletion studies is that phenotypes may be less pronounced for factors that are members of gene families and where functional redundancies may be present.
Exploiting super-enhancers
As previously discussed, super-enhancers have been shown to overlap with factors necessary for cell fate. As such, a logical progression of super-enhancer discovery is the concomitant discovery of the cell fate proteins these super-enhancers regulate. Indeed, several groups have successfully used this approach to demonstrate that super enhancers are associated with transcription factors necessary for cell fate specification (41, 83, 84). For example, in embryonic stem cells, super-enhancers covered genes that define cell identity such as Oct4, Sox2, and Nanog as well as several novel factors (41). Additionally, in a study of somatic copy number alterations in cancer pathogenesis, focally amplified lineage-specific super-enhancers in human epithelial cancers were targeted to known oncogenes such as Klf5, Usp12, Pard6B, and Myc, and this resulted in overexpression (84). An alternative method used Gene Ontology (GO) analysis on the top ranked super-enhancers. Several transcriptional regulators came up as regulators of these programs including Sox9, which was then confirmed as a crucial chromatin rheostat of hair follicle stem cell regulation and identity (83). Thus, transcription factors identified to be associated with super-enhancers in macrophage subsets are candidates for prioritization. For example, numerous known and putative transcription factors are associated with super-enhancers in microglia that have not been studied in this cell type, including Zfp691, Sall1, and Nfat1c (45).
Co-expression analysis
A common starting point for identifying potential cell fate-specific transcription factors is to perform RNA expression analyses that allow comparisons of gene expression across cell types and/or during cell differentiation. Temporal changes in expression of transcription factors during developmental transitions can suggest contributing transcription factors. For example, analysis of RNA expression during neutrophil differentiation from hematopoietic stem cells from human patients revealed changes in transcriptional activators that included GATA2, AML1/RUNX1, SCL/TAL1, C/EBPa and PU.1 (85). Various software tools have been developed to enable placing transcription factor expression into the context of networks and to predict cause-effect relationships, such as CYTOSCAPE, Ingenuity Pathway Analysis and Pathway Studio (86). For example, Ingenuity pathway tools were used to define physical and regulatory interactions that predict distinct roles of C/EBPs, Maf, NFE2, and nuclear receptor family members in directing the expression of 14 macrophage-associated modules (60).
Discovery of transcription factor motifs using ATAC-Seq
A recently reported method for identifying important transcription factors is through analysis of open chromatin identified using the transposase-accessible chromatin sequencing (ATAC-seq) method (87). ATAC-seq is analogous to DNase I hypersensitivity in defining open chromatin, but uses a transposase-assisted integration of DNA primer sequences. The practical importance of this difference is that open regions of chromatin can be identified in relatively small populations of cells (reportedly as few as 500) (87). It is thus amendable to analysis of the often limited numbers of cells that can be obtained from mouse tissues or clinical samples. Several groups have used this method to define open chromatin and then have leveraged this information to define the protein specific DNA binding motifs. For example, ATAC-seq was used in the identification of putative enhancers in tissue-resident macrophages (62). These analyses identified Mef2c and Gata6 as candidate LDTFs in microglia and peritoneal macrophages, respectively. This method has also been used to define several LDTFs including BAF and p63 mutual recruitment to almost 15,000 open chromatin regions in adherent human keratinocytes (88) and PDX1, NKX6.1 in β-cells and NKX2.2, MAFB, FOXA2in islet cells (89). Interestingly, this approach has also been used to define the LDTFs responsible for driving in vivo tumor development, specifically in Ras-dependent oncogenesis in mice (90). Here, open chromatin regions in Ras-driven tumors were found to contain both AP-1 and Stat92E. Further, the introduction of a loss of function Stat92E mutant rescued the tumor phenotype, confirming the applicability of using ATAC-seq in the novel discovery of LDTFs necessary for tumor development. A limitation of ATAC-seq experiments is that open regions of chromatin are not necessarily active regulatory regions. In addition, it is not yet clear what the ‘false negative’ rate is for this method. Despite these limitations, it appears that ATAC-Seq will be a powerful method for defining the most important transcription factor motifs within specific cell types.
Natural genetic variation as a ‘mutagenesis’ strategy
An emerging approach for novel LDTF and SDTF discovery uses the natural genetic variation provided by different strains of inbred mice. Natural genetic variation was initially used as a way to test the collaborative and hierarchical model of enhancer selection involving PU.1, C/EBP, and AP-1 as described above (57). The demonstration that mutations in PU.1 motifs led to loss of not only PU.1 binding but also nearby C/EBP binding suggested that natural genetic variation could be used to discover unknown collaborative binding partners. To test this approach, ChIP-Seq was used to define the binding sites of PU.1 in resident peritoneal macrophages and microglia in three inbred mouse strains providing 5 to 40 million single nucleotide polymorphisms (45). Genomic regions exhibiting differential binding of PU.1 were then analyzed for SNPs. Motifs altered by SNPs that were statistically correlated with strain-specific PU.1 binding were considered to represent collaborative binding partners of PU.1. This analysis led to the identification of several dozen motifs that were highly correlated with nearby binding of PU.1. Importantly, these included motifs for C/EBP factors, which were independently established as important collaborative binding partners for PU.1. In addition, GATA6 motifs were identified near PU.1 binding sites in large peritoneal macrophages and SMAD motifs in microglia, providing proof of principle for the utility of this approach as a discovery strategy. Many of the additional motifs are recognized by transcription factors that are expressed in macrophages, but which have not as yet been studied in this context.
These varying approaches to uncovering putative LDTFs and SDTFs exploit different angles and offer different advantages and disadvantages. For example, defining super-enhancers and performing RNA expression analysis provides candidates at a relatively low cost; however, these methods do not explain binding patterns and assume protein expression and activity. Using measures of open chromatin provides likely motifs, but these are correlative and cannot distinguish specific proteins from families of factors that bind similar motifs. Using SNPs in natural genetic variation provides genetic evidence for the importance of transcription factors recognizing a particular motif in driving collaborative binding interactions, but also does not specify the particular factor among a transcription factor family. Thus, an essential step is to validate roles of specific transcription factors through loss of function studies. These studies in themselves can be challenging to interpret due to transcription factor redundancy, requiring simultaneous loss of function strategies. In addition, loss of function studies may lead to early embryonic lethality, effects in multiple tissues, or loss of a progenitor cell, precluding analysis in specific macrophage populations. Thus, conditional methods for gene deletion are often required. Optimally, this can be achieved by Cre recombinases that are directed to the particular macrophage population of interest. However, a relatively limited set of macrophage-specific Cre drivers are available at present and new and highly specific drivers that target gene deletion in particular macrophage subsets would be valuable additions to this field.
Conclusions and future directions
A central aspect of macrophage biology is their ability to sense the environment through their expression of a multitude of cell surface receptors that control the expression and/or activity of downstream transcription factors. The emergent picture is that specific phenotypes arise from the combinatorial actions of broadly expressed signal dependent factors and more restricted lineage-determining factors at macrophage specific enhancers. Thus, the complement of enhancers within each macrophage is proposed to correspond to tens of thousands of distinct analogue genomic sensors that integrate diverse signaling inputs to regulate the expression of target genes. Although these principles appear to be general for the many cell types examined thus far, macrophages may be particularly specialized as sensors and responders to environmental signals.
Integration of findings from gain and loss of function experiments and epigenetic profiling suggest a revised model for selection and activation of tissue-specific macrophage enhancers (Figure 4). In this model, a core set of lineage determining transcription factors, exemplified by PU.1 and C/EBP factors, select primed enhancers in most or all macrophage populations. These enhancers can be acted upon by environment-specific signals to induce direct target genes of those signals. For example, retinoic acid is present in high concentrations in the peritoneal cavity compared to the adult brain, and thus retinoic acid responsive enhancers and their target genes are preferentially activated in peritoneal macrophages. Conversely, TGFβ is more abundant in the adult brain than within the peritoneal cavity, resulting in preferential activation of Smad-dependent enhancers and genes in microglia. Among the spectrum of genes induced by these enhancers are genes encoding transcription factors that have the potential to collaborate with PU.1, and presumably other macrophage LDTFs, to select new enhancers that are environment-specific. For example, GATA6 expression is positively regulated by retinoic acid and functions as a collaborative partner of PU.1 to select peritoneal macrophage-specific enhancers. Thus, this model proposes that the full complement of tissue specific gene expression is driven by the combination of direct effects of environmental factors on common enhancers and induced expression of transcription factors that drive the selection and function of tissue-specific enhancers.
Figure 4.
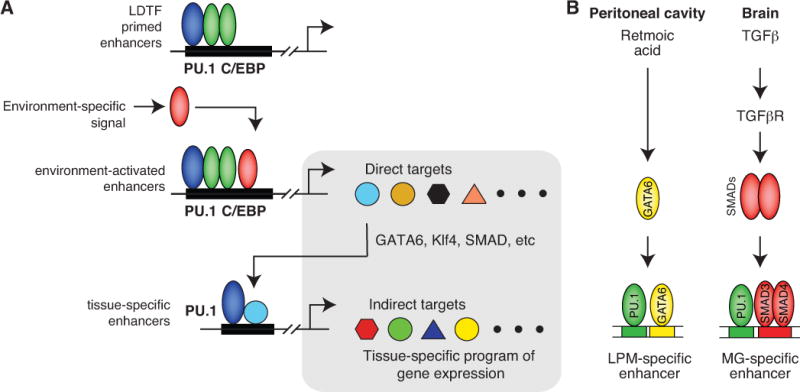
Selection and activation of tissue-specific macrophage enhancers. A. Generic model. A core set of macrophage LDTFs, exemplified by PU.1 and C/EBP factors, prime a common set of enhancers in many or all macrophage subsets. These enhancers can be acted upon by environment-specific signals to drive the expression of direct target genes. A subset of these genes includes transcription factors that can collaborate with macrophage LDTFs, such as PU.1, to select a secondary, tissue-specific set of enhancers that drive expression of additional target genes. The tissue-specific gene expression program thus results from both direct and indirect environmental effects. B. Examples of signals preferential for the peritoneal cavity (retinoic acid) or brain (TGFβ), resulting in expression of collaborative factors Gata6 or Smads, respectively.
CRISPR-Cas9 technology could be used to untangle many of the genetic and biochemical questions that still surround enhancer selection and activation. CRISPR-Cas9 is a bacterial based system that has been adapted as a tool for genome editing. It contains a single-guide (sgRNA) sequence that has a target sequence which binds to complimentary host DNA, and forms a complex with the DNA endonuclease protein Cas9. Once targeted to a genomic DNA sequence complimentary to the sgRNA, Cas9 causes a double stranded break in the target DNA, which may result in a deletion if the break is repaired by non-homologous end joining (30, 91). Thus, CRISPR-Cas9 system could be used to delete enhancers in order to ascertain which gene or genes a particular enhancer regulates (92, 93). Alternatively, a mutant form of Cas9, denoted dCas9, which is unable to induce a break in the DNA could be used to modify enhancer activity. This can be accomplished by adding an activation (VP16) or repression (Krüppel-associated box (KRAB) domain of KOX1) domain to the dCas9 (94, 95). This technique allows an activation or repression domain to be targeted to any enhancer in the genome. More specific modifications could be made using a combination of CRISPR-Cas9 and homologous recombination. Here, a homologous DNA sequence with small mutations would be co-introduced with CRISPR-Cas9. Recombination of this sequence would allow for subtle changes of specific DNA sequences to either alter transcription factor binding or remove eRNA start sites (96, 97). CRISPR-Cas9 could also be used for biochemical analysis of specific enhancers. Using enChIP, a tagged dCas9 based system, Cas9 can be specifically targeted to a specific enhancer and Chromatin Immunoprecipitation coupled with MASS-spectrometry protein sequencing would be used to determine the complexes bound at the target enhancer (98). Using CRISPR-Cas9 technologies should allow for a more thorough understanding of enhancer activity, specifically relating to enhancer targeting and redundancy through direct knockout or targeted activation/repression, transcription factor cooperation through base pair modification, and eRNA requirements and function, among many others.
A remaining limitation of this model is that it does not take into account the potential origin of macrophages as a determinant of enhancer landscapes, or the question of whether there are transcriptional circuits that are ‘hard wired’. The analysis of large and small peritoneal macrophages indicate that small peritoneal macrophages exhibit a program of gene expression that is distinct from the large peritoneal macrophages that live in the same environment. The basis for this difference is as yet unexplained. In addition, while many genes that are specific for large peritoneal macrophages and microglia fall dramatically when transferred from the in vivo environment to an in vitro environment, many other genes that are specific to these subsets do not change. The basis for this retention of tissue-specific programs of expression is also not understood. Going forward, genomic studies, including studies of DNA methylation, which could provide a more long lasting epigenetic mark, need to be combined with lineage tracing, to help establish the potential importance of origin in defining tissue-specific phenotypes.
In addition to providing insights into mechanisms that specify macrophage identity and function, the delineation of tissue-specific LDTFs and SDTFs has direct relevance to better understanding roles of macrophages in disease. First, knowledge of these factors is likely to facilitate efforts to reprogram patient-derived stem cells to specific macrophage phenotypes in vitro. Such reprogrammed cells are useful for studying cell-autonomous disease mechanisms and responses to drugs. Second, the ascertainment of the genome wide binding patterns of these factors and the effects of natural genetic variation will help inform interpretation of risk alleles identified by genome wide association studies. Third, the observation that macrophage enhancer landscapes are dependent on constant input from their environment implies that these landscapes will change in the context of disease. Because these changes can now be measured in relatively small populations of macrophages isolated from tissues, it should be possible to determine the corresponding transcription factors that are gained or lost using motif analysis. This information might then be used to ‘reverse engineer’ the potential signaling pathways that are gained or lost in the particular disease state and thereby gain insights in mechanisms driving macrophage phenotypic conversion.
Lastly, there is emerging interest in the potential to alter cellular phenotypes by targeting enhancers. The observation that at least some enhancer RNAs contribute to enhancer function has suggested the possibility of using anti sense oligonucleotides to knock down expression of cell-specific eRNAs as a means of altering gene expression in a cell-specific manner (34). In addition, a relatively new class of pharmaceuticals is targeted at proteins that are ‘readers, writers and erasers’ of the epigenetic code associated with regulation of gene expression. Such molecules include inhibitors of histone de-acetylases, histone methyltransferases, and histone tail mimetics. In addition to acting at promoters, these molecules also exert effects at enhancer elements. A striking demonstration of the potential of this general class of molecules to have translational potential is provided by histone tail mimetics that prevent the interaction of the extra terminal domain (BET) family of proteins with acetylated histones (99). These compounds disrupt chromatin complexes responsible for the expression of key inflammatory genes in activated macrophages, and confer protection against lipopolysaccharide-induced endotoxic shock and bacteria-induced sepsis. Subsequent studies demonstrated that super-enhancers are particularly susceptible to this class of compounds, further raising the possibility of targeting the enhancer landscape for therapeutic purposes (100). Intriguingly, this class of histone tail mimetics alters inflammation-induced enhancer selection in endothelial cells and inhibits the development of atherosclerosis in a mouse model (65).
In conclusion, the expanding appreciation of the tissue-specific homeostatic functions of macrophages and their various roles in human disease reinforces the importance of efforts to understand the mechanisms by which they achieve their distinct phenotypes. Genome wide approaches to defining macrophage enhancer selection and function are likely to provide a fruitful avenue of investigation towards this goal for the foreseeable future.
References
- 1.Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunological reviews. 2014;262(1):36–55. doi: 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton GM, Medzhitov R. Toll-like receptors and their ligands. Current topics in microbiology and immunology. 2002;270:81–92. doi: 10.1007/978-3-642-59430-4_5. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9(10):692–703. doi: 10.1038/nri2634. Epub 2009/10/28. nri2634 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Smale ST. Transcriptional regulation in the innate immune system. Current opinion in immunology. 2012;24:51–7. doi: 10.1016/j.coi.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Advances in experimental medicine and biology. 2005;560:11–8. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- 7.Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol. 2016;17(1):26–33. doi: 10.1038/ni.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–34. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lumeng CN. Lung Macrophage Diversity and Asthma. Annals of the American Thoracic Society. 2016;13(Suppl 1):S31–4. doi: 10.1513/AnnalsATS.201506-384MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurotaki D, Uede T, Tamura T. Functions and development of red pulp macrophages. Microbiol Immunol. 2015;59(2):55–62. doi: 10.1111/1348-0421.12228. [DOI] [PubMed] [Google Scholar]
- 11.Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res. 2016;118(4):653–67. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 13.Ju C, Mandrekar P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol research: current reviews. 2015;37:251–62. [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, Scott DK, O’Doherty RM. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59(2):347–57. doi: 10.2337/db09-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ransohoff RM, El Khoury J. Microglia in Health and Disease. Cold Spring Harb Perspect Biol. 2016;8(1):a020560. doi: 10.1101/cshperspect.a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81. doi: 10.1016/j.conb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Vlaeminck Y, González-Rascón A, Goyvaerts C, Breckpot K. Cancer-Associated Myeloid Regulatory Cells. Frontiers in Immunology. 2016;7:113. doi: 10.3389/fimmu.2016.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winter DR, Jung S, Amit I. Making the case for chromatin profiling: a new tool to investigate the immune-regulatory landscape. Nat Rev Immunol. 2015;15(9):585–94. doi: 10.1038/nri3884. [DOI] [PubMed] [Google Scholar]
- 20.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer-Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A, Rubin EM. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444(7118):499–502. doi: 10.1038/nature05295. Epub 2006/11/07. [DOI] [PubMed] [Google Scholar]
- 21.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, Walter K, Abnizova I, Gilks W, Edwards YJ, Cooke JE, Elgar G. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3(1):e7. doi: 10.1371/journal.pbio.0030007. Epub 2005/01/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2 Pt 1):299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 23.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology. 2015 doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman Meyer J, Song X, Zhang F, Ma Q, Ohgi Kenneth A, Krones A, Rosenfeld Michael G. Enhancer Activation Requires trans-Recruitment of a Mega Transcription Factor Complex. Cell. 2014;159:358–73. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 27.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, Trompouki E, Bowman Teresa V, Zon Leonard I, Yuan G-C, Orkin Stuart H, Xu J. Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Developmental Cell. 2016;36:9–23. doi: 10.1016/j.devcel.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright AV, Nunez JK, Doudna JA. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell. 2016;164(1–2):29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends in biochemical sciences. 2014;39:170–82. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17(4):207–23. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 33.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, Glass CK. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Molecular cell. 2013;51:310–25. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam MTY, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, Lee CY, Watt A, Grossman TR, Rosenfeld MG, Evans RM, Glass CK. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–5. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, Oh S, Kim H-S, Glass CK, Rosenfeld MG. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–20. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaukowitch K, Joo J-Y, Liu X, Watts Jonathan K, Martinez C, Kim T-K. Enhancer RNA Facilitates NELF Release from Immediate Early Genes. Molecular Cell. 2014;56:29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, Young RA. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350(6263):978–81. doi: 10.1126/science.aad3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai F, Gardini A, Zhang A, Shiekhattar R. Integrator mediates the biogenesis of enhancer RNAs. Nature. 2015 doi: 10.1038/nature14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Austenaa LM, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, Curina A, de Wit E, Bouwman BA, de Pretis S, Piccolo V, Termanini A, Prosperini E, Pelizzola M, de Laat W, Natoli G. Transcription of Mammalian cis-Regulatory Elements Is Restrained by Actively Enforced Early Termination. Mol Cell. 2015;60(3):460–74. doi: 10.1016/j.molcel.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 40.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, Black BL, Visel A, Pennacchio LA, Collins FS, National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program A, Authors NCSP Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110(44):17921–6. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, Young RA. Convergence of Developmental and Oncogenic Signaling Pathways at Transcriptional Super-Enhancers. Molecular cell. 2015;58:362–70. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hah N, Benner C, Chong L-W, Yu RT, Downes M, Evans RM. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E297–302. doi: 10.1073/pnas.1424028112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosselin D, Link VM, Romanoski Casey E, Fonseca Gregory J, Eichenfield Dawn Z, Spann Nathanael J, Stender Joshua D, Chun Hyun B, Garner H, Geissmann F, Glass Christopher K. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell. 2014;159:1327–40. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–5. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tronche F, Yaniv M. HNF1, a homeoprotein member of the hepatic transcription regulatory network. Bioessays. 1992;14(9):579–87. doi: 10.1002/bies.950140902. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 49.MacArthur S, Li XY, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keranen SV, Knowles DW, Stapleton M, Bickel P, Biggin MD, Eisen MB. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 2009;10(7):R80. doi: 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132(6):958–70. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–81. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandmann T, Jensen LJ, Jakobsen JS, Karzynski MM, Eichenlaub MP, Bork P, Furlong EE. A temporal map of transcription factor activity: mef2 directly regulates target genes at all stages of muscle development. Dev Cell. 2006;10(6):797–807. doi: 10.1016/j.devcel.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Scott EW, Simon MC, Anastasi J, Singh H. Science. Vol. 265. New York, NY: 1994. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages; pp. 1573–7. [DOI] [PubMed] [Google Scholar]
- 54.McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. The EMBO journal. 1996;15:5647–58. [PMC free article] [PubMed] [Google Scholar]
- 55.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hubner N, Evans RM. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49(1):158–71. doi: 10.1016/j.molcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, Glass CK. Effect of natural genetic variation on enhancer selection and function. Nature. 2013 doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–71. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 59.Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome C Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–28. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A. 2010;107(6):2568–73. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell. 2014;159:1312–26. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, Friedman N, Amit I. Chromatin state dynamics during blood formation. Science. 2014;345:943–9. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157(4):832–44. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown Jonathan D, Lin Charles Y, Duan Q, Griffin G, Federation AJ, Paranal Ronald M, Bair S, Newton G, Lichtman AH, Kung AL, Yang T, Wang H, Luscinskas Francis W, Croce KJ, Bradner James E, Plutzky J. NF-κB Directs Dynamic Super Enhancer Formation in Inflammation and Atherogenesis. Molecular Cell. 2014;56:219–31. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen Stephanie K, Lovell Claudia D, Dolgalev I, Yonekubo Y, Zhang G, Rusinova E, Gerona-Navarro G, Cañamero M, Ohlmeyer M, Aifantis I, Zhou M-M, Tsirigos A, Hernando E. Control of Embryonic Stem Cell Identity by BRD4-Dependent Transcriptional Elongation of Super-Enhancer-Associated Pluripotency Genes. Cell Reports. 2014;9:234–47. doi: 10.1016/j.celrep.2014.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navás D, Cordon-Cardo C, Clappier E, Soulier J, Ferrando AA. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature Medicine. 2014;20:1130–7. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreau-Gachelin F, Wendling F, Molina T, Denis N, Titeux M, Grimber G, Briand P, Vainchenker W, Tavitian A. Spi-1/PU.1 transgenic mice develop multistep erythroleukemias. Molecular and cellular biology. 1996;16:2453–63. doi: 10.1128/mcb.16.5.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenbauer F, Wagner K, Kutok JL, Iwasaki H, Le Beau MM, Okuno Y, Akashi K, Fiering S, Tenen DG. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nature genetics. 2004;36:624–30. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 70.Schönheit J, Kuhl C, Gebhardt ML, Klett FF, Riemke P, Scheller M, Huang G, Naumann R, Leutz A, Stocking C, Priller J, Andrade-Navarro MA, Rosenbauer F. PU.1 level-directed chromatin structure remodeling at the Irf8 gene drives dendritic cell commitment. Cell reports. 2013;3:1617–28. doi: 10.1016/j.celrep.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 71.Terry RL, Miller SD. Molecular control of monocyte development. Cellular immunology. 2014 doi: 10.1016/j.cellimm.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carotta S, Willis SN, Hasbold J, Inouye M, Pang SHM, Emslie D, Light A, Chopin M, Shi W, Wang H, Morse HC, Tarlinton DM, Corcoran LM, Hodgkin PD, Nutt SL. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. The Journal of experimental medicine. 2014;211:2169–81. doi: 10.1084/jem.20140425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, Sasaki H, Matsunaga S, Yoshinari M, Ryo A, Nakazawa M, Ozato K, Tamura T. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nature communications. 2014;5:4978. doi: 10.1038/ncomms5978. [DOI] [PubMed] [Google Scholar]
- 74.Mancino A, Termanini A, Barozzi I, Ghisletti S, Ostuni R, Prosperini E, Ozato K, Natoli G. A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev. 2015;29(4):394–408. doi: 10.1101/gad.257592.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, Frazier WA, Murphy TL, Murphy KM. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. 2009;457(7227):318–21. doi: 10.1038/nature07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alfaqeeh S, Oralova V, Foxworthy M, Matalova E, Grigoriadis AE, Tucker AS. Root and Eruption Defects in c-Fos Mice Are Driven by Loss of Osteoclasts. J Dent Res. 2015;94(12):1724–31. doi: 10.1177/0022034515608828. [DOI] [PubMed] [Google Scholar]
- 77.Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266(5184):443–8. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 78.Johnson RS, Spiegelman BM, Papaioannou V. Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell. 1992;71(4):577–86. doi: 10.1016/0092-8674(92)90592-z. [DOI] [PubMed] [Google Scholar]
- 79.Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360(6406):741–5. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- 80.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15(11):1026–37. doi: 10.1038/ni.3005. [DOI] [PubMed] [Google Scholar]
- 81.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110(3):416–27. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–43. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, Fuchs E. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature. 2015;521:366–70. doi: 10.1038/nature14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nature genetics. 2016;48:176–82. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sweeney CL, Teng R, Wang H, Merling RK, Lee J, Choi U, Koontz S, Wright DG, Malech HL. Stem cells. Dayton, Ohio: 2016. Molecular analysis of neutrophil differentiation from human iPSCs delineates the kinetics of key regulators of hematopoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics. 2010;4(5):353–60. doi: 10.1186/1479-7364-4-5-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, Khavari PA. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome biology. 2015;16:284. doi: 10.1186/s13059-015-0840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Molecular Metabolism. 2016;5:233–44. doi: 10.1016/j.molmet.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davie K, Jacobs J, Atkins M, Potier D, Christiaens V, Halder G, Aerts S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling. PLOS Genetics. 2015;11:e1004994. doi: 10.1371/journal.pgen.1004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–6. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–51. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lewis WR, Malarkey EB, Tritschler D, Bower R, Pasek RC, Porath JD, Birket SE, Saunier S, Antignac C, Knowles MR, Leigh MW, Zariwala MA, Challa AK, Kesterson RA, Rowe SM, Drummond IA, Parant JM, Hildebrandt F, Porter ME, Yoder BK, Berbari NF. Mutation of Growth Arrest Specific 8 Reveals a Role in Motile Cilia Function and Human Disease. PLoS Genet. 2016;12(7):e1006220. doi: 10.1371/journal.pgen.1006220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee JS, Grav LM, Pedersen LE, Lee GM, Kildegaard HF. Accelerated homology-directed targeted integration of transgenes in Chinese hamster ovary cells via CRISPR/Cas9 and fluorescent enrichment. Biotechnol Bioeng. 2016 doi: 10.1002/bit.26002. [DOI] [PubMed] [Google Scholar]
- 98.Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439(1):132–6. doi: 10.1016/j.bbrc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 99.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]