

ABSTRACT
Antibody-targeted nanoparticles have great promise as anti-cancer drugs; however, substantial developmental challenges of antibody modules prevent many candidates from reaching the clinic. Here, we describe a robust strategy for developing an EphA2-targeting antibody fragment for immunoliposomal drug delivery. A highly bioactive single-chain variable fragment (scFv) was engineered to overcome developmental liabilities, including low thermostability and weak binding to affinity purification resins. Improved thermostability was achieved by modifying the framework of the scFv, and complementarity-determining region (CDR)-H2 was modified to increase binding to protein A resins. The results of our engineering campaigns demonstrate that it is possible, using focused design strategies, to rapidly improve the stability and manufacturing characteristics of an antibody fragment for use as a component of a novel therapeutic construct.
KEYWORDS: Antibody-drug conjugate, antibody engineering, antibody fragment, developability, liposome, manufacturability, protein A binding, stability
Abbreviations
- scFv
single chain variable fragment
- CDR
complementarity-determining region
- EphA2
ephrin type-A receptor 2
- FACS
fluorescence-activated cell sorting
- DSF
differential scanning fluorimetry
- HTP
high throughput
- CLIA
chelated ligand-induced internalization assay
- ADN
antibody-directed nanotherapeutics
Introduction
Nanotherapeutics that combine an active drug molecule with a nanosized drug carrier offer many possibilities to beneficially modify the properties of the drug. One convenient and well-established nanocarrier platform is liposomes, which are vesicles formed by one or more lipid bilayers enclosing an aqueous interior. A small molecule is an effective chemotherapeutic, but can be limited in practice due to toxicity or rapid clearance from the circulation. Using liposomes as a drug delivery system for small molecules may be an efficient way to mitigate toxicity and increase efficacy of chemotherapy by improving pharmacokinetics and tumor localization.1,2 The first liposomal drug, pegylated liposomal doxorubicin (PLD; Doxil®; Johnson & Johnson), was approved in 1995.3 With the recent approval of a liposomal formulation of irinotecan (nal-IRI; ONIVYDE®) for the treatment of gemcitabine-refractory pancreatic cancer,4 more than 10 lipid-based nanoparticle drugs are now approved for clinical use (reviewed in ref.5). Using a targeting ligand such as an antibody fragment can improve the delivery of liposomal drug by increasing drug bioavailability subsequent to internalization and increasing the microdistribution within the tumor. 2,6,7 Antibody-directed nanotherapeutics (ADNs) are particularly attractive in oncology, where overexpressed cell surface receptors on tumors can be exploited to specifically deliver active chemotherapeutic agents. 8-11
Antibodies used as targeting agents for liposomes need to have specific biological characteristics to be effective in improving the delivery of the liposomal drug. Generally, an antibody fragment (e.g., Fab or scFv) lacking an Fc region is used so that effector functions are disallowed,12 and immunogenicity is decreased.13 To improve delivery of the payload, the antibody must be capable of mediating endocytosis after binding to its ligand.11 For liposomal targeting, antibody fragments with moderate affinity are generally the most effective at improving efficacy.14,15 Finally, to efficiently conduct preclinical toxicology studies and acquire accurate information on the effect of targeting on clearance and biodistribution of the ADN, it is desirable to have an antibody that is cross-reactive to a protein ortholog in rodent species.
In addition to the biological criteria, an antibody must be stable and support efficient purification for a robust and scalable manufacturing process. To meet the stability requirement to support insertion of antibody fragment into the liposome at high temperature, an unfolding temperature of greater than 70°C is desirable. A therapeutic antibody fragment, such as a scFv, must interact specifically with an affinity resin, such as Protein A, to enable reasonable purification yields at scale. For most therapeutic antibodies, Protein A purification is the first and most critical chromatography step because it selectively and efficiently binds antibodies from complex solutions such as harvested cell culture fluids. Protein A purification typically removes >99.5% of product impurities in a single step while also providing significant viral clearance.16 Stability and affinity resin-binding parameters require optimization in order to select a manufacturable antibody fragment appropriate for ADN construction.
Here we discuss rapid engineering of an anti-EphA2 antibody isolated from a phage library for development into a liposomal targeting scFv. EphA2 is a receptor tyrosine kinase that is commonly overexpressed on tumor cells including breast, prostate, and lung cancers.17 The originally identified antibody had strong internalization properties and biological activity in cell lines overexpressing EphA2.18 Further characterization of the scFv revealed, however, that it had a low unfolding temperature and did not bind well to affinity resins, and thus could not be advanced as a therapeutic targeting moiety. Given that suitable biophysical and manufacturing properties were absolutely required, 2 sequential engineering campaigns were completed to derive a scFv with improved thermal stability and protein A binding. We were able to design precise changes in both the framework and complementary-determining regions (CDRs) of the scFv to screen a small number of constructs that could be assayed in a short period. Using these focused engineering techniques, we were able to rapidly generate a scFv with the desired physicochemical properties that was then suitable for further development as a therapeutic targeting antibody fragment. The final scFv (scFv-3) is a component of MM-310, an EphA2-directed nanotherapeutic encapsulating a novel docetaxel prodrug that entered clinical trials earlier this year.
Results
Characterization of an anti-EphA2 antibody for liposomal conjugation
Anti-EphA2 antibodies were isolated with a multistep screening process using both phage and yeast selections.18 D2–1A7, one of the antibodies identified, is ligand blocking and shows strong internalization in the cell lines overexpressing EphA2.18 When profiled as a potential module for targeting nanoparticles, however, the scFv was unstable during the conjugation process, and, as a scFv, it bound poorly to protein A, with less than 20% being retained on the column. The scFv thus had good bioactivity, but was not viable as a therapeutic lead due to poor biophysical and manufacturing properties. Engineering campaigns were designed to identify scFvs with improvements in both the melting temperature and protein A binding capacity, while retaining bioactivity.
Framework stabilization of A7 to improve melting temperature
We hypothesized that the instability observed during the conjugation and subsequent insertion into liposomes of D2–1A7 was due to its modest melting temperature (65°C, Table 1). To increase the melting temperature, we grafted the CDRs of D2–1A7 onto different frameworks, followed by mutagenesis on the framework to retain affinity and improve stability. The framework of the heavy chain variable region of D2–1A7 is IGHV3–30. VH3s are considered very stable19 and typically have the ability to bind to protein A,20-22 making them a commonly used framework for therapeutic antibodies. We therefore decided keep the heavy chain within the VH3 family, but to graft the heavy chain of D2–1A7 onto a IGHV3–23 backbone, the most common isotype.23 Additionally, 2 stabilizing mutations were made within the IGHV3–23 framework, E6Q24 and S49A.25
Table 1.
Design and melting temperature of thermostable scFvs.
| scFv | Frameworks | Additional mutations | Tm (°C) |
|---|---|---|---|
| D2–1A7 | IGVH3–30 | 65 | |
| IGVλ3–19 | |||
| EphA2-TS1 | IGVH3–23 | VH: E6Q, S49A | 71 |
| IGVλ1–40 | Vλ: Q1S, V3E, G13V, R18T, S22T, L39K, F65S, K66S, A74T, L78A | ||
| EphA2-TS2 | IGVH3–23 | VH: E6Q, S49A | 65 |
| IGVλ1–40 | Vλ: Q1S, S22T, F65S, K66G |
A thermostable scFv was designed by altering the frameworks of the initially identified antibody, D2–1A7, and adding mutations to retain affinity or improve stability. Numbering is according to Kabat.46 Melting temperatures were measured using differential scanning fluorimetry.47
The framework of the light chain variable region of D2–1A7 is IGVλ3–19, which is not commonly found in the natural human antibody repertoire.23,26 Therefore, we decided to graft the CDRs to the more common framework IGVλ1–40, which is also found frequently paired with IGVH3–23.23 Two potentially thermostable variants, TS1 and TS2, were designed. Based on homology modeling, Positions 1 and 22 were thought to interact with the antigen, and so both variants were back-mutated to keep the original residues from D2–1A7 at these positions (Q1S, S22T). In TS1, we kept a hydrophobic pocket we thought might be important for stability (G13V, L78A), while adjusting charge on the surface (V3E, R18T). We compared the amino acid sequence to a repertoire of naïve library sequences and made additional changes, introducing L39K (TS1), F65S (both), K66S (TS1), K66G (TS2), and A74T (TS1). As shown in Table 1, EphA2-TS1 demonstrated a desired improvement in the thermostability, with an increased melting temperature of 6 degrees (to 71°C), and it was selected for further optimization and characterization. This work confirms that it is possible to use homology modeling to identify possible instabilities within the variable regions of an antibody.
Identification of region preventing protein A binding
EphA2-TS1 was engineered on a VH3–23 framework, but unexpectedly did not bind well to protein A resins, severely limiting its ability to be purified at an industrial scale. Inspection of the FR1, CDR-2 and FR3 sequence of the heavy chain, which have been shown to be involved in protein A binding, 20-22 was not obviously informative as to which amino acids were affecting the interaction between protein A and the scFv, as there were no deviations from germline sequences. Given this uncertainty, we decided to develop a high-throughput protein A binding assay to more closely identify the region of the scFv that most affected the ability to bind to protein A. To accomplish this, we used a previously identified scFv (F5 scFv) that bound to protein A,6,27 and designed a set of variants that differed in the pairing of the heavy and light chains of F5 and EphA2-TS1. In order to directly test the effect of the CDR-H2 sequence, we also made a scFv that replaced the CDR-H2 of EphA2-TS1 with the CDR-H2 of F5 (TS1*). We transiently expressed the variants in mammalian cells and loaded equal amounts of protein onto a protein A 96-well plate. We loaded the resin, determined the amount of scFv remaining in the flow-through, and calculated the percentage of scFv that bound to the protein A resin (Fig. 1). As expected, F5 had the highest percentage of protein bound, with 75% of the scFv binding to protein A. This is in contrast to the original scFv, D2–1A7, where only 11% of the scFv was recovered. EphA2-TS1 showed some improvement over D2–1A7 (29% vs. 11%), but was still not acceptable for development. Notably, the CDR-H2 graft of F5 into EphA2-TS1 showed significant improvement in protein A purification (65% recovery of protein), suggesting that the CDR-H2 of EphA2-TS1 was conferring a negative effect on protein A binding. However, this scFv did not bind to recombinant EphA2, and was not considered further (data not shown).
Figure 1.
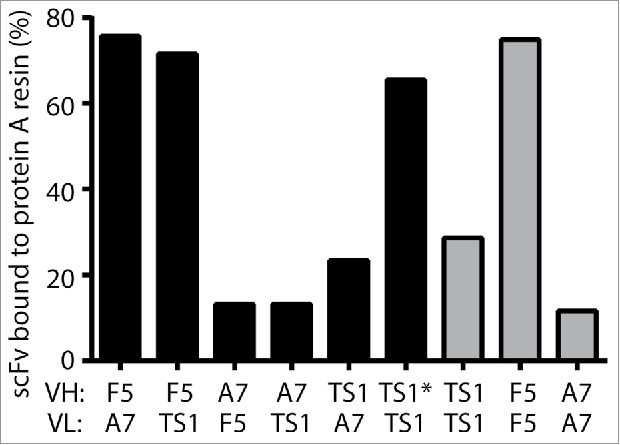
Protein A binding can be determined using a high-throughput assay. ScFvs were permuted based on the VH (variable region of the heavy chain) and VL (variable region of the light chain) and screened using a high-throughput assay to determine the amount of protein A binding. As expected, the heavy chain determined the amount of protein bound to protein A. F5, a scFv known to bind well to protein A, was used as a positive control. A7 refers to D1–2A7 and was used as a negative control. TS1 refers to EphA2-TS1. TS1* refers to a scFv with the EphA2-TS1 variable region and CDR-H2 of F5. This scFv had a high percentage of scFv bound to protein A resin, but lost the ability to bind to EphA2.
Library design and selection using yeast display
Given the overlap between protein A and EphA2 binding within CDR-H2, we decided to design a library and use yeast display to screen for clones that both retained EphA2 binding, and improved protein A binding. Our library introduced diversity into positions that we hypothesized to be important for protein A binding based on previous reports in the literature, 20-22 as well as positions we thought could be contributing to EphA2 binding. We then compared our library design with databases of antibody repertoire downloaded from the National Center for Biotechnology Information (NCBI), as well as next-generation sequencing of non-immune patient repertoire in order to preserve highly conserved residues. Ultimately, we selected residues VH49, VH50, VH52A, VH53, VH55, and VH57 as targets for mutagenesis. Again guided by the human repertoire, only a subset of amino acids was selected for most positions, allowing a reduction of the library size to only 60,000 variants (Table 2). This smaller size allowed us to rapidly screen the library using fluorescence-activated cell sorting (FACS). The scFvs were displayed on yeast and, for the first round of selection, we selected for both EphA2 and protein A binding. This was a very small percentage of the population, but was enriched ∼3X over the anti-protein A alone (Fig. 2A-B). To ensure we had identified a population that bound to recombinant EphA2, we amplified and reselected the clones that were positive for both binding to EphA2 and an anti-FLAG antibody (Fig. 2C-D). Enrichment over the negative control improved substantially, ∼20-fold. These results indicate that by using a focused, strategically designed library, it is possible to rapidly identify scFvs that exhibited both EphA2 and protein A binding.
Table 2.
Library design of CDR-H2.
| Position | EphA2-TS1 | F5 | Library |
|---|---|---|---|
| 49 | A | S | AGST |
| 50 | V | A | AVDEG |
| 51 | I | I | I |
| 52 | S | S | S |
| 52A | Y | G | CDGHNPRSTY or P (10%) |
| 53 | D | R | ADGHNPRST |
| 54 | G | G | G |
| 55 | S | D | ADGHNPRST |
| 56 | N | N | N |
| 57 | K | T | TNK |
| 58 | Y | Y | Y |
| 59 | Y | Y | Y |
| 60 | A | A | A |
Figure 2.
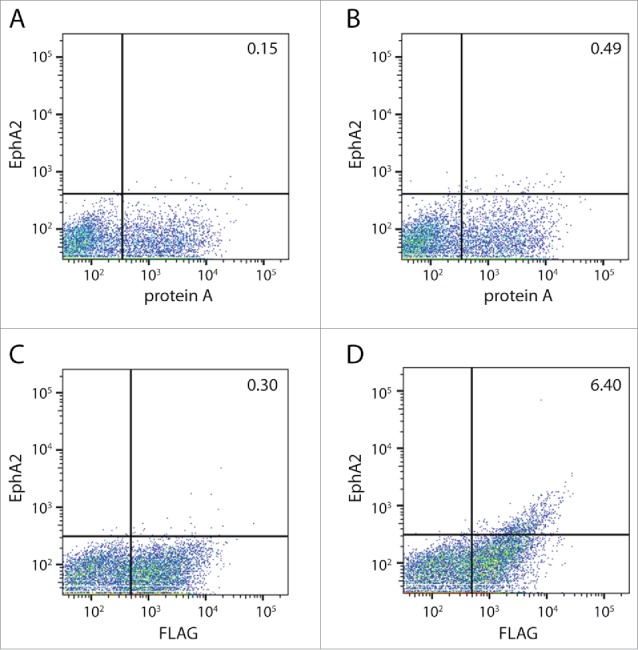
FACS of CDR-H2 library. The CDR-H2 library was generated using targeted mutagenesis as described in Table 2. (A) Yeast cells after one round of FACS stained with an antibody against protein A. (B) Yeast cells after one round of FACS, stained with antibodies against protein A (x-axis) and EphA2 (y-axis). The isolated clones were then amplified and sorted again. (C) Stained yeast cells using an antibody against the FLAG epitope to measure expression (x-axis) or (D) antibodies against the FLAG epitope (x-axis) and EphA2 (y-axis).
High-throughput characterization of soluble scFvs
We identified scFvs that were positive for protein A and EphA2 binding when displayed on yeast by sequencing and characterized them as soluble proteins for binding to protein A resin, melting temperature, and EphA2 binding activity. Using the same protein A binding assay as previously described, 15 of 22 variants had binding of at least 50%, a substantial improvement over EphA2-TS1 (Fig. 3). A majority of scFvs also had melting temperatures greater than 70°C (Table 3), suggesting the molecules were not destabilized by mutations in the CDR-H2 region. Activity of the scFv variants as ligands for directing the uptake of nanoparticles by EphA2-expressing cells was tested with a chelated ligand-induced internalization assay (CLIA).6,28 In this assay, histidine-tagged scFvs are attached in a releasable manner to fluorescent-labeled liposomes using the Ni-NTA chelation bond, so that the ligand-assisted total uptake and internalization of the liposomes by cells in culture can be measured by cytofluorimetry before and after treatment of cells with imidazole, which strips away liposomes that are bound to the surface. As shown in Fig. 4, most scFvs had binding and internalization similar to the EphA2-TS1 positive control (whose mean cell fluorescence was taken as 100%) when tested against 2 cancer cell lines with high expression levels of EphA2 (OVCAR-3 and U-251) and a murine cancer cell line (CT-26 colorectal) to demonstrate cross reactivity (Fig. 4). Nine scFvs were selected for further characterization based on the following criteria: protein A binding of greater than 50%, melting temperature of at least 69°C, and binding/internalization of at least 80% of the EphA2-TS1. Taken together, these results validate our library design and screening strategy, and identified promising candidates for further analysis of manufacturability.
Figure 3.
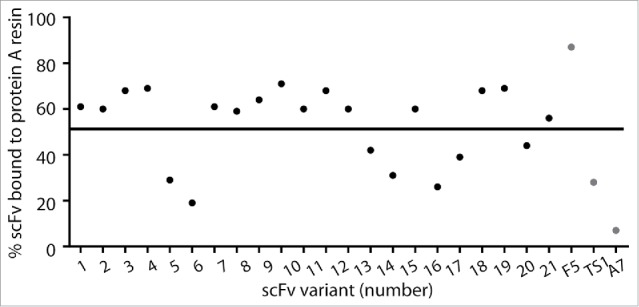
High-throughput protein A binding assay of anti-EphA2 scFvs. scFvs from the CDR-H2 library were expressed as soluble protein and binding was measured using the high-throughput protein A binding assay. F5 was used as a positive control, and A7 was used as a negative control.
Table 3.
scFvs identified from yeast display screening.
| scFv | CDR H2 sequence | Tm (°C) |
|---|---|---|
| scFv-1 | SVISPAGNNTYY | 71.5 |
| scFv-2 | SVISPAGRNKYY | 70.7 |
| scFv-3 | TVISPDGHNTYY | 71.7 |
| scFv-4 | TVISPHGRNKYY | 69.3 |
| scFv-5 | SVISRRGDNKYY | 66.7 |
| scFv-6 | SVISNNGHNKYY | 68.7 |
| scFv-7 | SVISPAGPNTYY | 67.5 |
| scFv-8 | TVISPSGHNTYY | 70.3 |
| scFv-9 | TVISPNGHNTYY | 70.9 |
| scFv-10 | SAISPPGHNTYY | 72.9 |
| scFv-11 | AVISHHGSNTYY | 70.1 |
| scFv-12 | AGISHPGDNTYY | 75.7 |
| scFv-13 | TVISPTGANTYY | 70.3 |
| scFv-14 | TVISPAGPNKYY | 61.9 |
| scFv-15 | SVISPHGSNKYY | 70.7 |
| scFv-16 | SVISNNGHNTYY | 69.5 |
| scFv-17 | TVISPPGSNKYY | 64.9 |
| scFv-18 | SVISPAGTNTYY | 72.5 |
| scFv-19 | SVISPPGHNTYY | 69.3 |
| scFv-20 | TVISHDGTNTYY | 68.3 |
| scFv-21 | TVISRHGNNKYY | 68.1 |
| scFv-22 | TVISPGGPNKYY | 62.3 |
| F5 | SAISGRGDNTYY | 78.9 |
| EphA2-TS1 | AVISYDGSNKYY | 72.5 |
scFvs were expressed as soluble proteins, sequenced, and the melting temperature was measured using DSF.
Figure 4.
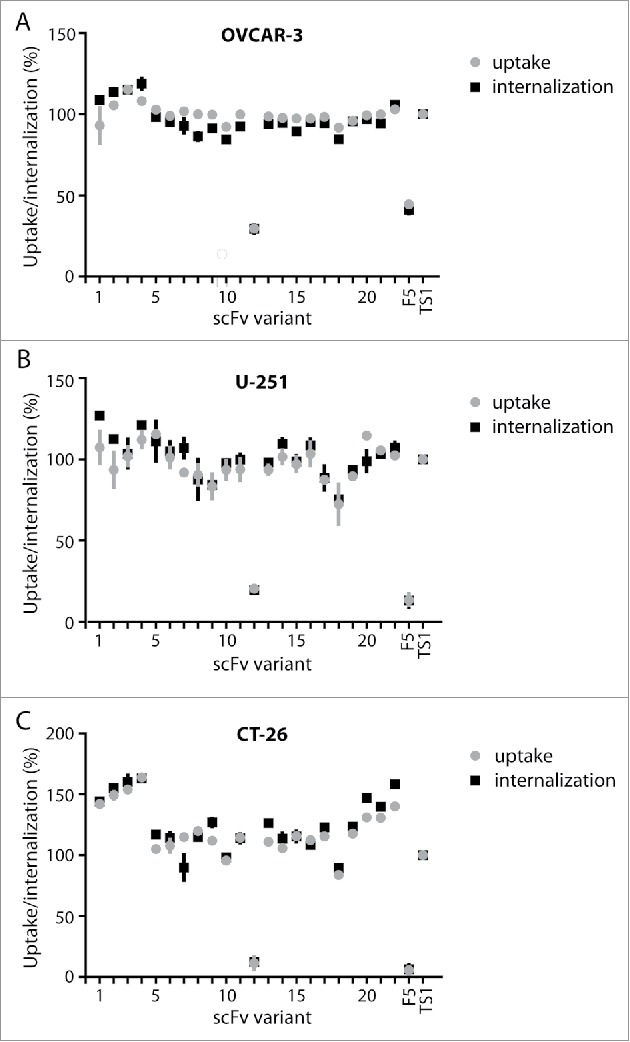
Uptake and internalization of liposomes by scFv variants on cell lines expressing EphA. ScFvs were evaluated for the ability to cause cellular uptake of liposomes in 3 cell lines using the CLIA high-throughput assay. Cell lines expressing high levels of human EphA2 (OVCAR-3, U-251) or murine EphA2 (CT-26) were used to evaluate scFvs. F5, which binds to ErbB2, was used as a negative control, and all data was normalized to the uptake and internalization of EphA2-TS1.
Analysis of variants for manufacturability
In order to assess the manufacturability of the different variants, we designed a screening strategy to identify molecules with high expression, high protein A binding by chromatography, and low aggregation (Table 4). The first criterion we evaluated was expression level of the scFvs at 1 L scale. Expression eventually needs to be sufficient to support an economically viable commercial process. Although expression levels at this point in development do not have to meet the final desired yields for manufacturing due to lack of cell line and cell culture optimization, ranking expression at this point in screening is helpful in removing poorly expressing scFvs or clones. Antibodies are typically manufactured in Chinese hamster ovary (CHO) cell lines; however, we have found that titers from transiently produced material are predictive of their behavior in CHO cells. As shown in Table 4, most of the variants had better expression titers (2–3-fold improvement over EphA2-TS1) with scFv-10 being the best. We next measured protein A binding capacity and purification yields for different variants using a chromatography technique that is routinely used for clinical molecules. Most variants showed 2–12-fold improvement in binding capacity, with again scFv-10 being the best (Table 4). In comparison to EphA2-TS1, the neutralization losses were higher for some variants, although the final purification yield was comparable or higher for all. Finally, for successful downstream process development, it is critical to have low aggregate content to reduce losses during subsequent processing steps. The post neutralization aggregate content was comparable and acceptable for all variants, with scFv-7 being the best. These new scFvs had much improved expression and protein A binding capacity, as well as decreased aggregation, and so were characterized for stability during the conjugation process.
Table 4.
Characterization of developability at 1L scale.
| scFv | Titers (mg/L) | Neutralization Loss (%) | Final Yield (mg) | Aggregates (%) | Fold Improvement in Protein A binding Capacity |
|---|---|---|---|---|---|
| scFv-2 | 12.4 | 21 | 9.8 | 7 | 5.5 |
| scFv-3 | 20.3 | 8 | 18.7 | 6 | 6.0 |
| scFv-4 | 13.1 | 7 | 12.2 | 7 | 5.8 |
| scFv-7 | 23.4 | 23 | 18.0 | 3 | 10.3 |
| scFv-8 | 19.3 | 15 | 16.4 | 6 | ND |
| scFv-9 | 19.3 | 15 | 16.4 | 4 | ND |
| scFv-10 | 27.7 | 17 | 23.0 | 11 | 12.3 |
| scFv-11 | 19.3 | 32 | 13.1 | 4 | 8.5 |
| scFv-13 | 21.0 | 8 | 19.3 | 4 | ND |
| EphA2-TS1 | 11.8 | 15 | 10.0 | 11 | 1.0 |
All titers were calculated using bio-layer interferometry (ForteBio). The final yield is from 1L of supernatant. The percentage of aggregates was measured using size-exclusion chromatography. The fold improvement in protein A binding capacity was measured as described in the Materials and Methods section.
Testing conjugation and insertion into liposomes
As the scFvs have been designed as part of a targeted nanoparticle, the selected scFvs were conjugated to liposomes using a micellar insertion method wherein scFvs containing a free C-terminal cysteine were first conjugated to a thiol-reactive lipopolymer, malemide-PEG-DPSE, in the form of micelles in an aqueous buffer.29,30 After purification, the conjugates were added to the liposomes and incubated above the transition temperature (Tm) of the liposome lipid bilayer to effect insertion of the conjugate into the liposome membrane. Because targeted drug delivery often employs liposomes with a high melting temperature, the scFv-PEG-DPSE conjugates were screened for their stability during thermal stress (65°C, 40 min) in a liposome aqueous medium. The EphA2-binding avidity of the conjugates before and after the thermal stress was measured as an EphA2 association rate using bio-layer interferometry, and the percentage of the activity remaining after thermal stress was compared with that of the conjugate made with the parental scFv (Table 5). At the same time, conjugates composed of the scFv with methoxy-PEG(2000)-distearoylphosphatidylethanolamine (PEG-DSPE) were inserted into the liposomes at a given protein/phospholipid ratio (10–12 g protein per mole of liposome phospholipid, or 30–36 scFv copies/particle) at 60°C for 30 min, purified from non-inserted conjugate, and their binding avidity to EphA2 was studied by bio-layer interferometry in the same manner (Table 5). As expected, the EphA2 binding of the liposomes prepared with variants was similar to the liposomes prepared using the parental scFv, confirming that the mutations made in these scFvs did not affect binding of the immunoliposomes to the antigen. The resistance of the PEG-DSPE-conjugated scFvs to thermal stress, however, was different across the variants. Unexpectedly, some of the scFvs variants (2, 3, and 10) had significantly improved thermal stability over EphA2-TS1.
Table 5.
Analysis of scFvs for EphA2 binding avidity after conjugation to PEG-DSPE linker and insertion into a liposome, and compared with binding after thermal stress.
| scFv |
Binding of liposome (% on-rate) |
Binding after thermal stress (%) |
| scFv-2 | 116.6 | 26.9 |
| scFv-3 | 119.1 | 46.9 |
| scFv-4 | 117.5 | 5.6 |
| scFv-7 | 119.2 | 4.8 |
| scFv-8 | 133.3 | 7.3 |
| scFv-9 | 118.1 | 12.2 |
| scFv-10 | 109.6 | 98.8 |
| scFv-11 | 120.5 | −0.09 |
| scFv-13 | 120.2 | 9.0 |
| scFv-16 | 113.4 | 4.6 |
| scFv-19 | 129.3 | 5.1 |
| EphA2-TS1 | 100 | 13.0 |
These assays allowed us to identify 3 candidate molecules for entry into the clinical translation phase. After thorough analysis of the stability of the molecules to be conjugated to a liposome, as well as evaluation of the manufacturing properties of the scFvs at a 1 L scale, we selected 2 scFvs for advancement as a possible nanoparticle targeting arm: scFv-3 and scFv-10. Both scFvs underwent more extensive testing in process development, as well as more extensive in vitro and in vivo biological and pharmacokinetic / pharmacodynamic assays, leading to the selection of scFv-3 as the best-suited module for a targeted therapeutic nanoparticle.
Discussion
Antibody engineering typically has 3 primary areas of focus: improving affinity, increasing stability, and reducing immunogenicity. Antibodies are initially selected for specific binding to the target of interest. If further affinity improvements are needed, well-established techniques that include mutagenesis of the CDRs or light chain shuffling can be employed. To date, improving the stability of an antibody has largely remained a trial and error effort, requiring iterative rounds of engineering. This can either lead to delays in lead selection, or result in the selection of manufacturable antibodies with inferior bioactivity. This is especially true for antibody fragments, which are typically less stable than traditional IgGs.
Antibody modules are commonly used in the design of ADNs which offer substantial promise in improving the delivery and therapeutic index of both small molecule and nucleic acid-based therapeutics.2,31-33 A high-Tm, small unilamellar liposome drug carrier platform has been well established for this purpose due to good drug retention properties, low clearance from the circulation and certain selectivity to tumor tissues due to differential vascular permeability (EPR effect).34 Liposome nanotherapeutics also afford micellar post-insertion, a versatile and robust “click” method, to be used for attachment of targeting ligands (including proteins) to the particles6). The antibody-targeting ligands are convenient and useful for modulating the internalization and overall microdistribution of the nanoparticle at the site of disease.2,11 Typically, construction of the final ADN on the high-Tm liposome drug carrier platform using a membrane post-insertion method requires a high temperature post-insertion or membrane capture step (60–65°C) to incorporate the targeting ligand efficiently,29,30,35 providing an opportunity for denaturation and inactivation of the antibody during the process. Identifying antibody fragments with the requisite stability thus becomes imperative. Maintaining the ability to not only bind to target cells, but also induce internalization in order to enable intracellular processing and drug release, is also essential.10,11,36
Achieving all these characteristics within the confines of the scFv format remains a challenge. Here, we addressed this challenge by describing the rapid engineering of a therapeutic antibody fragment that we employed in the context of developing an EphA2-targeted nanoparticle. We started with an antibody with poor manufacturability, and proceeded to generate scFvs that retained the desired biological activity while demonstrating improved thermal stability and binding to protein A without negatively impacting project timelines. Stability can be difficult to engineer when characterizing dozens of scFvs, given that most standard assays, including differential scanning calorimetry or column purifications, use significant amounts of material. Therefore, high-throughput assays were used as a surrogate to predict protein A binding and thermostability. Moreover, because the ultimate function of the targeting scFv was to affect target-specific uptake of the drug nanocarrier by the target cell, a high-throughput functional assay was also included. scFvs with improved characteristics were identified using 3 high-throughput assays: protein A, differential scanning fluorimetry (DSF), and CLIA. Using these assays, we were able to screen dozens of scFvs using a minimal amount of material, typically less than 300 µg/scFv. Further characterization of selected scFvs at larger scale confirmed their improved thermostability and protein A binding, validating the assays that were used to identify them. Relying on these methods also greatly reduced the time required to identify lead-optimized clones; new scFvs were designed and characterized within 3 months of initial lead identification.
It is always desirable to have antibodies with robust bioactivity and stability, and ideally, identification of a lead molecule would include selections for both. Recent work has shown that the somatic hypermutation process in vivo selects for mutations that improve both affinity and stability,37 underscoring the importance of stability in antibodies. Many current selections focus on bioactivity, and then antibodies with poor stability or manufacturability are discarded. We have described a method of rescuing antibodies with poor manufacturability, using very targeted designs and screens. The assays described here should be generally applicable to other antibodies that are currently poorly manufacturable, allowing for a greater diversity of antibodies to be developed for clinical use.
Materials and methods
Design of thermostable scFvs
To design the thermostable variants of D2–1A7, a homology model was first made as described.38 Briefly, MolIDE was used to identify and build a zero-gapped template for the variable regions. The model was then energy minimized using SCWRL,39-41 DeepView,42 and Eris.43 The resulting model was visually inspected in PyMOL to identify any potential side chain clashes. The sequence of D2–1A7 was then compared with databases of VH and VL protein sequences from the NCBI Entrez protein database to evaluate frequency of each residue position, using consensus sequences that were defined by Kabat.44 The linker ASTGGGGSGGGGSGGGGSGGGGS and a VH-VL orientation was used in all scFvs tested.
High-throughput expression of soluble scFvs and quantitation of titer
Variants were subcloned into a pCEP4 vector (Thermo Fisher Scientific, Waltham, MA) that has a C-terminal hexahistidine tag. All clones were transiently expressed using the Expi293 system (Thermo Fisher Scientific). Cells were grown using Expi293 media in 5% CO2 to a density of 2.5 million cells/mL in a 24-well plate and then transfected with 1 µg of DNA/mL of cells. After six days, the soluble scFvs were harvested by centrifuging the cells at 4000 × g. To quantitate titer, supernatants were diluted 1:10 in phosphate-buffered saline (PBS) and loaded onto 96-well plates. Anti-Penta-HIS sensor tips (Pall ForteBio, Menlo Park, CA) were hydrated for 10 min in 1X PBS prior to the assay. Quantitation assays were run using the Octet® software and concentrations were calculated using a standard curve of purified F5 and D2–1A7. Data was analyzed and processed with Octet® Data Analysis software.
High-throughput protein A binding assay
Supernatants containing 250 µg of protein were loaded into a 96-well protein A HP Multitrap plate (GE Healthcare) previously washed with 1X PBS. The wells were then washed with 600 µL of 1X PBS. To elute the scFv, 200 µL of 0.1 M acetic acid was added to each well and incubated at room temperature for several minutes. The plate was centrifuged at 100x g for 2 minutes, and 20 µL of 1 M Tris, pH 8.0 was added to neutralize the scFvs. To determine the percentage of protein A binding, the concentration of scFv remaining in the flow-through was compare with the amount of protein (250 µg) loaded originally using bio-layer interferometry (ForteBio).
Differential scanning fluorimetry
The DSF assay was performed as described.38 Briefly, 10 μM of scFv and 1X Sypro Orange in 1X PBS was mixed to a final volume of 25 μl and heated from 20°C to 90°C at a rate of 1°C/min using the IQ5 real time detection system (Bio-Rad; Hercules, CA). The resulting fluorescence data were collected and transferred to GraphPad Prism for additional analysis. The melting temperature reported is the temperature at the maximum value of the first derivative.
Yeast display for EphA2 and protein A binding
Library growth, induction and selection were completed as previously described.38 Briefly, the selection was performed using FACS sorting on the BD Aria system. Induced yeast cells were incubated with 200 nM EphA2 with a hexahistidine (His6) tag (produced at Merrimack) and 10 μg/mL protein A antibody (Thermo Fisher Scientific, P21462). Cells were washed twice with FACS buffer (1x PBS, 0.5% BSA, pH 7.4) to remove unbound antigen. The antigen bound cells were then incubated with 2 μg/mL M2 anti-FLAG antibody (Sigma, F1804) labeled with Alexa647 (Life Technologies, A-30679) and 2 μg/mL anti-His6 antibody (R&D Systems, MAB050) labeled with Alexa 488 (Thermo Fisher Scientific, A-30676) for 30 minutes. After two washes, the cells were re-suspended in FACS buffer and the fluorescent signal was measured using a FACSCalibur (Becton Dickinson; Franklin Lakes, NJ). The median fluorescent intensities (MFI) were determined using FlowJo software. EphA2 binding MFI (anti-His6-Alexa 488) was normalized to expression MFI (anti-Flag-Alexa 647).
Transient expression at 1 L scale
Genes were synthesized (DNA 2.0; Newark, CA) and subcloned into a pCEP4 vector containing the sequence “GGSGGC” at the C-terminus for liposome conjugation. All single chains were expressed using the 293F transient transfection system (Thermo Fisher Scientific) using F17 media supplemented with 4 mM L-glutamine and 0.1% pluronic F-68. 1L of 293F cells were grown (37°C, 5% CO2) in F17 media to a density of 1.5–2.0 million/mL in a baffled shake flask. On the day of transfection, 1 mg of DNA and 2.5 mL of polyethylenimine solution (1 mg/mL) were premixed in 100 mL of cell culture media, briefly vortexed, incubated for 15 min, and added to cell culture. After a week, the cells were harvested by centrifugation (4000x g, 20 min), and the supernatant filtered using a 0.22 µm filter.
Protein A chromatography and binding capacity determination
The filtered supernatant was loaded onto a protein A column (MabSelect, GE Healthcare Life Sciences), equilibrated with PBS, and washed with high conductivity buffer (PBS with 500 mM NaCl) to reduce non-specific interactions. The bound protein was eluted with 20 mM sodium citrate (pH 3.2), held at low pH for 1 hour, neutralized with 1 M Tris to a pH of 6.0, and filtered with a 0.2 μM filter. The purification losses at each step were estimated from the change in volume and absorbance (A280) values. The binding capacity of each scFv to protein A resin was determined by passing the harvested supernatant through a MabSelect column at a flow-rate of 500 cm/h and 3 min residence time until 10% break-through was achieved. The column was then washed with 1X PBS and bound material eluted with 20 mM sodium citrate (pH 3.2). The eluted peaks were pooled and their absorbance (A280) measured to estimate binding capacity (μg of protein bound/mL of resin).
Size-exclusion chromatography
Sample (50 µg) was injected on a TSKgel SuperSW3000 column using 10 mM sodium phosphate, 300 mM NaCl, pH 7.0 as running buffer. All measurements were performed on Agilent 1100 HPLC equipped with an auto sampler, binary pump and diode array detector. Data was analyzed using Chemstation software.
Preparation of test liposomes and chelated ligand-induced internalization assay
DiI5-NTA liposomes were prepared from hydrogenated soy phosphatidylcholine (HSPC), cholesterol, PEG-DSPE, an NTA lipid Ni2+-DOD-tris-NTA, 45 and a fluorescent lipid dye, DilC18(5)-DS, at the molar ratio of 100:66.7:5:0.5:0.3, respectively, using a lipid film hydration-polycarbonate membrane (100 nm) extrusion method. This protocol produces unilamellar vesicles with the z-average size of 100–110 nm and polydispersity index less than 0.1. The extruded liposomes were purified by gel-chromatography on a Sephadex® G-75 (GE Healthcare) eluted with 5 mM HEPES, 144 mM NaCl, pH 6.5, and sterilized by passage through a 0.2-µm filter. The concentration of liposomal phospholipid was then determined after acid digestion using spectrophotometric phosphomolybdate method. Prior to incubation with cells the liposomes were diluted to 0.4 mM phospholipid in Hank's Balanced Salt Solution, and NiSO4 was added to 0.1 mM. The hexahistidine scFvs were diluted into cell culture medium (see below) to the concentration of 25 μg/mL of scFv and mixed with equal volume of the liposome solution (“fluorescent NTA-liposome/scFv mixture”). OVCAR-3, U-251, and CT-26 were grown in the adherent state in RPMI-640 cell culture medium supplemented with 10% fetal bovine serum, 1x penicillin/streptomycin, and L-glutamine to 90% confluence and harvested using 0.25% trypsin- ethylenediaminetetraacetic acid (EDTA). The cell suspension was dispensed at 100,000 cells per well into a 96-well “V” bottom shape polypropylene cell culture plate, washed with 1X PBS, and re-suspended in 100 μL of fluorescent NTA-liposome/scFv mixture. The plate was sealed and incubated for 4 h at 37°C in the atmosphere of 5% CO2 on a shaker while protected from light. The cells were pelleted by centrifugation, the supernatants were aspirated, and the cells were washed twice either in 200 μL/well of PBS to remove any unbound extracellular liposomes, or with 200 μL/well of PBS containing 0.25 M imidazole (pH 7.5) to remove unbound extracellular liposomes and surface-bound liposomes, but not internalized liposomes. The relative amounts of cell-associated liposomes were evaluated by FACS (Cy5 fluorescence channel).
Conjugation of scFvs to a lipopolymer linker
Purified scFvs were treated with 15 mM L-cysteine, 5 mM EDTA, adjusted to pH 6.0–6.2, and incubated for one hour at 30°C to reduce/activate the thiol group of C-terminal cysteine residue of each scFv. Excess cysteine was removed by gel-chromatography on a Sephadex column eluted with conjugation buffer (5 mM citrate, 1 mM EDTA, 140 mM NaCl, pH 6.0). Next, the scFvs were conjugated to the lipopolymer linker, maleimido-PEG(2000)-DSPE (mal-PEG-DSPE, Avanti Polar Lipids), as follows. Mal-PEG-DSPE was added to the treated scFvs to achieve a lipid/protein molar ratio of 4:1. The mixture was incubated with stirring for 2–4 h at room temperature and the reaction was stopped by quenching non-reacted maleimides with cysteine at a final concentration of 0.5 mM for 5 minutes. The quenched reaction mix was chromatographed on an Ultrogel AcA 34 gravity column and eluted with S10C-6.5 buffer (100 g/L low endotoxin sucrose, 10 mM citric acid USP, adjusted to pH 6.5 with NaOH. The first (void volume) protein peak, containing purified scFv-PEG-DSPE conjugate, was collected. The purity of scFv-PEG-DSPE conjugates was assessed by SDS-PAGE.
Preparation of test liposomes and insertion of scFv lipopolymer conjugates into liposome membrane
Egg sphingomyelin (NOF), cholesterol (Avant Polar Lipids), methoxy-PEG(2000)-distearoylglycerol (PEG-DSG, NOF), and DiIC18(5)-DS at molar ratio 100:66.7:8:0.3, respectively, were dissolved at 70°C in absolute ethanol and diluted with stirring at 70°C into 10 volumes of CS-250 buffer (250 mM aqueous NaCl, 5 mM citrate, pH 5.5) to a final concentration of 100 mM phospholipid. The lipid suspension was extruded at 70°C through 200 nm and 100 nm polycarbonate membrane filters and the extruded material was cooled to room temperature. The liposomes were purified by tangential flow diafiltration using a MiniKros hollow fiber cartridge (Spectrum Laboratories, MWCO 500 kD) via 10 volume exchanges of CS-250 buffer, and passed through a 0.2-µm sterilizing filter. This protocol typically produces unilamellar vesicles with the z-average size of 100–110 nm and polydispersity index less than 0.1 (“Dil5-SM liposomes”). Before membrane insertion of the scFv-PEG-DSPE conjugates, the liposomes were exchanged into dextrose-citrate buffer (17% aqueous dextrose, 20 mM citrate, pH 5.7) using size-exclusion chromatography on a Sephadex G-25 column (PD-10, GE Healthcare).
The scFv-PEG-DSPE conjugates were then mixed with Dil5-SM liposomes in dextrose-citrate buffer to achieve a protein/phospholipid ratio of 10–12 g/mol. The mixtures were quickly heated to 60°C and maintained at this temperature for 30 minutes with stirring. Then the mixtures were chilled on ice and the liposomes with membrane-inserted scFv-PEG-DSPE conjugates were separated from any residual non-inserted conjugate by gel-chromatography on a column of Sepharose CL-4B (GE Healthcare) eluted with citrate saline buffer. The scFv-linked liposomes were collected in the void volume of the column and analyzed for antigen-binding avidity using bio-layer interferometry and, for the cell uptake, using cytofluorimetry, each as described below. The presence of scFv on the liposomes was confirmed and quantified by SDS-PAGE (data not shown).
Bio-layer interferometry analysis of EphA2-targeted conjugates or liposomes
The liposomes were assayed for EphA2-binding avidity (association rate) using the ForteBio Octet Red 96 system (Pall ForteBio). Anti-His5 sensors were first coated with 10 µg/ml of his-tagged recombinant, human EphA2 (produced at Merrimack). For scFv-PEG-DSPE conjugates, the sensors were then incubated with 2.5 µg/ml of the conjugate in PBS, pH 7.4, and the slopes of association curves were determined between 3–13 seconds of incubation, corrected for the buffer-only background, and compared across the variants. ScFv-linked liposomes were incubated with the sensors in PBS at 25 µM of liposome phospholipid, the slopes of the association curves were determined between 3–20 seconds of incubation, corrected for the buffer-only background, and compared across the variants.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev 1999; 51:691-743; PMID:10581328 [PubMed] [Google Scholar]
- 2.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci 2008; 97:4696-740; PMID:18351638; http://dx.doi.org/ 10.1002/jps.21358 [DOI] [PubMed] [Google Scholar]
- 3.Gill PS, Wernz J, Scadden DT, Cohen P, Mukwaya GM, von Roenn JH, Jacobs M, Kempin S, Silverberg I, Gonzales G, et al.. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 1996; 14:2353-64; PMID:8708728; http://dx.doi.org/ 10.1200/jco.1996.14.8.2353 [DOI] [PubMed] [Google Scholar]
- 4.Wang-Gillam A, Li C, Bodoky G, Dean A, Shan Y, Jameson G, Macarulla T, Lee K, Cunningham D, Blanc JF, et al.. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet (London, England) 2016; 387:545-57; PMID:26615328; http://dx.doi.org/ 10.1016/S0140-6736(15)00986-1 [DOI] [PubMed] [Google Scholar]
- 5.Kraft JC, Freeling JP, Wang Z, Ho RJY. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci 2014; 103:29-52; PMID:24338748; http://dx.doi.org/ 10.1002/jps.23773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirpotin DB, Noble CO, Hayes ME, Huang Z, Kornaga T, Zhou Y, Nielsen UB, Marks JD, Drummond DC. Building and characterizing antibody-targeted lipidic nanotherapeutics. Methods Enzymol 2012; 502:139-66; PMID:22208985; http://dx.doi.org/ 10.1016/B978-0-12-416039-2.00007-0 [DOI] [PubMed] [Google Scholar]
- 7.Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG, Noble CO, Lücker PB, Zandstra PW, Drummond DC, et al.. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol Appl Pharmacol 2012; 262:1-10; PMID:22676972; http://dx.doi.org/ 10.1016/j.taap.2012.04.008 [DOI] [PubMed] [Google Scholar]
- 8.Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res 2005; 65:11631-8; PMID:16357174; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1093 [DOI] [PubMed] [Google Scholar]
- 9.Cheng WW, Allen TM. The use of single chain Fv as targeting agents for immunoliposomes: an update on immunoliposomal drugs for cancer treatment. Expert Opin Drug Deliv 2010; 7:461-78; PMID:20331354; http://dx.doi.org/ 10.1517/17425240903579963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC. Development of ligand-targeted liposomes for cancer therapy. Expert Opin Ther Targets 2004; 8:335-53; PMID:15268628; http://dx.doi.org/ 10.1517/14728222.8.4.335 [DOI] [PubMed] [Google Scholar]
- 11.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 2006; 66:6732-40; PMID:16818648; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4199 [DOI] [PubMed] [Google Scholar]
- 12.Koning GA, Morselt HW, Gorter A, Allen TM, Zalipsky S, Scherphof GL, Kamps JA. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res 2003; 20:1249-57; PMID:129480239271260 [DOI] [PubMed] [Google Scholar]
- 13.Harding JA, Engbers CM, Newman MS, Goldstein NI, Zalipsky S. Immunogenicity and pharmacokinetic attributes of poly(ethylene glycol)-grafted immunoliposomes. Biochim Biophys Acta 1997; 1327:181-92; PMID:9271260; http://dx.doi.org/ 10.1016/S0005-2736(97)00056-4 [DOI] [PubMed] [Google Scholar]
- 14.Cao Y, Marks JD, Huang Q, Rudnick SI, Xiong C, Hittelman WN, Wen X, Marks JW, Cheung LH, Boland K, et al.. Single-chain antibody-based immunotoxins targeting Her2/neu: design optimization and impact of affinity on antitumor efficacy and off-target toxicity. Mol Cancer Ther 2012; 11:143-53; PMID:22090420; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0519 [DOI] [PubMed] [Google Scholar]
- 15.Rudnick SI, Lou J, Shaller CC, Tang Y, Klein-Szanto AJ, Weiner LM, Marks JD, Adams GP. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res 2011; 71:2250-9; PMID:21406401; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghose S, McNerney T, Hubbard B. Protein A Affinity Chromatography for Capture and Purification of Monoclonal Antibodies and Fc-Fusion proteins: Practical Considerations for Process Development In: Shukla A, Etzel M, Gadam S, editors. Process Scale Bioseparations for the Biopharmaceutical Industry. 2006. page 464-84. [Google Scholar]
- 17.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets 2011; 15:31-51; PMID:21142802; http://dx.doi.org/ 10.1517/14728222.2011.538682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Zou H, Zhang S, Marks JD. Internalizing cancer antibodies from phage libraries selected on tumor cells and yeast-displayed tumor antigens. J Mol Biol 2010; 404:88-99; PMID:20851130; http://dx.doi.org/ 10.1016/j.jmb.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ewert S, Huber T, Honegger A, Plückthun A. Biophysical Properties of Human Antibody Variable Domains. J Mol Biol 2003; 325:531-53; PMID:12498801; http://dx.doi.org/ 10.1016/S0022-2836(02)01237-8 [DOI] [PubMed] [Google Scholar]
- 20.Potter KN, Li Y, Pascual V, Capra JD. Staphylococcal protein A binding to VH3 encoded immunoglobulins. Int Rev Immunol 1997; 14:291-308; PMID:9186782; http://dx.doi.org/ 10.3109/08830189709116521 [DOI] [PubMed] [Google Scholar]
- 21.Graille M, Stura E a, Corper a L, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A 2000; 97:5399-404; PMID:10805799; http://dx.doi.org/ 10.1073/pnas.97.10.5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roben PW, Salem AN, Silverman GJ. VH3 family antibodies bind domain D of staphylococcal protein A. J Immunol 1995; 154:6437-45; PMID:7759880 [PubMed] [Google Scholar]
- 23.Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y, et al.. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs 2013; 5:445-70; PMID:23571156; http://dx.doi.org/ 10.4161/mabs.24218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langedijk AC, Honegger A, Maat J, Planta RJ, van Schaik RC, Plückthun A. The nature of antibody heavy chain residue H6 strongly influences the stability of a VH domain lacking the disulfide bridge. J Mol Biol 1998; 283:95-110; PMID:9761676; http://dx.doi.org/ 10.1006/jmbi.1998.2064 [DOI] [PubMed] [Google Scholar]
- 25.Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IBJK, et al.. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity . MAbs 2011; 3:273-88; PMID:21393993; http://dx.doi.org/ 10.4161/mabs.3.3.15188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, Giesecke C, Dörner T, Andrews SF, et al.. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol 2013; 31:166-9; PMID:23334449; http://dx.doi.org/ 10.1038/nbt.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poul MA, Becerril B, Nielsen UB, Morisson P, Marks JD. Selection of tumor-specific internalizing human antibodies from phage libraries. J Mol Biol 2000; 301:1149-61; PMID:10966812; http://dx.doi.org/ 10.1006/jmbi.2000.4026 [DOI] [PubMed] [Google Scholar]
- 28.Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol 2006; 7:24; PMID:17014727; http://dx.doi.org/ 10.1186/1471-2172-7-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nellis DF, Ekstrom DL, Kirpotin DB, Zhu J, Andersson R, Broadt TL, Ouellette TF, Perkins SC, Roach JM, Drummond DC, et al.. Preclinical manufacture of an anti-HER2 scFv-PEG-DSPE, liposome-inserting conjugate. 1. Gram-scale production and purification. Biotechnol Prog 2005; 21:205-20; PMID:15903260; http://dx.doi.org/23036225 10.1021/bp049840y [DOI] [PubMed] [Google Scholar]
- 30.Nellis DF, Giardina SL, Janini GM, Shenoy SR, Marks JD, Tsai R, Drummond DC, Hong K, Park JW, Ouellette TF, et al.. Preclinical manufacture of anti-HER2 liposome-inserting, scFv-PEG-lipid conjugate. 2. Conjugate micelle identity, purity, stability, and potency analysis. Biotechnol Prog 21:221-32; PMID:15903261; http://dx.doi.org/23036225 10.1021/bp049839z [DOI] [PubMed] [Google Scholar]
- 31.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 2013; 65:36-48; PMID:23036225; http://dx.doi.org/ 10.1016/j.addr.2012.09.037 [DOI] [PubMed] [Google Scholar]
- 32.Hayes ME, Drummond DC, Kirpotin DB, Zheng WW, Noble CO, Park JW, Marks JD, Benz CC, Hong K. Genospheres: self-assembling nucleic acid-lipid nanoparticles suitable for targeted gene delivery. Gene Ther 2006; 13:646-51; PMID:16341056; http://dx.doi.org/ 10.1038/sj.gt.3302699 [DOI] [PubMed] [Google Scholar]
- 33.Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C, Carosio R, Loi M, Pagnan G, Emionite L, Cilli M, et al.. Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol Ther 2011; 19:2201-12; PMID:21829174; http://dx.doi.org/ 10.1038/mt.2011.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev 2015; 91:3-6; PMID:25579058; http://dx.doi.org/ 10.1016/j.addr.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 35.Iden DL, Allen TM. In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach. Biochim Biophys Acta 2001; 1513:207-16; PMID:11470092; http://dx.doi.org/ 10.1016/S0005-2736(01)00357-1 [DOI] [PubMed] [Google Scholar]
- 36.Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res 2003; 42:439-62; PMID:12814645; http://dx.doi.org/ 10.1016/S0163-7827(03)00032-8 [DOI] [PubMed] [Google Scholar]
- 37.Wang F, Sen S, Zhang Y, Ahmad I, Zhu X, a Wilson I, Smider V V, Magliery TJ, Schultz PG. Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Natl Acad Sci U S A 2013; 110:4261-6; PMID:23440204; http://dx.doi.org/ 10.1073/pnas.1301810110 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Xu L, Kohli N, Rennard R, Jiao Y, Razlog M, Zhang K, Baum J, Johnson B, Tang J, Schoeberl B, et al.. Rapid optimization and prototyping for therapeutic antibody-like molecules. MAbs 2013; 5:237-54; PMID:23392215; http://dx.doi.org/ 10.4161/mabs.23363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canutescu A, Dunbrack RL. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci 2003; 12:963-72; PMID:12717019; http://dx.doi.org/ 10.1110/ps.0242703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canutescu A, Dunbrack RL. MollDE: a homology modeling framework you can click with. Bioinformatics 2005; 21:2914-6; PMID:15845657; http://dx.doi.org/ 10.1093/bioinformatics/bti438 [DOI] [PubMed] [Google Scholar]
- 41.Canutescu A, Shelenkov A, Dunbrack RL. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci 2003; 12:2001-14; PMID:12930999; http://dx.doi.org/ 10.1110/ps.03154503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18:2714-23; PMID:9504803; http://dx.doi.org/ 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- 43.Yin S, Ding F, Dokholyan N V. Eris: an automated estimator of protein stability. Nat Methods 2007; 4:466-7; PMID:17538626; http://dx.doi.org/ 10.1038/nmeth0607-466 [DOI] [PubMed] [Google Scholar]
- 44.Wu TT, Kabat EA, Bilofsky H. Some sequence similarities among cloned mouse DNA segments that code for lambda and kappa light chains of immunoglobulins. Proc Natl Acad Sci U S A 1979; 76:4617-21; PMID:116235; http://dx.doi.org/ 10.1073/pnas.76.9.4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Z, Park JI, Watson DS, Hwang P, Szoka FC. Facile synthesis of multivalent nitrilotriacetic acid (NTA) and NTA conjugates for analytical and drug delivery applications. Bioconjug Chem 17:1592-600; PMID:17105240; http://dx.doi.org/ 10.1021/bc0602228 [DOI] [PubMed] [Google Scholar]
- 46.Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 1970; 132:211-50; PMID:5508247; http://dx.doi.org/ 10.1084/jem.132.2.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He F, Hogan S, Latypov RF, Narhi LO, Razinkov VI. High throughput thermostability screening of monoclonal antibody formulations. J Pharm Sci 2010; 99:1707-20; PMID:19780136; http://dx.doi.org/ 10.1002/jps.21955 [DOI] [PubMed] [Google Scholar]