

ABSTRACT
Excessive transforming growth factor (TGF)-β is associated with pro-fibrotic responses in lung disease, yet it also plays essential roles in tissue homeostasis and autoimmunity. Therefore, selective inhibition of excessive and aberrant integrin-mediated TGF-β activation via targeting the α-v family of integrins is being pursued as a therapeutic strategy for chronic lung diseases, to mitigate any potential safety concerns with global TGF-β inhibition. In this work, we reveal a novel mechanism of inhibiting TGF-β activation utilized by an αvβ8 targeting antibody, 37E1B5. This antibody blocks TGF-β activation while not inhibiting cell adhesion. We show that an N-linked complex-type Fab glycan in H-CDR2 of 37E1B5 is directly involved in the inhibition of latent TGF-β activation. Removal of the Fab N-glycosylation site by single amino acid substitution, or removal of N-linked glycans by enzymatic digestion, drastically reduced the antibody's ability to inhibit latency-associated peptide (LAP) and αvβ8 association, and TGF-β activation in an αvβ8-mediated TGF-β signaling reporter assay. Our results indicate a non-competitive, allosteric inhibition of 37E1B5 on αvβ8-mediated TGF-β activation. This unique, H-CDR2 glycan-mediated mechanism may account for the potent but tolerable TGF-b activation inhibition and lack of an effect on cellular adhesion by the antibody.
KEYWORDS: αvβ8, allosteric inhibition, fab glycosylation, integrin, ITGB8, mAb
Introduction
The transforming growth factor (TGF)-β family of cytokines play essential roles in mammalian development and normal tissue homeostasis.1-3 The effects of TGF-β signaling can be beneficial or deleterious, and they are cell-type and context specific.4 For example, in normal epithelial cells, TGF-β acts as a mediator of cell cycle arrest and differentiation, whereas in fibroblasts, TGF-β promotes differentiation of fibroblasts into pro-fibrotic myofibroblasts.4,5 Moreover, excess TGF-β activity is associated with disease conditions such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF).6 However, TGFΒ1 gene-deficient animals die as embryos,7 suggesting a key role for this pathway in development, and a global blockade of TGF-β is associated with impaired regulatory T cell (Treg) function, which may promote autoimmunity.8 Therefore, regulating aberrant and excessive TGF-β activity is an attractive therapeutic approach, which would aim to limit damaging TGF-β activity, yet maintain the normal homeostatic levels of this potent growth factor.
One approach to dampen excessive TGF-β signaling is by targeting specific TGF-β activation pathways. TGF-β is expressed as a pro-protein and processed by a furin-like convertase into a latency-associated peptide (LAP) and mature TGF-β1 protein.9,10 The LAP remains bound to the mature TGF-β1, preventing receptor binding.11 There are a number of TGF-β activation pathways that have been described, including pH dependent activation12 and exposure to reactive oxygen species.13 However, αv-integrin-mediated TGF-β-activation offers an attractive therapeutic approach because these integrin pathways have been shown to be elevated in chronic lung disease and are more tractable to therapeutic intervention.9,14-17 The αv subunit heterodimerizes with a number of β integrin subunits.18 The β6 and β8 subunits appear to be dominant with regards to TGF-β activation in vivo, as ITGB6 −/− and ITGB8 −/− recapitulate the TGFB1 knockout mouse phenotype.19 Targeting the αv subunit may not be an ideal therapeutic approach because this integrin has a number of physiological functions, including cell adhesion, mediated by αv:vitronectin interactions.20 However, targeting chronic aberrant TGF-β activation discretely via β8 blockade may be more therapeutically viable.
Both β6 integrin and β8 integrin subunits uniquely hetero-dimerize with αv, and both have been shown to be elevated in lung disease.21,22 In preclinical mouse models, therapeutic targeting of the integrin results in decreased TGF-β activation and subsequent efficacy.21,23 Latent TGF-β complex binds to either αvβ6 or αvβ8 via the arginine-glycine-aspartate (RGD)-peptide located within LAP, and αvβ8 shows binding specificity to latent TGF-β compared to other RGD-containing proteins.24,25 The mechanism of TGF-β activation by the two β subunits is hypothesized to be different. Alphav-β6-mediated TGF-β activation is via cytoskeletal reorganization,26 whereas αvβ8-mediated TGF-β-activation is postulated to occur via metallo-proteolytic cleavage of the latent TGF-β, which then frees TGF-β from the pre-protein complex upon binding.23 Additionally, αvβ8 has been shown to predominantly adopt a constitutively activated conformation upon interaction with latent TGF-β.23
Minagawa and colleagues have recently developed an αvβ8-selective targeting monoclonal antibody, 37E1B5, which inhibits IL-1β-induced airway remodeling via inhibiting TGF-β activation, but does not significantly inhibit αvβ8-mediated cell adhesion.23 Mutagenesis analyses indicated that the binding epitope of 37E1B5 in β8 is located distant from the RGD binding pocket, and the conclusion is supported by the electron microscopy of an αvβ8-37E1B5 Fab complex, which revealed that 37E1B5 Fab bound at the β8 subunit head-hybrid junction orientated away from the ligand binding pocket.23 Furthermore, electron microscopy imaging shows that 37E1B5 binding induced β8 head-hybrid domain angle changes, supporting an allosteric inhibition mechanism by the antibody.23
The 37E1B5 antibody contains N-linked glycosylation sites in the heavy chain variable region sequences. In this study, we have shown that the H-CDR2 glycan of the antibody is critical for TGF-β inactivation, but not for αvβ- binding. Interestingly, removing the glycosylation site by a single amino acid substitution, or removal of the N-glycans by enzymatic digestion drastically reduces the ability of 37E1B5 to inhibit TGF-β activity, while αvβ8 binding ability was conserved. We further show that 37E1B5 inhibitory activities are directly associated with the H-CDR2 glycoforms as we demonstrated the relationships between the glycan structure and the TGF-β inhibitory activity. Coinciding with structural studies by Minagawa et al., the novel glycan-mediated mechanism elucidated from this work suggests that 37E1B5 may have improved safety profiles compared to those that inhibit both integrin and TGF-β functions or others that broadly affect TGF-β activation.
Results
37E1B5 H-CDR2 is occupied with a complex glycan
The murine antibody 37E1B5 encodes two variable region N-linked glycosylation sites, NLS and NYT, one located at positions 19-21 in the heavy chain framework 1 (FW-1), and the other, a germline gene encoded site, at the H-CDR2 positions 59-61 (Fig. 1). Humanization removed the NLS glycosylation site in the FW-1 region, generating antibody “Beta8,” which did not adversely affect binding or biological activity of the antibody (data not shown). To assess the H-CDR2 glycosylation site, we first determined the relative occupancy of Fc and H-CDR2 N-glycosylation sites in the Chinese hamster ovary (CHO) cell-expressed Beta8 by LC-MS peptide mapping. The results showed that the Fc N-glycosylation site at amino acid 297 was fully occupied as expected. Furthermore, high levels of site occupancy (>85 %) were observed at H-CDR2 site N59 (data not shown), suggesting that amino acid N59 in H-CDR2 is adequately exposed for glycosylation to occur. We then determined the complexity of the H-CDR2 glycan by profiling the released, labeled oligosaccharides of CHO-expressed Beta8. This method provides a profile of the combined N-glycosidase F released glycan pool (Fc and Fab glycans), whereby the released glycans are fluorescently labeled with 2-aminobenzamide (2AB) and separated by hydrophilic interaction chromatography (HILIC). The results showed that a range of Beta8 glycoforms, predominantly of the complex-type, were found at these N-linked glycosylation sites (Fig. 2A). Subsequent site-specific glycan analysis by reduced antibody LC-MS, confirmed the predominant Fc glycoform to be a biantennary complex-type neutral glycan, FA2, which is typical for N297 in CHO-expressed IgG. However, the H-CDR2 site contained a range of glycoforms, distinct from the Fc, with predominantly the biantennary, charged (sialylated) glycoform FA2G2S2 present (Fig. 2B). We further created a H-CDR2 aglycosylated variant, Beta8-AG, by asparagine (N) to glutamine (Q) substitution at position 59. Reduced antibody LC-MS analysis of the aglycosylated variant Beta8 N59Q showed no H-CDR2 glycan, but the glycosylations at the canonical N297 site in the Fc region (Fig. 2C).
Figure 1.

The amino acid sequences of the heavy chain variable region of murine 37E1B5. The CDR regions are shown in italic and underlined. The N-linked glycosylation signals, NLS and NYT at the positions 19-21 and 59-61, are high-lighted as yellow, respectively.
Figure 2.

Characterization of Beta8 glycosylation. Relative quantitation of released glycans (Fc and Fab) by 2-aminobenzamide (2-AB)-labeled oligosaccharide profiling (A). Site-specific glycan analysis by reduced antibody LC-MS of heavy chain combined, de-convoluted mass spectra for Beta8 (B) and the aglycosylated Beta8-AG (N59Q) variant (C).
The H-CDR2 glycan is critical for 37E1B5 activity but not for αvβ8 binding
We initially assessed the activities of Beta8 and the H-CDR2 aglycosylated variant, Beta8-AG, in enzyme-linked immunosorbent assays (ELISAs). In the αvβ8 binding ELISA, Beta8-AG shows nearly identical binding activity to αvβ8 compared to its H-CDR2 glycosylated parental antibody Beta8 (Fig. 3A), indicating that the Fab glycan is not involved in antigen binding. However, when assessed in a LAP-αvβ8 binding inhibition assay where Beta8 antibodies were co-incubated with recombinant αvβ8 before being added to the plate-immobilized LAP, Beta8-AG shows greatly reduced inhibitory activity, in comparison to H-CDR2 glycosylated Beta8 (Fig. 3B). This indicates that the H-CDR2 glycan plays a critical role in Beta8 biological activity.
Figure 3.
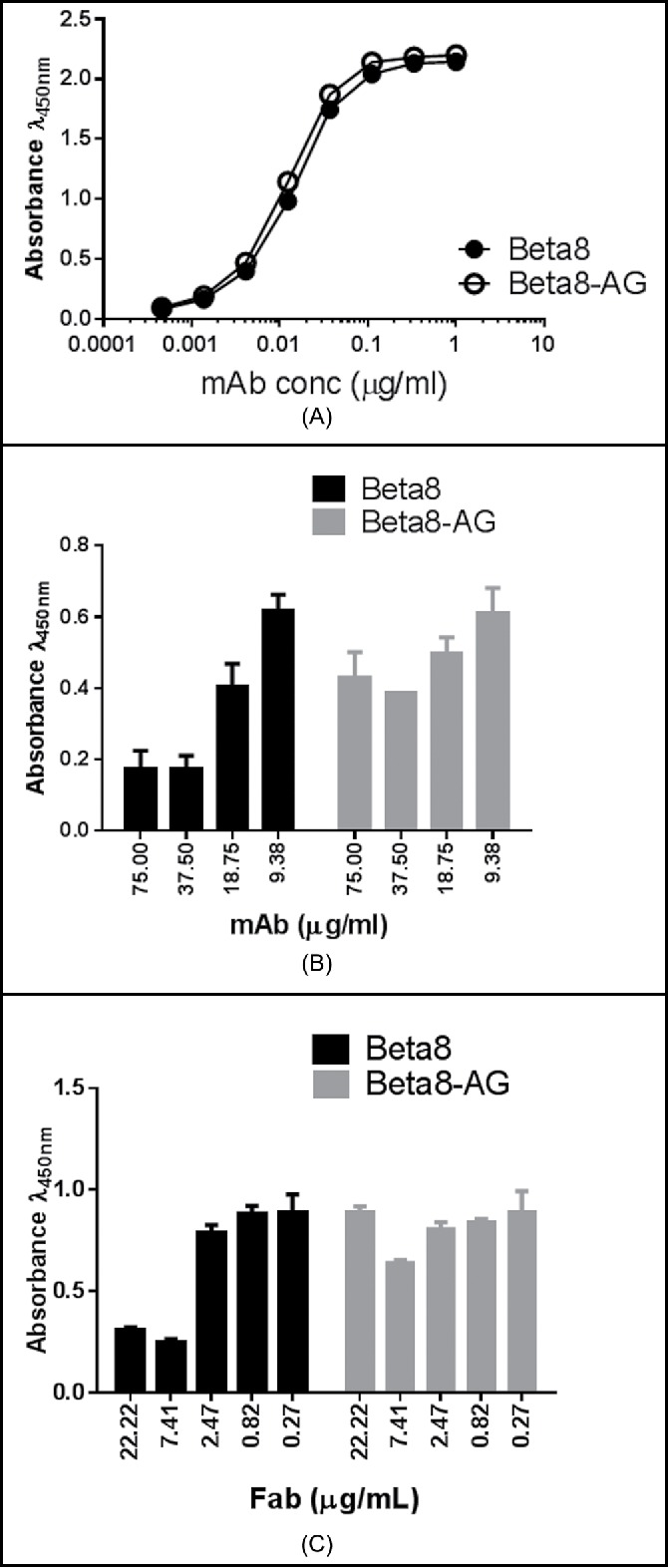
Binding of Beta8 and Beta8-AG to αvβ8 (A). Recombinant αvβ8 was coated to the ELISA wells. Antibody binding was detected using a HRP-conjugated goat anti-human Fc antibody. Inhibition of αvβ8 binding to LAP ((B) and C). Recombinant LAP was coated to the ELISA wells. The amount of the recombinant αvβ8 protein that generated 75% maximum binding signals was mixed with serially diluted Beta8 and Beta8-AG (B) or Fabs of Beta8 and Beta8-AG (C), and the reaction was incubated at room temperature. The ability of αvβ8 binding was detected by an HRP-conjugated anti-αvβ8 antibody.
To determine if antibody valency plays a role in the LAP binding inhibition of Beta8, we tested Fab formats of Beta8 and Beta8-AG. There was a modest reduction in binding of the Fab format of Beta8 to αvβ8 (Fig. S1), showing multivalent IgG improves binding. In the LAP-αvβ8 binding inhibition assay, however, Beta8 Fab still inhibited αvβ8 binding to LAP effectively, in comparison to Beta8 IgG (Fig. 3C). Moreover, as with the IgG, Beta8-AG Fab was inactive in the LAP binding assay (Fig. 3C). Taken together, these results indicate that antibody valency, although augmenting binding, has little or no effect on antibody inhibition of LAP-αvβ8 interaction. In contrast, the H-CDR2 glycan is crucial for Beta8 biological activity, but not for αvβ8 binding.
A complex-type Fab glycan in H-CDR2 is essential for the full bio-activity of Beta8
To further characterize the role of H-CDR2 glycosylation, we generated a number of additional distinct antibody glycoforms by enzymatic digestion of CHO-cell expressed Beta8, or by expressing Beta8 in different CHO host cell lines. A schematic overview of the approaches we used to generate the different antibody glycol forms for the characterizations is shown in Fig. 4A. N-glycosidase F digestion was used to produce deglycosylated Beta8 antibody where both H-CDR2 and Fc N-linked glycans were removed. Neuraminidase digestion was used to produce desialylated Beta8 glycoforms lacking terminal N-acetylneuraminic acid (NeuAc), instead terminating in galactose, also with low levels of glycoforms terminating in N-acetylglucosamine (GlcNAc). Expression of Beta8 in a Lec8 CHO mutant cell line, with defects in UDP-galactose transporter (UGT) activity, was used to produce antibody glycoforms lacking terminal galactose, instead terminating in GlcNAc with bi-, tri- and tetra-antennary forms produced. Expression of murine-human chimeric 37E1B5 in a CHOK1-derived MGAT1 (mannosyl (α-1,3-)-glycoprotein β-1,2-N-acetylglucosaminyltransferase) “knock out” cell line, was used to produce oligomannose glycoforms, specifically Man-5.22,27 The profile of the glycan variants are confirmed by reduced antibody LC-MS analysis (Fig. S2A-2D).
Figure 4.

Inhibition of TGF-β activation by 37E1B5 requires a complex glycan. Schematic overview of the different antibody glycoforms assessed for bioactivity and PK properties, illustrated for the binantennary glycan FA2G2S2 (A). TMLC-HeLa cell co-culture TGF-β signaling reporter assay. Relative luciferase units (RLU) from TMLC reporter cells show baseline luciferase reporter activity (“TMLC”). Relative luciferase units (RLU) from TMLC and HeLa αvβ8+ cell co-culture show maximal luciferase reporter activity (“TMLC + HeLa); and the anti-TGF-β mAb 1D11 (“anti-TGFb-1D11,” 7.5 µg/mL) effect on the TGF-β dependent luciferase reporter activity in the TMLC and HeLa αvβ8+ co-culture. Mouse IgG1 (“mIgG1,” 7.5 µg/mL) was used as the isotype control. (B). Maximum response of TGF-β dependent luciferase reporter activity by 37E1B5 variants in the TMLC co-culture assay with HeLa αvβ8+ cells. (C). Data shown is the mean ±SEM from 3 independent experiments.
To assess the functional role of the H-CDR2 glycan of Beta8, we used a TGF-β-dependent reporter assay to examine TGF-β activation. In this assay (Fig. 4B, C), αvβ8+ HeLa cells are co-cultured with TMLC (mink lung epithelial cells) that express luciferase under the control of a PAI-1 promoter, thus sensitively detecting TGF-β signaling.28 Prior to co-culture, HeLa cells were enriched for αvβ8 expression using flow cytometric sorting (“HeLa-B8”). TMLCs cultured alone and in co-culture with HeLa-B8 cells represent 0% and 100% maximal response, respectively (Fig. 4B). A TGF-β neutralizing antibody, 1D11, inhibited HeLa-B8 cell-mediated TGF-β induced reporter activity to that of background levels in the assay (Fig. 4B). The reporter responses were maximally inhibited by CHO-G22 cell expressed Beta8, which contains predominantly complex-type biantennary, sialylated H-CDR2 glycoforms, with reporter activity being reduced to 39 ± 9% reporter activity (Fig. 4C). Beta8 H-CDR2 glycoforms lacking sialic acid, terminating instead in GlcNAc (Lec8 expression) or galactose (desialylated) demonstrated a reduced ability to inhibit TGF-β activation compared to Beta8, and showed 52 ± 5% and 48 ± 4% reporter activities, respectively (Fig. 4C). The Beta8 oligomannose (CHOK1-derived MGAT1 cell expression) and deglycosylated Beta8 (N-glycosidase F digestion) glycoforms show further reduced reporter activities of 69 ±1 % and 80 ±4 %, respectively (Fig. 4C). These data clearly demonstrate the critical role of the H-CDR2 glycan in inhibiting TGF-β activation and that complex-type, sialylated H-CDR2 glycoforms are required for maximal biological activity of Beta8.
Role of glycan in the pharmacokinetics properties of Beta8 in mice
Antibody glycosylation has been hypothesized to affect antibody pharmacokinetics (PK).29 To examine whether desialylation (DE) and H-CDR2 aglycosylation by N59Q mutation (AG) had any effect on the PK properties of Beta8 (WT), we determined the PK parameters of the antibodies using humanized αvβ8-BAC mice.17 The mice were dosed with 1 and 10 mg/kg of each antibody and the level of plasma antibody concentrations were quantified using a human Fc capture ELISA. A non-linear, dose-dependent clearance was observed and accounted for the target-mediated elimination at the 1 mg/kg dose and assumed both target-mediated and FcRn-mediated clearances at the 10 mg/kg dose of the antibodies. A PK model was iteratively developed using the observed serum concentrations and including inter-compartmental distribution, non-specific clearance (CL) and target-mediated elimination. The nonlinear mixed effects software NONMEM (Icon Development Solutions, Version 7.3) was used to derive the PK parameters. Based on a comparison of the 5 simulated models, the WT ≠ DE = AG clearance model was the best fit to the data, with a ΔOFV of 4.906 (p < 0.05, 1 DF) to the base model. Although the WT ≠ DE ≠ AG clearance model had a ΔOFV of 7.971 (p < 0.05, 2 DF) to the base model, its additional degree of freedom does not result in a significant improvement with respect to the WT ≠ DE = AG model. Based on the WT ≠ DE = AG clearance model, WT Beta8 was cleared at a slower rate than the DE and AG variants at a rate of 0.353 mL/hr/kg, compared to 0.486 mL/hr/kg, respectively. The PK clearance derived from models WT ≠ DE = AG and WT ≠ DE ≠ AG, and the software NONMEM is shown in Table 1. A more complete PK clearance analysis of all 5 models is shown in Supplementary Table 1.
Table 1.
Model-based pharmacokinetic parameter estimates for comparison of the wild type (WT), fully glycosylated (AG) and the desialylated (DE) Beta8 in mice. PK parameters derived from the non-linear mixed effects software NONMEM (Icon Development Solutions, Version 7.3).
| Parameter | CL model WT ≠ DE = AG | CL model WT ≠ DE ≠ AG |
|---|---|---|
| Number of estimated parameters | 11 | 12 |
| Δ Objective with base model | 4.906 | 7.971 |
| CL-WT (mL/hr/kg) | 0.353 | 0.392 |
| CL-AG (mL/hr/kg) | 0.486 | 0.617 |
| CL – DE (mL/hr/kg) | 0.486 | 0.508 |
Discussion
The integrin β8-specific antibody 37E1B5 has been shown to inhibit TGF-β activity via an allosteric, non-competitive mechanism.23 In our study, we report the discovery and characterization of an H-CDR2 encoded, N-linked glycosylation site in 37E1B5 that plays an essential role in conferring antibody activity. Removal of the Fab N-linked glycans by mutation or enzymatic digestion abolished the ability of the antibody to inhibit both LAP binding to αvβ8 and TGF-β activation. On the other hand, the H-CDR2 encoded, N-linked glycan is not required for binding of the antibody to αvβ8. Fab glycan characterization, by enzymatic digestion or by expression in carbohydrate-engineered host cells, showed that maximal TGF-β inhibition is achieved with the variant that carries a mature, complex-type, sialylated glycan, whereas truncated (e.g., Lec8 derived), or immature (e.g., oligomannose) glycoforms showed the least activity in the TGFβ reporter assay. Removal of the H-CDR2 N-linked glycosylation by N59Q substitution or N-glycosidase F digestion abolished Beta8 TGF-β inhibitory activity.
Minagawa et al., reported that the binding epitope of 37E1B5 is located at the βI domain of β8, which is ∼28 Å distal to the LAP/RGD ligand binding site, based on modeling of the β3 integrin subunit, a structure homologous with β8.23 The spatial distance suggests that a direct interaction between the LAP/RGD binding pocket in β8 and the antibody backbone is unlikely. Electron microscopy of 37E1B5 Fab and β8 complex indicated that antibody binding appears to induce a conformational change in β8, whereby 37E1B5 induces a bend in the βI domain, holding the integrin in a bent formation.23 Our results indicated that, in addition to the binding-induced conformation change, the H-CDR2 glycan of the antibody sterically hinders and prevents LAP and αvβ8 interaction. The apparent correlations between the ability to inhibit LAP:αvβ8 interaction and the Fab glycan complex-type further support the steric hindrance effect, although we could not rule out the possibility that the same steric hindrance could also prevent the recruitment of a metalloproteinase to the integrin:LAP complex, thereby preventing the enzymatic liberation of active TGF-β. Additionally, conferring LAP-αvβ8 binding inhibition and TGFβ activation inhibitory activities by introducing an N-linked glycosylation site to H-CDR2 of a 37E1B5 binding competing, non-neutralizing anti-αvβ8 antibody 2A10 strongly support the critical role of the Fab-glycan in the functionality of the antibody.30 As the electron-microscopy of the glycan-engineered 2A10 and β8 complex revealed a similar structural change similar to 37E1B5 and β8 complex,30 these results strongly argue that the H-CDR2 glycan is also responsible for the induced conformational change of β8 upon binding.
In the TGF-β reporter assay, we consistently observed incomplete inhibition of TGF-β activation by Beta8. This could be due to the unique mechanism by which Beta8 binds to the integrin or due to other TGF-β activating pathways, such as other αv integrins being present on the HeLa cells or reporter cells or soluble plasma proteins in the assay. We tried to establish a primary cell assay using human lung fibroblasts; however, unlike previously published reports,22 we did not detect any αvβ8 expressed on multiple cell lines. Instead, we predominantly detected αvβ1, which was primarily responsible for the activation of TGF-β in this co-culture system (data not shown). We did find a subpopulation of HeLa cells with an enhanced level of αvβ8 expression, which we were able to selectively use for our studies to establish a suitable αvβ8-dependent functional assay system. Even though we did not observe complete TGFβ inhibition by Beta8, we were able to use HeLa cells as an endogenous source of biologically active αvβ8 and generate robust data that could discriminate subtle glycan effects. Future studies using other blocking approaches to these pathways may elucidate the pathways responsible for full TGF-β activation in this cell system.
The observed differences in clearance between Beta8 and desialylated Beta8 (Beta8-DE) is consistent with the previously described mechanism of carbohydrate-binding receptor mediated clearance through the asialo-glycoprotein receptor.31 However, the observed faster clearance of Fab aglycosylated Beta8-AG compared to Beta8 is unexpected, and it may be accounted for by other factors affecting the PK of the antibody, such as Beta8-AG being more susceptible to proteolysis or chemical degradation in vivo by removing the bulky Fab glycan.32
The phenomenon of variable region glycosylation affecting antibody function is rare, yet a few have been reported. For example, Jacquemin et al. has shown that a Factor VIII (FVIII) inhibitory antibody derived from patients treated with recombinant FVIII uses a variable region glycan to modulate the anti-FVIII antibody's inhibitory activity.33 By modeling the FVIII: FVIII antibody complex, the authors suggested that the Fab glycan is on an exposed loop of CDR1 away from the antibody binding site, and that only the outer arms of the glycan (not the core) could make contact with the antigen.33 Beta8 appears to use a similar mechanism to inhibit TGF-β activation. Inhibition of αvβ8-mediated TGF-β activation through a glycan-mediated allosteric mechanism can be effective, as the inhibition will occur without needing to saturate target binding. Additionally, sparing the important cellular functions of the integrin by using a non-competitive, allosteric mechanism specific to αvβ8, and not broadly to all αv integrins, could result in Beta8 being an attractive candidate for therapeutic development.
Materials and methods
Reagents
37E1B5 antibody was provided by Dr. Stephen Nishimura of University of California at San Francisco. All enzymes were purchased from New England Biolabs (Ipswich, MA) and Clontech (Mountain View, CA). Recombinant human integrin αvβ8 (Cat# 4135-AV) and TGF-β1 latency associated peptide (Cat# 246-LP/CF) were purchased from R&D Systems (Minneapolis, MN). The goat anti-mouse IgG Fc-HRP (Cat# 115-035-071, Lot# 92577) and the mouse anti-human IgG-Fc-HRP (Cat# 209-035-098, Lot# 115868) were purchased from Jackson ImmunoResearch (West Grove, PA). Biotinylated anti-human CD51 (αv) (Cat# 327906) and the HRP conjugated streptavidin (Cat# 405210) were purchased from BioLegend (San Diego, CA). TMB peroxidase substrate was purchased from KPL (Gaithersburg, MD). Blocker™Casein was purchased from Thermal Scientific (Walkersville, MD). Antibody positions are listed according to the Kabat EU numbering convention.34
Expression host cells
A CHOK1 cell derivative, CATSMGAT KO, was generated to produce Man-5 antibody glycoforms.35 Briefly, the MGAT1 gene which encodes mannosyl (−1,3-)-glycoprotein-1,2-N-acetyl-glucosaminyl-transferase was knocked out from the Chinese hamster ovary (CHO) K1 cells by a zinc-finger nuclease pair targeting the coding region of MGAT1. ZFN plasmids were transfected into host cells by nucleofection using standard protocols. MGAT1 KO cells were enriched by treatment with phyto-hemagglutinin (PHA) for 2 passages. PHA-resistant cells were stained with fluorescent Galanthus nivalis lectin (GNA)-FITC to detect high-mannose glycosylation of cell-surface proteins. Strongly staining cells were then sub-cloned by FACS into 96-well plates. Genotype of the MGAT-1 knockout was confirmed by sequencing. A CHO Lec8 cell line, deficient in the UDP-galactose transporter (UGT) activity, was obtained from ATCC (ATCC CRL-1737). The Lec8 cell line was used to produce antibody glycoforms lacking terminal galactose. CHO (G22) is a high-yielding CHO transient expression cell line created by engineering the suspension-adapted CHO-K1 host cells to express the Epstein-Barr virus (EBV) nuclear antigen-1 (EBNA-1) with and without the co-expression of the gene for glutamine synthetase (GS).36
Transfection and protein purification
Approximately 1 × 106 CATSMGATKO cells/ml were seeded in CDCHO +6 mM glutamine media (Life Technologies/Thermo Fisher) 24 hr pre-transfection, and then transfected with 37E1B5 in pEN vector.37 Adherent Lec8 cells (ATCC) were grown in Alpha MEM with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were grown to 80% confluency in a Corning Cell Stack 10-Stack chamber with 6360 cm2 cell growth area (Sigma Aldrich) and transfected with pEFCMVOE vector using 1 μg/ml DNA and Lipofectamine 2000 (Thermo Fisher Scientific) at a 1:2.5 ratio (w/v) and according to the manufacturer instructions. Conditioned media was harvested for protein purification at Day 8 post transfection. The cells were removed by centrifugation and the supernatant was filtered through a 0.2 micron filter. The secreted antibody proteins were purified from the conditioned media directly on 1 mL HiTrap protein A columns (GE Healthcare, NJ), according to the manufacturer's instructions. Purified antibody samples, typically >95% homogeneity, were dialyzed against phosphate-buffered saline (PBS), flash frozen, and stored at −70°C. Monomer content after purification was determined by analytical size-exclusion chromatography (SEC). As needed, preparative SEC was used to remove soluble aggregate until >98% monomeric protein is obtained. Deglycosylated Beta8 antibody was prepared by overnight incubation at 37°C with 2U/mg of N-glycosidase F (Sigma-Aldrich). Beta8 antibody lacking terminal N-acetylneuraminic acid (NeuAc) was prepared by incubation at 25°C for 2 hours with 0.02 mU/mg of neuraminidase (Roche). Following enzymatic digestion, antibody proteins were re-purified using Protein A columns, as described above. To generate Fab, 30 mgs of IgG was digested for 5 hours at 37C with 0.75 mL of immobilized papain (Thermo Scientific) under the recommended buffer conditions. The cleaved mixture was dialyzed against PBS and Fab purified from Fc by passage over a 1 mL rProtein A FF column (GE Healthcare).
Determination of N-glycosylation site occupancy by LC-MS peptide mapping
Glycosylated and deglycosylated antibody samples were prepared by overnight incubation at 1 mg/mL in 10 mM Tris HCl, pH 8 with and without 2 units of N-glycosidase F (Sigma-Aldrich). For each sample, 100 µg of protein at 1.6 mg/ml was denatured and reduced in 5.2 M Guanidine, 85 mM Tris, 0.7 mM EDTA, 16 mM DTT, pH 7.6 for 30 minutes at 37°C. Following reduction, samples were incubated with 40 mM iodoacetamide at room temperature in the dark for 30 minutes. Reduced and alkylated protein was buffer-exchanged into 2 M urea, 100 mM Tris pH 7.6 and incubated at 37°C for 4 hours with trypsin at a 1:20 enzyme:substrate ratio. Protein digests were analyzed by RP LC-MS using an Acquity UPLC coupled to a Synapt G2 QToF mass spectrometer (Waters, Milford, US). For each sample, 5 µg of tryptic digest was injected onto a 150 mm × 2.1 mm, 1.7 µm particle size BEH300 C18 analytical column held at 55°C (Waters, Milford, US). Peptides were eluted at a constant flow rate of 0.2 mL/min using a 75 min binary gradient; solvent B was increased from 0% to 35%. The column was cleaned prior to the subsequent injection, by oscillating between high (95%) and low (5%) solvent B for 5 min. Solvent A (water) and B (acetonitrile) were supplemented with 0.02% (v/v) trifluoroacetic acid. Spectra were acquired between 50-2500 m/z using a data-independent mode of acquisition and processed using MassLynx (Waters, Milford, USA). Relative intensities of isotope- and charge-deconvoluted spectra for non-glycosylated and deglycosylated peptides, differing in mass by 1 Da, were used to determine relative glycosylation site occupancy for both Fab (H-CDR2) and Fc N-glycosylation sites.
Reduced antibody LC-MS
Reverse-phase LC-MS analysis of reduced antibody samples was performed using an Acquity UPLC coupled to a Synapt G1 quadruple time-of-flight mass spectrometer (Waters, Milford, US). Purified protein at 1 mg/ml was reduced by incubation at 37°C for 30 minutes in 10 mM DTT, 10 mM Tris HCl, pH 8. Two µg of reduced protein was injected onto a 50 mm × 2.1 mm, 1.7 µm particle size BEH C4 analytical column held at 65°C (Waters, Milford, US). Protein was eluted at a constant flow rate of 0.15 mL/min using a 15 minute binary gradient; solvent B was initially held at 5% for 3 minutes, increased to 25% over the next minute, then to 45% over 10 minutes before increasing to 95% over the final minute. The column was cleaned prior to the next injection by oscillating between high (95%) and low (5%) solvent B for 7 minutes. Solvent A (water) and B (acetonitrile) were supplemented with 0.01% (v/v) trifluoroacetic acid and 0.1% (v/v) formic acid. Spectra were acquired between 500–4500 Th. Key instrument parameters included +ve ionization mode, source voltage: 3.4 kV, sample cone voltage: 50 V, source temperature: 140°C, desolvation temperature: 400°C. Heavy chain spectra were processed using MassLynx (Waters, Milford, USA) to determine relative ion intensities for the predominant Fab (H-CDR2) and Fc antibody glycoforms.
Released, 2-aminobenzamide-labeled oligosaccharide profiling
N-glycan release, fluorescent labeling with 2AB and relative quantitation by UPLC-FLR were performed as detailed in the literature.38 Briefly, N-glycans were released at 5 mg/ml in 50 mM Tris HCl, pH 7.8 with 7.5 units of N-glycosidase F by overnight incubation at 37°C. Released glycans were labeled with 2-AB using (Prozyme 2AB labeling kit). Excess 2AB was removed using HILIC cartridges and clean up station base plate (Prozyme) operated via a vacuum manifold. Analysis of the 2AB-labeled N-glycans was performed on a Waters Acquity UPLC BEH Glycan column (2.1 mm ×150 mm, 1.7 μm particle). 2AB-labeled dextran ladder standard containing glucose unit (GU) oligomers (Prozyme) was used for glycan retention time normalization converting retention times into glucose units. Instrumentation used included an Acquity UPLC with a fluorescence detector (Waters, Milford, MA, USA).
αvβ8 binding ELISA
Recombinant human αvβ8 diluted in PBS (2 μg/ml) was coated on 96-well Maxisorp plates (Nunc) overnight at 4°C. Plates were then blocked with 3% bovine serum albumin for 1 hour at room temperature. After washing, titrated antibodies were added and allowed to incubate for 1 hour. After three washes in PBS, peroxidase-conjugated anti-human Fc (1:20,000) was used as the detection antibody. The wells were washed 3 times and TMB substrate solution (KPL) was added. The reaction was stopped with 2 N H2SO4 and the absorbance at 450 nm was measured. In the αvβ8 competition binding ELISA, a fixed amount of mouse 37E1B5 (0.05 μg/ml) was mixed with serial diluted Beta8 IgG (10 μg/ml) or Beta8 Fab (3.33 μg/ml) and then added to the αvβ8-coated wells. After 1 hour incubation at room temperature, the plate was washed 3 times in PBS. Mouse 37E1B5 binding was detected with peroxidase-conjugated anti-mouse Fc (1:20,000).
LAP and αvβ8 binding inhibition
Recombinant human LAP diluted in PBS (4 μg/ml) was coated on 96-well Maxisorp plates (Nunc) overnight at 4°C. Plates were then blocked with 3% casein (Thermo Scientific) for 2 hours at room temperature. After washing, a fixed amount of αvβ8 (50 μg/ml) was mixed with serially diluted Beta8 antibodies and added to the LAP-coated wells. After 2 hours incubation at room temperature, the plate was washed 3 times in PBS. The αvβ8 binding was detected by adding the biotinylated-anti-αv antibody (1:1000) and incubated at room temperature for 1 hour. After washing, peroxidase-conjugated streptavidin (1:3000) was added and plates were incubated for 30 min. The wells were then washed 3 times and TMB substrate solution (KPL) was added. The reaction was stopped with 2 N H2SO4 and the absorbance at 450 nm was measured.
TGF-β Bioassay
TGF-β activation was measured using transformed mink lung epithelial cells (TMLC) stably transfected with a portion of the plasminogen activated inhibitor 1 (PAI-1) promoter linked to a luciferase reporter (cells provided by Daniel Rifkin, New York University) and cultured as described previously.28 HeLa-B8 cells (1.5 × 104 cells/well) were co-cultured with TMLCs (1.5 × 104 cells/well) in a 96-well plate overnight in DMEM high glucose (Life Technologies/Thermo Fisher) supplemented with 10 % FBS and 10 U/ml Penicillin G, 10 μg/mL streptomycin G sulfate with or without test antibody. After 16 hours, supernatants were removed and cells were lysed in 100 µL of cell lysis buffer (Promega) and luciferase activity determined using the luciferase assay system (Promega) by transferring 80 µL of lysate and mixing with 80 µL of substrate in a white walled clear bottom 96-well plate. Samples were read immediately on a luminometer and shown as either relative luciferase units (RLU) or percent maximal response, determined by using TMLCs alone as the baseline or 0 % control and TMLCs co-cultured with HeLa-B8 cells as maximal or 100% response in the assay.
Generation of the HeLa-B8 cell line
HeLa-B8 cells is a derivative of the HeLa cell line (ECACC). Briefly, confluent HeLa cells maintained in MEM (Life Technologies/Thermo Fisher) supplemented with 10 % FBS, 1 % non-essential amino acids (Life Technologies/Thermo Fisher) and 10 U/ml Penicillin G (Life Technologies/Thermo Fisher), 10 μg/mL streptomycin G sulfate and prior to use, cells were removed using accutase and resuspended in PBS at 1 × 106 cells/mL. LIVE/DEAD fixable aqua dead cell stain (Life Technologies/Thermo Fisher, 1:1000) was added to the cells on ice for 20 minutes. Cells were pelleted and washed in cold flow cytometry staining buffer (eBioscience). Recombinant 37E1B5-mIgG1 or isotype-mIgG1 (100 µg/ml of 1 × 106 cells/ ml) was added to the cells and incubated on ice for 30 minutes. Cells were pelleted and washed and a secondary anti-mouse-Alexa-647 (Jackson ImmunoResearch, 1:200) added to the cells and incubated on ice for 30 minutes. Cells were pelleted and washed and resuspended at 10 × 106 cells/ml in HeLa cell medium containing 1% FBS. Cells were sorted on a BD FACSAria III cell sorter (BD Biosciences) using Chi-37E1B5 antibody. High αvβ8+ sorted cells were then cultured in complete HeLa cell medium, expanded and banked for future use. Cells remained positive for high αvβ8 expression for at least 1 month of culture.
Mouse pharmacokinetic studies
The PK profiles of the Beta8 (WT) and its glycan variants, aglycosylated (AG) and desialylated (DE) (1 and 10 mg/kg, intravenous administration) were evaluated in plasma from humanized avB8-BAC mice using a capillary micro-sampling technique. This method allows up to 7 repeated longitudinal bleeds to be taken at given time points from the same animal (n = 4 mice per group). Levels of plasma antibody exposure were quantified using a human Fc capture ELISA. Briefly, Nunc Maxisorp plates were coated overnight at 4°C (0.3 µg/well) with a goat anti-human IgG, Fc fragment specific capture antibody (Jackson ImmunoResearch, UK). Following washing and blocking of plates with 2% w/v Marvel (M-PBS) in PBS for 1h, dilutions of samples and standards (0.001-1 μg/ml) in M-PBS were added to the plate (all at 100 μl per well) and incubated at room temperature for 1 h. Following a washing step, a sheep anti-human kappa light chain Affinity Purified HRP secondary antibody (1:15000 dilution, The Binding Site, UK) in M-PBS was added and incubated at room temperature for 1 h. Following washing, TMB was added and, after color development was stopped with an equal volume of 2N H2SO4, absorbance was read at 450 nm.
Mouse pharmacokinetics analysis
A PK model was iteratively developed using the observed serum concentrations in animals dosed with 1 mg/kg and 10 mg/kg of tested antibodies. The nonlinear mixed effects software NONMEM (Icon Development Solutions, Version 7.3) was used for this purpose. The NONMEM “Objective Function Value” (OFV), which is proportional to twice the negative log-likelihood of the data,39 was used to assess improvements to the goodness of fit provided by the model, as compartments and interactions were added. The difference in OFV between 2 nested models is approximately χ2-distributed and can be used to carry out a likelihood ratio test. An OFV improvement of 3.84 can correspond to a nominal significance level of <0.05 (for one differing parameter, corresponding to 1 degree of freedom).39 The addition of more parameters increases the magnitude of OFV improvement required for a significant change (e.g., 5.99 for 2 differing parameters, corresponding for 2 degrees of freedom). The PK of the WT, deglycosylated (DE) and aglycosylated (AG) antibodies were best described with a mixed effects model including inter-compartmental distribution, non-specific clearance (CL) and target-mediated elimination phases, together with their biological variability. Once the model was set up, combinations of differences between non-specific clearances of the Beta8 variants were tested. Specifically, we analyzed models where WT, the deglycosylated (DE) and the aglycosylated (AG) clearances were all different and all constrained to be the same, and where only 2 out of 3 were the same (5 models in total).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to acknowledge Andrew Meng and Neil Mody for assistance in Mass-Spec and structural characterization of antibody glycan variants; Dr. Daniel Rifkin of New York University for reagent sharing and Dr. Stephen Nishimura of UCSF for data sharing and helpful discussions.
References
- 1.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012; 180:1340-55; PMID:22387320; http://dx.doi.org/ 10.1016/j.ajpath.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 1992; 359:693-9; PMID:1436033; http://dx.doi.org/ 10.1038/359693a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thannickal VJ, Lee DY, White ES, Cui Z. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 2003; 278(14):12384-9; PMID:12531888; http://dx.doi.org/ 10.1074/jbc.M208544200 [DOI] [PubMed] [Google Scholar]
- 4.Sonnylal S, Shi-Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, Leask A, Abraham D, Bou-Gharios G, de Crombrugghe B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum 2010; 62(5):1523-32; PMID:20213804; http://dx.doi.org/ 10.1002/art.27382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varga J, Whitfield ML. Transforming growth factor-β in systemic sclerosis (scleroderma). Front Biosci (Schol Ed) 2009; 1:226-35; PMID:19337284; http://dx.doi.org/ 10.2741/s22 [DOI] [PubMed] [Google Scholar]
- 6.Aschner Y, Downey GP. Transforming growth factor-β: Master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol 2016; 54(5):647-55; PMID:26796672; http://dx.doi.org/ 10.1165/rcmb.2015-0391TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A 1993; 90:770-4; PMID:8421714; http://dx.doi.org/ 10.1073/pnas.90.2.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantel PY, Schmidt-Weber CB. Transforming growth factor-β: recent advances on its role in immune tolerance. Methods Mol Biol 2011; 677:303-38; PMID:20941619; http://dx.doi.org/ 10.1007/978-1-60761-869-0_21 [DOI] [PubMed] [Google Scholar]
- 9.Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor β1–an intimate relationship. Eur J Cell Biol 2008; 87(8-9):601-15; PMID:18342983; http://dx.doi.org/ 10.1016/j.ejcb.2008.01.012 [DOI] [PubMed] [Google Scholar]
- 10.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol 2007; 179(6):1311-1323; PMID:18086923; http://dx.doi.org/21677751 10.1083/jcb.200704042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature 2011; 474:343-9; PMID:21677751; http://dx.doi.org/ 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J Cell Biol 1988; 106:1659-65; PMID:2967299; http://dx.doi.org/8885242 10.1083/jcb.106.5.1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-β 1. Mol Endocrinol 1996; 10:1077-83; PMID:8885242; http://dx.doi.org/ 10.1210/mend.10.9.8885242 [DOI] [PubMed] [Google Scholar]
- 14.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012; 11(10):790-811; PMID:23000686; http://dx.doi.org/ 10.1038/nrd3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV Jr, Sheppard D. Inactivation of the integrin β 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 1996; 133:921-8; PMID:8666675; http://dx.doi.org/ 10.1083/jcb.133.4.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margadant C, Sonnenberg A. Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Reports 2010; 11(2):97-105; PMID:20075988; http://dx.doi.org/ 10.1038/embor.2009.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schürpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J 2011; 30:4712-27; PMID:21946563; http://dx.doi.org/ 10.1038/emboj.2011.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res 2010; 339:269-80; PMID:19693543; http://dx.doi.org/19118215 10.1007/s00441-009-0834-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 2009; 122:227-32; PMID:19118215; http://dx.doi.org/ 10.1242/jcs.035246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sims MA, Field SD, Barnes MR, Shaikh N, Ellington K, Murphy KE, Spurr N, Campbell DA. Cloning and characterisation of ITGAV, the genomic sequence for human cell adhesion protein (vitronectin) receptor α subunit, CD51. Cytogenet Cell Genet 2000; 89:268-71; PMID:10965141; http://dx.doi.org/ 10.1159/000015631 [DOI] [PubMed] [Google Scholar]
- 21.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, et al.. Partial inhibition of integrin α(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 2008; 177:56-65; PMID:17916809; http://dx.doi.org/ 10.1164/rccm.200706-805OC [DOI] [PubMed] [Google Scholar]
- 22.Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, et al.. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin alphavbeta8-mediated activation of TGF-β. J Clin Invest 2011; 121:2863-75; PMID:21646718; http://dx.doi.org/ 10.1172/JCI45589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minagawa S, Lou J, Seed RI, Cormier A, Wu S, Cheng Y, Murray L, Tsui P, Connor J, Herbst R, et al.. Selective targeting of TGF-β activation to treat fibroinflammatory airway disease. Sci Transl Med 2014; 6(241):241ra79; PMID:24944194; http://dx.doi.org/ 10.1126/scitranslmed.3008074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA. The integrin α v β 6 binds and activates latent TGF β 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96(3):319-28; PMID:10025398; http://dx.doi.org/ 10.1016/S0092-8674(00)80545-0 [DOI] [PubMed] [Google Scholar]
- 25.Ozawa A, Sato Y, Imabayashi T, Uemura T, Takagi J, Sekiguchi K. Molecular basis of the ligand binding specificity of alphavbeta8 integrin. J Biol Chem 2016; 291:11551-65; PMID:27033701; http://dx.doi.org/ 10.1074/jbc.M116.719138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson NC, Sheppard D. Integrin-mediated regulation of TGFbeta in fibrosis. Biochim Biophys Acta 2013; 1832:891-6; PMID:23046811; http://dx.doi.org/15509794 10.1016/j.bbadis.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi S, Williams SA, Seppo A, Kurniawan H, Chen W, Ye Z, Marth JD, Stanley P. Inactivation of the Mgat1 gene in oocytes impairs oogenesis, but embryos lacking complex and hybrid N-glycans develop and implant. Mol Cell Biol 2004; 24:9920-9; PMID:15509794; http://dx.doi.org/ 10.1128/MCB.24.22.9920-9929.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem 1994; 216:276-84; PMID:8179182; http://dx.doi.org/ 10.1006/abio.1994.1042 [DOI] [PubMed] [Google Scholar]
- 29.Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci 2015; 104:1866-84; PMID:25872915; http://dx.doi.org/ 10.1002/jps.24444 [DOI] [PubMed] [Google Scholar]
- 30.Nishimura SU, Lou JU, Cormier AU, Baron JLU, Marks JDU, Murray LG, et al.. IMPROVED ALPHA-V BETA-8 ANTIBODIES In: Office UP, ed. USA, 2015 [Google Scholar]
- 31.Rice KG, Lee YC. Oligosaccharide valency and conformation in determining binding to the asialoglycoprotein receptor of rat hepatocytes. Adv Enzymol Relat Areas Mol Biol 1993; 66:41-83; PMID:8430516 [DOI] [PubMed] [Google Scholar]
- 32.Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J 2012; 14:554-8; PMID:22610647; http://dx.doi.org/ 10.1208/s12248-012-9369-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquemin M, Radcliffe CM, Lavend'homme R, Wormald MR, Vanderelst L, Wallays G, Dewaele J, Collen D, Vermylen J, Dwek RA, et al.. Variable region heavy chain glycosylation determines the anticoagulant activity of a factor VIII antibody. J Thromb Haemost 2006; 4:1047-55; PMID:16689758; http://dx.doi.org/ 10.1111/j.1538-7836.2006.01900.x [DOI] [PubMed] [Google Scholar]
- 34.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. NIH Publ. No91-3442, 1991 [Google Scholar]
- 35.Oganesyan V, Mazor Y, Yang C, Cook KE, Woods RM, Ferguson A, Bowen MA, Martin T, Zhu J, Wu H, et al.. Structural insights into the interaction of human IgG1 with FcgammaRI: no direct role of glycans in binding. Acta Crystallogr D Biol Crystallogr 2015; 71:2354-61; PMID:26527150; http://dx.doi.org/ 10.1107/S1399004715018015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daramola O, Stevenson J, Dean G, Hatton D, Pettman G, Holmes W, Field R. A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog 2014; 30:132-41; PMID:24106171; http://dx.doi.org/ 10.1002/btpr.1809 [DOI] [PubMed] [Google Scholar]
- 37.Xu L, Jin X, Rainey GJ, Wu H, Gao C. A mammalian expression system for high throughput antibody screening. J Immunol Methods 2013; 395(1-2):45-53; PMID:23831609; http://dx.doi.org/ 10.1016/j.jim.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 38.Ahn J, Bones J, Yu YQ, Rudd PM, Gilar M. Separation of 2-aminobenzamide labeled glycans using hydrophilic interaction chromatography columns packed with 1.7 microm sorbent. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878:403-8; PMID:20036624; http://dx.doi.org/ 10.1016/j.jchromb.2009.12.013 [DOI] [PubMed] [Google Scholar]
- 39.Wahlby U, Jonsson EN, Karlsson MO. Assessment of actual significance levels for covariate effects in NONMEM. J Pharmacokinet Pharmacodyn 2001; 28:231-52; PMID:11468939; http://dx.doi.org/ 10.1023/A:1011527125570 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.