

ABSTRACT
Bispecific antibodies (BsAbs) are emerging as an important class of biopharmaceutical. The majority of BsAbs are created from conventional antibodies or fragments engineered into more complex configurations. A recurring challenge in their development, however, is the identification of components that are optimised for inclusion in the final format in order to deliver both efficacy and robust biophysical properties. Using a modular BsAb format, the mAb-dAb, we assessed whether an ‘in-format’ screening approach, designed to select format-compatible domain antibodies, could expedite lead discovery. Human nerve growth factor (NGF) was selected as an antigen to validate the approach; domain antibody (dAb) libraries were screened, panels of binders identified, and binding affinities and potencies compared for selected dAbs and corresponding mAb-dAbs. A number of dAbs that exhibited high potency (IC50) when assessed in-format were identified. In contrast, the corresponding dAb monomers had ∼1000-fold lower potency than the formatted dAbs; such dAb monomers would therefore have been omitted from further characterization. Subsequent stoichiometric analyses of mAb-dAbs bound to NGF, or an additional target antigen (vascular endothelial growth factor), suggested different target binding modes; this indicates that the observed potency improvements cannot be attributed simply to an avidity effect offered by the mAb-dAb format. We conclude that, for certain antigens, screening naïve selection outputs directly in-format enables the identification of a subset of format-compatible dAbs, and that this offers substantial benefits in terms of molecular properties and development time.
KEYWORDS: Bispecific, monoclonal antibody, domain antibody, biopharmaceutical, phage display, in-format screening, nerve growth factor, vascular endothelial growth factor
Abbreviations
- BsAb
(bispecific antibody)
- mAb
(monoclonal antibody)
- dAb
(domain antibody)
- scFv
(single-chain variable fragment)
- CDR
(complementarity-determining region)
- NGF
(nerve growth factor)
- VEGF
(vascular endothelial growth factor)
- TrkA
(Tropomyosin receptor kinase A)
- SPR
(surface plasmon resonance)
- ITC
(isothermal calorimetry)
Introduction
Conventional mono-specific monoclonal antibodies (mAbs) are a well-validated treatment modality. Many human diseases, however, are multi-factorial, dependent on the interaction of numerous, frequently redundant targets; consequently, targeting a single antigen may be insufficient to achieve the desired therapeutic benefit. Although a strategy involving combinations of mAbs has been pursued, 1-3 this approach is often challenging to develop commercially because of increased manufacturing costs and development time, in addition to more complex clinical development as a result of the requirement to assess the safety and efficacy of each mAb separately and in combination. An alternative approach is to combine multiple pharmacologies into one molecule by generating a bispecific antibody (BsAb).
BsAbs are single biological drugs capable of engaging 2 molcular targets, either ligands or receptors present on the same or different cell types, in order to modulate their bioactivity. BsAbs have been generated using a multitude of different approaches over the past 25 y. First generation BsAbs, such as those derived from hybrid hybridomas, quadromas, or by chemical cross-linking of mAbs, have served to demonstrate the promise of the approach, both in preclinical animal models and in the clinic. 4-5 The challenges faced when manufacturing large homogeneous batches of material, and poor clinical efficacy, often due to the emergence of immunogenicity induced by rodent-derived antibodies, as for other biopharmaceuticals with a large proportion of non-human sequence, proved to be major limitations for progression of these types of molecules. 6,7
More recently, alternative genetically-engineered BsAb formats have been described. Bispecific T-cell Engager (BiTE™) antibodies are a fusion of 2 single chain variable fragments (scFv), that induce immune cell killing of cancer cells via their ability to specifically target T cells to tumor cells.8,9 While clinical development of BiTE™ molecules as anti-cancer therapeutics appears promising, their short half-life suggests this format may be unsuitable for use in diseases where there is a rationale to neutralise the activity of soluble ligands or inhibit signaling of cell surface receptors.
In order to retain the favorable pharmacokinetic properties of full-length IgG, a number of BsAb formats based on an immunoglobulin scaffold have been described, including mAb-scFv and DVD-Ig™ formats,10-12 although many of these are not without limitations. In some cases, the engineering complexity required to generate bispecificity is challenging, in others, lead molecules with favorable biophysical characteristics cannot be generated consistently. As a result, these limitations often hamper the development of BsAbs from an initial promising format into a lead discovery platform capable of delivering developable therapeutic biopharmaceuticals. Recently described BsAb formats, including CrossMab and common light chain technologies, are much closer to the canonical IgG structure as compared to the mAb-scFv and DVD-Ig™ formats.13
Domain antibodies (dAbs), the smallest known antigen-binding fragments derived from an antibody, occur naturally in camelids14,15 and sharks.16 Fully human recombinant forms representing the variable regions of the immunoglobulin heavy and light chains (VH and VL, respectively) are bioactive as monomers and can be highly expressed in E. coli; 17 such dAbs may be reformatted into larger molecules to create drugs with prolonged serum half-lives or other pharmacological activities.18,19
We previously demonstrated the ability to generate BsAbs that couple domain antibody (dAb) binding domains to an intact mAb scaffold via a flexible linker sequence to produce a ‘mAb-dAb’ molecule capable of binding bivalently 2 discrete targets, or 2 different epitopes on the same target.20,21 A recurring challenge in the development of BsAbs, however, is the identification of components that are optimised for inclusion in the final format in order to be able to deliver both efficacy and robust biophysical properties. Therefore, in order to further develop the mAb-dAb as a robust BsAb lead discovery platform, we assessed whether an ‘in-format’ screening approach could expedite the identification of format-compatible dAb moieties. These studies were performed in the context of wider activities in the pursuit of therapeutic biopharmaceuticals, including the use of other in vitro, and in vivo, antibody discovery platforms.
We report here that, for a subset of dAbs, the mAb-dAb format affords a significant improvement in potency, by comparison of formatted dAbs with the corresponding dAb monomers. Furthermore, different binding modes yield similar potency enhancements, as inferred from stoichiometric analyses of mAb-dAbs binding to 2 different antigens. These observations highlight the extent to which the activity of the dAb in this more complex architecture is influenced by its environment. We conclude that this advantageous property can be exploited in the creation of therapeutically-relevant BsAbs.
Results
During the development of the mAb-dAb format, the need to retrospectively refine molecules comprising dAbs that had undergone extensively affinity maturation as unformatted dAbs led us to investigate screening systems that would allow us to identify dAbs that were ‘fit-for-format’ from the outset. To investigate this approach, we selected nerve growth factor (NGF) as the antigen of therapeutic interest and performed conventional dAb selections using both using soluble and passive selection methods.
For reasons unknown, selections performed with passively coated NGF did not generate any target binders, as judged by surface plasmon resonance (SPR) analysis of E. coli-expressed unformatted dAbs; however, selections performed with soluble biotinylated NGF yielded a panel of 205 NGF-binding dAbs, representing 93 unique sequences (55 Vκ dAbs and 38 VH dAbs). Although binding affinities of the dAbs were not fully quantitated, binding activity was estimated by calling report points during the association and dissociation phases of each binding event, which allowed ranking of each dAb as ‘slow’, ‘medium’, or ‘fast’ in each phase. This analysis indicated that ∼50% of the unformatted dAbs had a rapid off-rate and none fell into the ‘fast on, slow off’ category that could represent a high affinity lead (data not shown). This technique was configured to enable screening of several hundred hits, and it should be noted that this assay format is unable to estimate true affinity because of the unknown concentration of the dAb sample being tested.
To assess whether the same binding profiles of unformatted dAbs were retained when the same dAbs were formatted as mAb-dAbs, all 93 NGF-binding dAbs were sub-cloned into the mAb-dAb format using a mAb with favorable biophysical properties as the mAb component (anti-interleukin(IL)-4 pascolizumab22). A short flexible linker (GSTVAAPSGS) was used to link the mAb heavy chain to the dAb, and mAb-dAbs were expressed in HEK293/6E cells and assessed for the ability of the dAb portion to bind to NGF by SPR. In this case, to avoid undesirable avidity effects using a bivalent format with immobilised target antigen, each mAb-dAb was captured on a Protein A sensor chip and NGF passed through the flow cells. Full kinetics were derived using Proteon SPR and a number of the dAbs that had fast or very fast off-rates as monomers were observed to show exceptionally high comparative (pM) binding affinity when analyzed in-format (Table 1). For other dAbs, there was no significant change in observed affinity as a consequence of reformatting as a mAb-dAb, while other dAbs lost binding altogether; these molecules were not pursued further.
Table 1.
SPR analysis of anti-NGF dAb monomers and corresponding mAb-dAbs. To determine the affinity of unformatted anti-NGF dAbs, NGF was immobilised on SPR sensor chips and dAb supernatants passed through the flow cells; dAbs that bound to immobilised antigen were ranked by dissociation rate. To determine the affinity of mAb-dAbs incorporating the corresponding dAbs, mAb-dAbs were captured on Protein A sensor chip and NGF passed through the flow cells; binding was measured by ProteOn™ SPR. Curves were fitted and evaluated using the 1:1 Langmuir binding model.
| BIAcore™ SPR for the unformatted dAb | ProteOn™ SPR for the dAb in mAb-dAb format |
|||
|---|---|---|---|---|
| dAb | kd (1/s) | ka (M-1) | kd (1/s) | KD (nM) |
| DT03-K-001 | 9.98 × 10−1 | 1.58 × 106 | 8.86 × 10−5 | 0.056 |
| DT03-K-018 | 7.22 × 10−3 | 1.79 × 106 | 1.30 × 10−4 | 0.073 |
| DT03-K-038 | 2.39 × 10−3 | 7.96 × 105 | 6.13 × 10−5 | 0.077 |
| DT03-H-018 | 1.91 × 10−3 | 3.38 × 105 | 1.91 × 10−4 | 0.565 |
| DT03-H-019 | 3.99 × 10−3 | 2.16 × 105 | 3.44 × 10−4 | 1.59 |
Next, we assessed whether the subset of ‘format-compatible’ dAbs were able to prevent binding to NGF receptors or to neutralise the function of NGF in a series of in vitro assays. The five lead mAb-dAbs as described in Table 1 were assessed for their ability to inhibit NGF binding to its receptors (TrkA or p75), and also for inhibition of NGF-induced responses in non-recombinant cell lines (TF-1 cell proliferation and PC-12 neurite outgrowth). In all assays, the panel of molecules was compared with an anti-NGF tool mAb of known affinity and potency. mAb-dAbs containing Vκ dAbs (DT03-K-001, DT03-K-018, and DT03-K-038), were of particular interest in these studies because they demonstrated the greatest apparent gain in binding affinity by inclusion into the mAb-dAb format, relative to the VH dAbs selected (DT03-H-018 and DT03-H-019). Consistent with this observation, mAb-dAbs incorporating format-compatible Vκ dAbs were consistently the most potent molecules across the majority of the selected assay formats (Table 2).
Table 2.
Quantitative and qualitative analyses of the potency of NGF-targeting mAb-dAbs in NGF receptor binding and cell-based assays. Each mAb-dAb, or anti-NGF tool mAb or isotype control, was assessed for inhibition of NGF binding to NGF receptors (TrkA or p75), and also for inhibition of cellular responses to recombinant NGF (TF-1 proliferation and PC-12 neurite outgrowth); in all cases, NGF-targeting molecules or isotype control were pre-incubated with NGF before assessing receptor binding or NGF-stimulated responses in non-recombinant cell lines. IC50 values represent the average of at least 2 independent experiments.
| NGF Receptor Binding Assay |
||||
|---|---|---|---|---|
| Molecule | TrkA IC50 (nM) | p75 IC50 (nM) | TF-1 cell proliferation IC50 (nM) | Neurite Outgrowth (Qualitative Analysis) |
| Pascolizumab-DT03-H-018 | 0.197 | 0.615 | 1.141 | Partial inhibition observed at 28 nM |
| Pascolizumab-DT03-H-019 | 0.315 | 1.188 | 12.326 | Inactive at all concentrations tested |
| Pascolizumab-DT03-K-001 | 0.089 | 0.444 | 0.308 | Full inhibition observed at 28 nM |
| Partial inhibition observed at 2.8 nM | ||||
| Pascolizumab-DT03-K-018 | 0.037 | 0.391 | 0.220 | Full inhibition observed at 28 nM |
| Partial inhibition observed at 2.8 nM | ||||
| Pascolizumab-DT03-K-038 | 0.289 | 1.060 | 0.377 | Full inhibition observed at 28 nM |
| Partial inhibition observed at 2.8 nM | ||||
| Anti-NGF tool mAb | 0.0134 | 0.1746 | 0.156 | Full inhibition observed at 2.8 nM |
| Partial inhibition observed at 0.28 nM | ||||
| hIgG1 isotype mAb | No curve | No curve | No curve | Inactive at all concentrations tested |
In order to further understand the potency enhancement afforded by formatting dAbs, the potencies of 3 format-compatible dAbs as unformatted monomers were compared with the corresponding mAb-dAbs in a TrkA (NGF receptor) binding assay. Consistent with the observed low affinity of dAb monomers observed previously (Table 1), such molecules exhibited partial or no detectable inhibition even at the highest concentrations tested (1 µM); in contrast, the same dAbs formatted as mAb-dAbs were capable of fully inhibiting binding of NGF to TrkA, with IC50 values in the sub-nM range for all molecules tested (Fig. 1). For example, VH dAb DT03-H-018 showed no detectable inhibition in this assay format as a dAb monomer, compared with an IC50 of 166 pM for the corresponding dAb in-format. Similar observations were made for the 2 Vκ dAbs assessed in this manner; DT03-K-018 and DT03-K-001 exhibited IC50 values in the low-µM range as unformatted dAbs, in contrast to sub-nM IC50 values when formatted (Fig. 1). This represents an improvement in potency of ∼3 log units for the in-format dAb compared with the dAb monomer as quantified by IC50 measurement in a receptor binding assay. This improvement is far in excess of the expected avidity effect offered by the mAb-dAb format as a result of incorporating 2 dAb monomers per molecule.
Figure 1.

Inhibition of NGF binding to NGF receptor TrkA by anti-NGF dAbs and corresponding mAb-dAbs. Inhibition of NGF binding to TrkA was assessed using the relevant purified recombinant proteins in a standard ELISA format; dAbs, or corresponding mAb-dAbs, were pre-incubated with NGF before transferring to an ELISA plate coated with TrkA. An anti-NGF tool mAb was used as a positive control. Receptor-bound NGF, and inhibition thereof, was detected using a non-neutralising anti-NGF mAb.
Next, we sought to investigate the effect of the linker used to fuse dAb to mAb on the potency of the dAb moiety. mAb-dAbs incorporating both VH and Vκ anti-NGF dAbs were created by fusion with a ‘long’ linker (GSTVAAPSTVAAPSTVAAPSGS) and compared in NGF-induced functional assays with mAb-dAbs incorporating the original ‘short’ linker (GSTVAAPSGS). In both TF-1 and PC-12 assays, the longer linker enhanced the potency of formatted VH dAbs, with a concomitant reduction in the potency of the corresponding formatted Vκ dAbs (Fig. 2), suggesting a critical role for the linker between dAb and mAb in the mAb-dAb architecture that is dependent on subtle differences between Vκ and VH dAbs and the manner in which they interact with the antigen.
Figure 2.
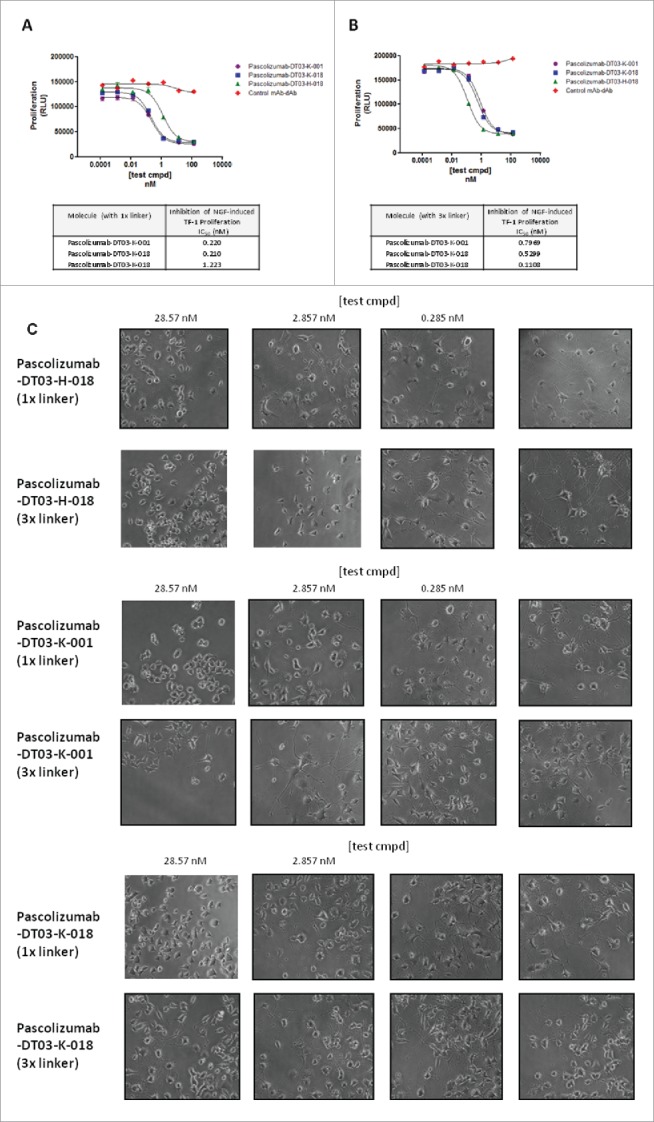
The effect of linker length on potency of mAb-dAbs. Linkers between anti-NGF dAbs and pascolizumab were classed as ‘short’ (GSTVAAPSGS) or ‘long’ (GSTVAAPSTVAAPSTVAAPSGS). mAb-dAbs were generated in HEK293/6E cells by transient transfection and purified using Protein A resin. Potency was assessed through quantitative analysis of TF-1 proliferation (Fig. 2A and 2B) or qualitative analysis of PC-12 cell neurite outgrowth (Fig. 2C). For neurite outgrowth, a thick border on each image denotes concentrations of test compound that partially or fully inhibited outgrowth.
The observation that both affinity and potency could be enhanced when NGF-targeting dAbs were formatted as mAb-dAbs led us to investigate potential mechanisms that might underlie the observed enhancement. We determined the stoichiometric ratio of mAb-dAbs bound to NGF by isothermal titration calorimetry (ITC) to infer potential binding mechanisms. Two additional mAb-dAbs incorporating naïve anti-vascular endothelial growth factor (VEGF) dAbs, also exhibiting significantly higher potency in-format than as monomers (data not shown), were assessed in the same manner. All mAb-dAbs incorporated the ‘short’ linker (GSTVAAPSGS). These analyses were designed to test the hypothesis that the mAb-dAb architecture was capable of creating an additional avidity effect by ‘trapping’ a single homodimeric molecule of NGF or VEGF between the dAb moieties, as may be inferred from a 1:1 stoichiometric ratio, or whether contacts between the CH3 or CH2 domains of the mAb were important for the enhanced target binding, as may be inferred from a 1:2 stoichiometric ratio.
Intriguingly, we observed that mAb-dAb molecules containing NGF-targeting molecules exhibited a 1:2 stoichiometric ratio, whereas in-format VEGF-targeting dAbs exhibited a 1:1 stoichiometric ratio (Table 3). From these data we hypothesize that the NGF dimer is bound between each dAb moiety and the CH3 domain of the mAb. In contrast, for mAb-dAbs targeting VEGF, we infer that an avidity effect is created by trapping the VEGF dimer between the 2 dAb moieties. This suggests that, although mAb-dAbs are capable of different modes of target engagement, both binding modes are fortuitously able to afford the same potency enhancement.
Table 3.
Stoichiometric ratio and apparent affinity for the interaction between VEGF or NGF and relevant mAb-dAbs. Each antibody was titrated with each antigen to give a final excess of >1 .fold5-. Data from the titrations were analyzed using isothermal titration calorimetry and fitted within Origin→ using the 1:1 model (MicroCal version).
| Molecule | Target Antigen | Stoichiometric Ratio | Apparent Affinity (nM) |
|---|---|---|---|
| Pascolizumab-DT03-H-018 | NGF | 1:2 | <3.45 |
| Pascolizumab-DT03-K-018 | NGF | 1:2 | <1.0 |
| Pascolizumab-DT02-H-098 | VEGF | 1:1 | <3.2 |
| Pascolizumab-DT02-K-044 | VEGF | 1:1 | <26.0 |
Discussion
We describe here the finding that ‘in-format’ screening of domain antibodies (dAbs)—specifically, direct cloning of dAbs from naïve selections and subsequent screening of molecules in the mAb-dAb format—identifies a subset of dAbs that display greatly enhanced potency compared to the corresponding unformatted molecules. This was exemplified in NGF binding affinity/avidity and receptor inhibition assays where anti-NGF dAbs as unformatted monomers were compared with the same molecules in mAb-dAb format (Table 1 and Fig. 1). Such formatted dAbs also exhibited potencies (IC50) in the low-nM or sub-nM range in cell-based functional assays using NGF-responsive non-recombinant cell lines (Table 2) despite having undergone no affinity maturation. mAb-dAbs were generated by fusing the dAb moiety to pascolizumab22 via a short flexible linker (GSTVAAPSGS); subsequent studies indicated the importance of this linker, with an apparent distinction between VH or Vκ dAbs being observed for the targets studied in this work (Fig. 2). Finally, we inferred different target binding modes for mAb-dAbs with either anti-NGF or anti-VEGF dAb moieties by ITC; specifically, binding ratios of 1:2 and 1:1 were observed for NGF- and VEGF-targeting mAb-dAbs, respectively.
Initially, we anticipated that the process of in-format screening would be an effective way to exclude from further analysis any dAbs that exhibited undesirable or detrimental properties such as low expression, compromised biophysical properties, or where antigen engagement was negatively affected by the molecular architecture. Given that the dAb CDRs in a mAb-dAb are orientated toward the C-terminus of the mAb scaffold, the potential for steric inhibition of antigen binding is great. However, we also observed that a number of leads, once formatted, not only maintained the ability to interact with the target antigen, but exhibited improved affinity and potency, sometimes by several orders of magnitude.
This led to the development of the concept of ‘in-format screening’, whereby formatting could be used as a strategy to not only identify dAbs with favorable biophysical properties in the final format, but also to achieve high potency lead dAbs without the need for additional rounds of affinity maturation. For campaigns where the ultimate goal was to produce a mAb-dAb, these observations led us to dispense with primary screening of unformatted dAb monomers in favor of in-format screening of naïve dAb selection outputs. As a consequence, subsequent studies did not compare unformatted dAbs with the same molecules formatted as mAb-dAbs as described here. However, the ability to identify high potency naïve dAbs directed against other targets, including VEGF, indicated that the observed effect was not limited to a single target and, more recently, additional discovery campaigns on other more complex therapeutic targets including LRP6 (a co-receptor of Frizzled in the WNT signaling pathway) have resulted in similar observations,23 suggesting that the reported potency enhancement is not limited to target antigens such as NGF and VEGF, which naturally exist in a dimeric form.
Comparison of our original and refined lead discovery processes exemplifies how this approach circumvents time-consuming, iterative rounds of affinity maturation of dAb hits and subsequent optimisation of formatted mAb-dAbs (Fig. S1). In-format screening ensures that the dAb is chosen specifically for activity in the context of the BsAb format, thereby by-passing dAb optimisation to meet both potency and developability criteria post facto.
During the course of our studies, we consistently generated data that suggested a subset of dAbs were particularly suited to inclusion at the C-terminus of the IgG CH3 domain. The attributes of dAbs suited to this format are likely to be antigen specific rather than reflecting a specific, target-independent characteristic. This led us to postulate a number of hypotheses as to why this molecular architecture may yield such an effect. Fusion of the N-terminus of the dAb to the mAb CH3 domain results in the dAb CDRs being orientated toward the loops at the bottom of the CH3 domain; intuitively, this would suggest that most dAbs would lose, as opposed to gain, target antigen contacts due to steric hindrance. Indeed, our screening did suggest many dAbs lost target antigen binding in this format. However, the affinity/avidity gains observed for other leads indicate that additional binding energy has been gained through an increased number of interactions between mAb-dAb and target for a subset of dAbs that we thus consider to be highly ‘format compatible’.
Both of the target antigens described here—NGF (PDB ID: 1BET24), and VEGF (PDB ID: 1VPF25)—are homodimers that form either parallel (NGF) or anti-parallel (VEGF) dimers; therefore, the mAb-dAb format (which contains 2 dAb moieties per molecule) might be predicted to favor an avidity-driven 1:1 stoichiometry, whereby the mAb-dAb binds a single homodimer of target antigen in a ‘clamp’ arrangement, subject to sufficient degrees of freedom for the necessary interactions to form.
To test this concept, ITC experiments were undertaken to determine the stoichiometry of binding for both target antigens. These experiments strongly suggested that in-format dAbs bound to their target antigens in different configurations. While the VEGF-targeting mAb-dAbs under evaluation complexed target antigen in the predicted 1:1 binding mode as described above, in-format anti-NGF dAbs very clearly bound target antigen in a 1:2 complex, with each mAb-dAb molecule binding to 2 NGF dimers. We therefore propose that VEGF is bound at an equivalent point in the duplicated antigen structure by each dAb, with the 2 dAbs acting in the manner of a ‘clamp’ to bind VEGF, thus affording a significant additional avidity effect (Fig. 3); indeed, this binding mechanism is consistent with that described previously for an alternative BsAb format incorporating related anti-VEGF dAbs,26 although for this ‘dual dAb’ architecture, binding is likely to be achieved by ‘side-on’ engagement of 2 dAbs resulting in an exceptionally strong avid binding complex.
Figure 3.
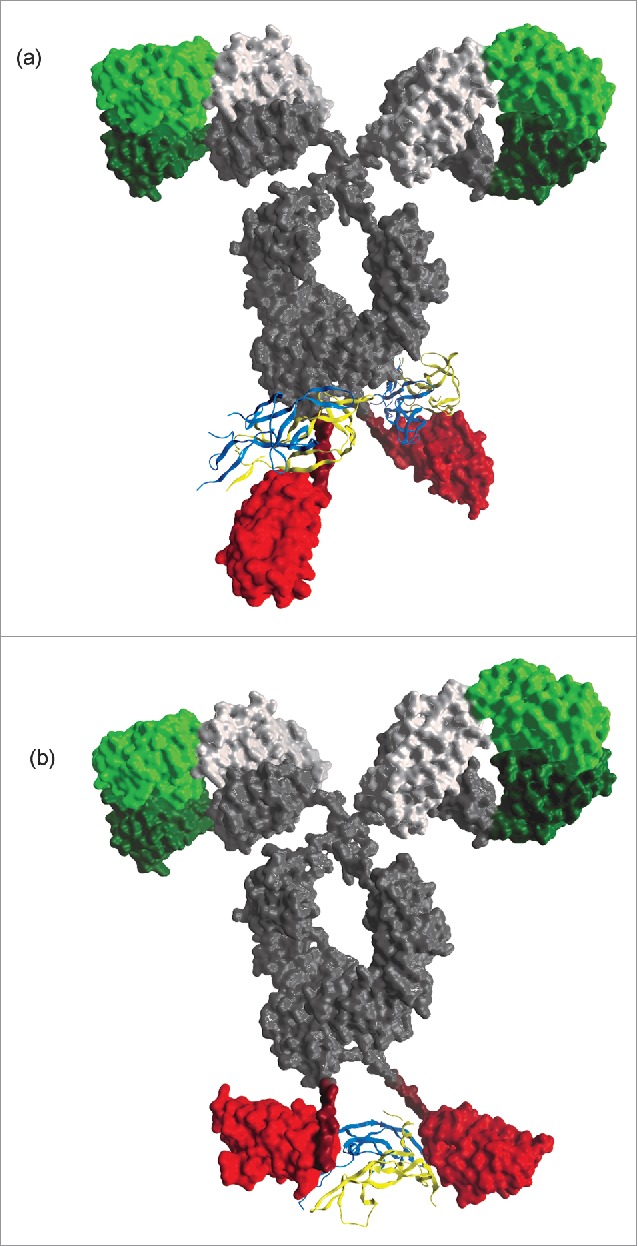
Schematic diagrams generated using MOE™ (Chemical Computing Group, Montreal, Canada) representing proposed binding modes of anti-NGF (Fig. 3A) and -VEGF (Fig. 3B) mAb-dAbs. Protein models were generated approximately to scale using stoichiometric data for mAb-dAbs incorporating anti-NGF or anti-VEGF dAbs which indicated a 1:2 or 1:1 binding mode, respectively. In both cases, a standard mAb crystal structure was used as the mAb moiety (PDB ID: 1IGT, Harris et al. 1997). A ligand-bound anti-NGF Fab co-crystal structure (PDB ID: 4EDX34) was used to generate the remainder of the structure for the mAb-dAb incorporating an anti-NGF dAb; similarly, a ligand-bound anti-VEGF dAb co-crystal structure (generated in-house) was used to recreate the remainder of the proposed structure for the mAb-dAb incorporating an anti-VEGF mAb-dAb. In both cases, the schematic structural model took into account whether the target was an anti-parallel (VEGF) or parallel (NGF) dimer.
The binding of 2 NGF dimers by a single mAb-dAb poses a more difficult question; it is possible that in this instance the target antigen is interacting with dAb CDR residues and, serendipitously, with residues within the mAb CH3 domain (Fig. 3). This mode of binding may not have been anticipated or discovered without the use of the in-format screening approach. Further structural studies on Fc-dAb/target antigen co-complexes are required to fully dissect the binding mechanism and explain the potency gain seen for a subset of dAbs when formatted as mAb-dAbs. Recently, the in-format screening approach described herein has been used to select high affinity/potency naïve dAbs against the LRP6 protein.23 Here again, the in-format screening approach was effective at identifying highly potent, format compatible dAbs that required little or no maturation to deliver pM binding affinity to this complex antigen. This illustrates that the dimeric status of the target antigens used in this work is not wholly responsible for the observed affinity gains reported here, and we have subsequently extended this approach successfully to all dAb discovery for mAb-dAb bispecific programmes.
For the examples cited here, it has been conclusively demonstrated that the in-format screening approach was critical in the identification of format-compatible dAb leads. By exploiting advances in high-throughput mammalian transfection, thousands of naïve dAbs can be readily screened in-format and diverse lead panels created that require relatively minor affinity maturation, if any, to achieve target potency. This has the dual benefits of reducing lead discovery cycle times and minimising the accumulation of mutations acquired during maturation, which often have a negative effect on downstream developability criteria.
Crucially, had in-format screening not been rigorously extended to all dAbs irrespective of the potency as an unformatted dAb, it is likely that many of the identified dAb leads would have been overlooked or ignored in primary screening, given that many of the molecules showed little or no evidence of target binding or neutralisation; similarly, those dAbs that did show evidence of target neutralisation as an unformatted dAb monomers would have required iterative rounds of affinity maturation to attain therapeutically useful affinity and potency, with no guarantee of retaining favorable functional or biophysical properties when reformatted. In summary, we believe that the in-format screening approach described here is a powerful new strategy in the pursuit of therapeutically relevant BsAbs.
Materials and methods
Cells and reagents
TF-1 cells were sourced from ATCC. HEK293/6E cells were kindly provided by National Research Council, Canada. PC-12 cells were kindly provided by Guy's Hospital, London, UK. Recombinant human NGF (β-NGF) was sourced from R&D Systems (cat no. 256-GF, lot no. HS3209051). VEGF (VEGF-165), and NGF receptors TrkA and p75 (both expressed as Fc-fusion proteins), were generated in-house. The non-neutralising anti-NGF mAb was sourced from Abcam (cat no. 21558, lot no. 901565). The horseradish peroxidase (HRP)-labeled secondary antibody was sourced from Southern Biotech, Birmingham, AL, USA (cat no. 1070–05).
Phage selection of domain antibodies
Domain Antibody Libraries27 were selected against target antigens using passive and soluble selection methodologies as previously described.28,29 Briefly, 3 rounds of selection were performed, with decreasing target antigen concentration in successive rounds. Passive selections involved coating target antigen onto a solid phase (immunotubes or EIA plates, Nunc), exposing the phage to the target antigen, and then washing and eluting antigen-specific binders. Alternatively, soluble selections used biotinylated target antigen, followed by capture of target antigen-specific binders using paramagnetic beads coated with streptavidin or neutravidin (DynalBiotech UK, Thermo Scientific), before washing and elution of target antigen-specific binders.
Cloning, expression, and purification of dAb monomers
Antigen binders were sub-cloned, expressed, and purified as described previously.30 Briefly, dAb sequences were sub-cloned into a pUC-based ampicillin resistant vector, where periplasmic expression of proteins is driven by the lac promoter. After transformation into E. coli, colonies were inoculated into OnEx lactose-based auto induction media (Novagen) for protein expression at 37°C for 24 h. dAbs were screened either as unpurified supernatants or in purified form from 50 ml cultures. dAbs were purified by capture on either protein L (for Vκ dAbs) or protein A (for VH dAbs) resins. After capture, dAbs were eluted with 0.1 M glycine pH 2.2 and neutralised with 1 M Tris-HCl pH 8.0, following by buffer exchange to phosphate-buffered saline (PBS).
Cloning, expression, and purification of mAb-dAbs
A mammalian expression vector containing the heavy chain of an anti-IL4 antibody (pascolizumab22) was modified by removal of the stop codon at the end of the heavy chain open reading frame (ORF) and incorporation of an in-frame linker segment. Linkers used in these studies coded for amino acid sequences classed as ‘short’ (GSTVAAPSGS) or ‘long’ (GSTVAAPSTVAAPSTVAAPSGS); a number of other linkers have also been assessed and found to work well in linking the mAb and dAb component, including those derived from fibronectin, such as GSTGLDSPT. Appropriate restriction sites were included at the end of the linker sequence to accommodate the dAb ORF. DNA sequences encoding the dAbs of interest were amplified by PCR, digested and cloned into the modified heavy chain expression vector at the end of the linker sequence. A DNA fragment coding for the cognate VL region from the pascolizumab was cloned into a mammalian expression vector containing the human kappa constant region.
mAb-dAbs were expressed in HEK293/6E (National Research Council, Canada). Briefly, HEK cells were co-transfected with 293fectin (Invitrogen) and heavy and light chain expression plasmids, then placed in a shaking incubator at 37°C, 5% CO2 with 95% relative humidity. Tryptone feeding media (5% w/v sterile stock of Tryptone N1 (Organotechnie)) was added at 24 hours and the cells grown for a further 3 d Supernatants were harvested by centrifugation and filter sterilised. The expressed mAb-dAbs were purified by affinity chromatography using immobilised Protein A columns; the purity of the sample was determined by size exclusion chromatography and the protein concentration was determined by spectrophotometry. Where necessary, aggregates were removed by preparative size exclusion chromatography and the yield then reassessed.
Surface plasmon resonance
All SPR binding experiments were carried out using a BIAcore A100 (GE Healthcare), BIAcore T100 (GE Healthcare), or ProteOn (Biorad), at 25°C. Samples were run in HBS-EP buffer (GE Healthcare) or HBS-N buffer (GE Healthcare) for BIAcore and ProteOn experiments respectively. Antibodies and antigens were passed through flow cells at a rate of 30µl/min and buffer alone was used to reference the binding curves. NGF (R&D Systems) was used as antigen for SPR, immobilised directly to a sensor chip to enable screening of several hundred hits and direct comparison between dAbs and corresponding mAb-dAbs.
To determine dAb kinetics of unformatted dAb molecules in E. coli supernatants, antigen was immobilised on a CM5 sensor chip (GE Healthcare) at a range of concentrations with capturing RUs between 200–2700. Alternatively, biotinylated antigen was immobilised on a Streptavidin chip (GE Healthcare) and E. coli supernatant solutions containing the expressed dAbs passed through the flow cells; dAbs that bound to directly immobilised antigen or streptavidin-immobilised antigen were ranked by dissociation rate.
To analyze binding kinetics for dAbs formatted as mAb-dAbs, Protein A (Pierce) was immobilised on either a CM5 sensor chip or GLC chip (Biorad) by primary amine coupling for BIAcore and ProteOn binding analysis, respectively. Protein A coupled surfaces were then used to capture mAb-dAb molecules and antigens passed over at various concentrations. Binding mAb-dAbs were then evaluated for antigen binding-affinity and ranked according to the KD. Binding curves were fitted and evaluated using the 1:1 Langmuir binding model using BIAcore/ProteOn proprietary software.
NGF receptor (TrkA and p75) binding assays
In vitro inhibition of NGF binding to the NGF receptors TrkA or p75 was assessed using purified recombinant protein in a standard ELISA. Briefly, 96-well MaxiSorp® ELISA plates (Nunc) were coated overnight with 0.125 μg/ml Fc-tagged TrkA or p75 washed with PBS containing 0.05% Tween®-20 (Sigma) to remove unbound receptor, and blocked with 4% BSA. Anti-NGF molecules and appropriate controls were pre-incubated in a polypropylene plate (Nunc) for 1 hour with NGF (R&D Systems) at a final concentration of 0.8 ng/ml (59 pM) before adding to the ELISA plate; receptor-bound NGF was detected using a non-neutralising anti-NGF mAb followed by HRP-labeled secondary antibody. Data was analyzed using GraphPad Prism v5.04 (GraphPad Software Inc., San Diego, CA). Neutralisation of NGF binding to NGF receptors was expressed as the concentration of antibody required to inhibit binding of NGF by 50% (IC50).
NGF TF-1 cell bioassay
TF-1 cells are a haematopoietic cell line that proliferate in response to a number of different cytokines, including NGF. These cells were used to measure the ability of antibodies to neutralise NGF, similar to methods described previously.31,32
Briefly, various concentrations of anti-IL4/anti-NGF mAb-dAbs and appropriate controls were pre-incubated for 15 minutes at room temperature with NGF (R&D Systems) in RPMI 1640 media (Invitrogen). The antibody/ligand mixture was then added to TF-1 cells (10,000 cells/well) in RPMI 1640 media in a 96-well tissue culture treated plate (Costar®, Cambridge, MA, USA) and incubated for 4 d at 37°C in a humidified incubator containing 5% CO2. The final concentration of NGF in each assay was 5 ng/ml (370 pM). Cell proliferation was assessed using the CellTitre 96® Non-Radioactive Cell Proliferation Assay kit or CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer's instructions. Data were analyzed using GraphPad Prism v5.04 (GraphPad Software Inc., San Diego, CA). Neutralisation of NGF-induced TF-1 cell proliferation was expressed as the concentration of antibody required to inhibit proliferation by 50% (IC50).
NGF-induced neurite outgrowth assay
Inhibition of NGF-stimulated neurite outgrowth was performed using a previously-described NGF-responsive cell line.33 Briefly, PC-12 cells in RPMI 1640 media (Invitrogen) were plated at 500 cells/well in 96-well CellCoat® Collagen Type I-coated plates (Greiner) and incubated overnight at 37°C in a humidified incubator containing 5% CO2. Anti-NGF molecules and appropriate controls were pre-incubated for 1 hour with NGF (R&D Systems) at a final concentration of 10 ng/ml (740 pM) and then added to the cell plate. Neurite outgrowth was monitored qualitatively using an IncuCyte™ live cell imaging system (Essen BioScience, Ann Arbor, MI, USA).
Isothermal titration calorimetry
ITC titrations were carried out using a MicroCal VP-ITC in PBS at 25°C. 100 µM of each antibody was titrated into 10 µM (assuming monomer) of each antigen to give a final excess of >1.5-fold. Data from the titrations were fitted within Origin® using the 1:1 model (MicroCal version) that assumes a single binding affinity for all multiple sites found.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Simon Chell, Michael Mullin, David Granger, and Ian Catchpole, for reviewing the manuscript. We thank Chun-wa Chung for assistance with ITC. We thank Alan Lewis for schematic diagrams of mAb-dAbs binding target antigens.
References
- 1.Press MF, Lenz HJ. EGFR, HER2 and VEGF pathways: validated targets for cancer treatment. Drugs 2007; 67(14):2045-75; PMID:17883287; http://dx.doi.org/ 10.2165/00003495-200767140-00006 [DOI] [PubMed] [Google Scholar]
- 2.Demarest SJ, Hariharan K, Dong J. Emerging antibody combinations in oncology. MAbs 2011 Jul-Aug; 3(4):338-51; PMID:21697653; http://dx.doi.org/26035486 10.4161/mabs.3.4.16615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diamant E, Torgeman A, Ozeri E, Zichel R. Monoclonal Antibody Combinations that Present Synergistic Neutralizing Activity: A Platform for Next-Generation Anti-Toxin Drugs. Toxins 2015; 7:1854-81; PMID:26035486; http://dx.doi.org/ 10.3390/toxins7061854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. mAbs 2010; 2:129-36; PMID:20190561; http://dx.doi.org/ 10.4161/mabs.2.2.11221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kontermann R. Dual targeting strategies with bispecific antibodies. mAbs 2012; 4:182-97; PMID:22453100; http://dx.doi.org/ 10.4161/mabs.4.2.19000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau WBMD, Heije K, Neefjes JJ, Oosterwegel M, Rozemuller E, Bast BJEG. Absence of preferential homologous H/L chain association in hybrid hybridomas. J Immunol 1991; 146:906-14; PMID:1899099 [PubMed] [Google Scholar]
- 7.Manzke O, Tesch H, Lorenzen J, Diehl V, Bohlen H. Locoregional treatment of low-grade B-cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. II. Assessment of cellular immune responses. Int J Cancer 2001; 91:516-22; PMID:11251975; http://dx.doi.org/ 10.1002/1097-0215(200002)9999:9999%3c::AID-IJC1069%3e3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- 8.Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941-44; PMID:19509221; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0547 [DOI] [PubMed] [Google Scholar]
- 9.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol 2015; 93:290-6.; PMID:25367186; http://dx.doi.org/ 10.1038/icb.2014.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coloma MJ and Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol 1997; 15:159-63; PMID:9035142; http://dx.doi.org/ 10.1038/nbt0297-159 [DOI] [PubMed] [Google Scholar]
- 11.Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L, Davis-Taber R, et al.. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nature Biotech 2007; 25:1290-7; PMID:17934452; http://dx.doi.org/25637431 10.1038/nbt1345 [DOI] [PubMed] [Google Scholar]
- 12.Gu J, Yang J, Chang Q, Liu Z, Ghayur T, Gu J. Identification of Anti-EGFR and Anti-ErbB3 Dual Variable Domains Immunoglobulin (DVD-Ig) Proteins with Unique Activities. PLoS One 2015; 10:1-12; PMID:25997020; http://dx.doi.org/25637431 10.1371/journal.pone.0124135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; http://dx.doi.org/ 10.1016/j.molimm.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 14.Arbabi-Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett 1997; 414:521-6; PMID:9323027; http://dx.doi.org/ 10.1016/S0014-5793(97)01062-4 [DOI] [PubMed] [Google Scholar]
- 15.Lauwereys M, Arbabi-Ghahroudi M, Desmyter A, Kinne J, Hölzer W, De Genst E, Wyns L, Muyldermans S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J 1998; 17:3512-20; PMID:9649422; http://dx.doi.org/ 10.1093/emboj/17.13.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roux KH, Greenberg AS, Greene L, Strelets L, Avila D, McKinney EC, Flajnik MF. Structural analysis of the nurse shark (new) antigen receptor (NAR): molecular convergence of NAR and unusual mammalian immunoglobulins. P Natl Acad Sci USA 1988; 95:11804-9; PMID:9751746; http://dx.doi.org/2677748 10.1073/pnas.95.20.11804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989; 341:544-6; PMID:2677748; http://dx.doi.org/ 10.1038/341544a0 [DOI] [PubMed] [Google Scholar]
- 18.Holt LJ, Herring C, Jespers LS, Woolven BP and Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol 2003; 21:484-90; PMID:14573361; http://dx.doi.org/ 10.1016/j.tibtech.2003.08.007 [DOI] [PubMed] [Google Scholar]
- 19.Walker A, Chung C-W, Neu M, Burman M, Batuwangala T, Jones G, Tang C-M, Steward M, Mullin M, Tournier N et al.. Novel Interaction Mechanism of a Domain Antibody-based Inhibitor of Human Vascular Endothelial Growth Factor with Greater Potency than Ranibizumab and Bevacizumab and Improved Capacity over Aflibercept*. J Biol Chem 2016; 261:5500-11; PMID:26728464; http://dx.doi.org/12296858 10.1074/jbc.M115.691162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashman C, Batuwangala T, Burden NM, Clegg SJ, De Wilt RM, Ellis JH, Hamblin PH, Hussain F, Jespers L, Lewis A. et al. Patent publication number WO2009/068649. 2009
- 21.Ashman C, Pouliquen IJ.Conference Abstract: American Association ofPharmaceutical Scientists National Biotechology Conference; 2013. [Google Scholar]
- 22.Hart TK, Blackburn MN, Brigham-Burke M, Dede K, Al-Mahdi N, Zia-Amirhosseini P and Cook RM. Preclinical efficacy and safety of pascolizumab (SB 240683): a humanized anti-interleukin-4 antibody with therapeutic potential in asthma. Clin Exp Immunol 2002; 130:93-100; PMID:12296858; http://dx.doi.org/ 10.1046/j.1365-2249.2002.01973.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson H, Granger D, Jones G, Anderson L, Friel S, Rycroft D, Fieles W, Tunstead J, Steward M, Wattam T, Walker A, Griggs J, Al-Hajj M, Shelton C. Novel Bi-specific domain antibody to LRP6 Inhibits Wnt and R-spondin ligand-induced Wnt signaling and tumor growth. Mol Cancer Res 2016; 23(859-868) [DOI] [PubMed] [Google Scholar]
- 24.McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature 1991; 354:411-4.; PMID:1956407; http://dx.doi.org/ 10.1038/354411a0 [DOI] [PubMed] [Google Scholar]
- 25.Muller YA, Christinger HW, Keyt BA, de Vos AM. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: multiple copy flexibility and receptor binding. Structure 1997; 5:1325-38.; PMID:9351807; http://dx.doi.org/ 10.1016/S0969-2126(97)00284-0 [DOI] [PubMed] [Google Scholar]
- 26.Walker A, Chung C-W, Neu M, Burman M, Batuwangala T, Jones G, Tang C-M, Steward M, Mullin M, Tournier N et al.. Novel Interaction Mechanism of a Domain Antibody-based Inhibitor of Human Vascular Endothelial Growth Factor with Greater Potency than Ranibizumab and Bevacizumab and Improved Capacity over Aflibercept*. J Biol Chem 2016; 261:5500-11; PMID:26728464; http://dx.doi.org/22886245 10.1074/jbc.M115.691162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ignatovich O, Jespers L, Tomlinson IM, de Wildt RM. Creation of the large and highly functional synthetic repertoire of human VH and Vκ domain antibodies. Methods Mol Biol 2012; 911:39-63; PMID:22886245; http://dx.doi.org/ 10.1007/978-1-61779-968-6_4 [DOI] [PubMed] [Google Scholar]
- 28.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol 1994; 12:433-55; PMID:8011287; http://dx.doi.org/ 10.1146/annurev.iy.12.040194.002245 [DOI] [PubMed] [Google Scholar]
- 29.Chames P, Hoogenboom HR, Henderikx P. Selection of antibodies against biotinylated antigens. Methods Mol Biol 2002; 178:147-57; PMID:11968483 [DOI] [PubMed] [Google Scholar]
- 30.Enever C, Pupecka-Swider M, Sepp A. Stress selections on domain antibodies: ‘what doesn't kill you makes you stronger’. Protein Eng Des Sel 2015; 28:59-66; PMID:25655396; http://dx.doi.org/ 10.1093/protein/gzu057 [DOI] [PubMed] [Google Scholar]
- 31.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, Piao Y-F, Miyazono K, Urabe A, Takaku F. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol 1989; 140:323-34; PMID:2663885; http://dx.doi.org/ 10.1002/jcp.1041400219 [DOI] [PubMed] [Google Scholar]
- 32.Thom G, Cockroft AC, Buchanan AG, Candotti C, Cohen S, Lowne D, Monk P, Shorrock-Hart CP, Jermutus L, Minter RR. Probing a protein-protein interaction by in vitro evolution. P Natl Acad Sci USA 2006; 103:7619-24; PMID:16684878; http://dx.doi.org/24830649 10.1073/pnas.0602341103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. P Natl Acad Sci USA 1976; 73:2424-8; PMID:1065897; http://dx.doi.org/24830649 10.1073/pnas.73.7.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.La Porte SL, Eigenbrot C, Ultsch M, Ho WH, Foletti D, Forgie A, Lindquist KC, Shelton DL, Pons J. Generation of a high-fidelity antibody against nerve growth factor using library scanning mutagenesis and validation with structures of the initial and optimized Fab-antigen complexes. MAbs 2014; 6:1059-68; PMID:24830649; http://dx.doi.org/ 10.4161/mabs.28677 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.