

Abstract
The synthetic steroid mifepristone blocks the growth of ovarian cancer cells, yet the mechanism driving such effect is not entirely understood. Unbiased genomic and proteomic screenings using ovarian cancer cell lines of different genetic backgrounds and sensitivities to platinum led to the identification of two key genes upregulated by mifepristone and involved in the unfolded protein response (UPR): the master chaperone of the endoplasmic reticulum (ER), glucose regulated protein (GRP) of 78 kDa, and the CCAAT/enhancer binding protein homologous transcription factor (CHOP). GRP78 and CHOP were upregulated by mifepristone in ovarian cancer cells regardless of p53 status and platinum sensitivity. Further studies revealed that the three UPR‐associated pathways, PERK, IRE1α, and ATF6, were activated by mifepristone. Also, the synthetic steroid acutely increased mRNA translation rate, which, if prevented, abrogated the splicing of XBP1 mRNA, a non‐translatable readout of IRE1α activation. Moreover, mifepristone increased LC3‐II levels due to increased autophagic flux. When the autophagic–lysosomal pathway was inhibited with chloroquine, mifepristone was lethal to the cells. Lastly, doses of proteasome inhibitors that are inadequate to block the activity of the proteasomes, caused cell death when combined with mifepristone; this phenotype was accompanied by accumulation of poly‐ubiquitinated proteins denoting proteasome inhibition. The stimulation by mifepristone of ER stress and autophagic flux offers a therapeutic opportunity for utilizing this compound to sensitize ovarian cancer cells to proteasome or lysosome inhibitors.
Keywords: Ovarian cancer, Bortezomib, Mifepristone, Chloroquine, ER stress, Autophagic flux, mRNA translation, Unfolded protein response, Ubiquitin proteasome system
Highlights
Mifepristone triggers the unfolded protein response (UPR) in ovarian cancer cells.
Mifepristone‐induced UPR is mediated by increased mRNA translation rate.
Mifepristone increases autophagic flux in ovarian cancer cells.
Mifepristone is lethal to ovarian cancer cells when combined with chloroquine.
Mifepristone kills ovarian cancer cells in combination with proteasome inhibitors.
1. Introduction
The lethality of ovarian cancer can be attributed to its late diagnosis and a lack of long‐term effective treatment approaches. Since the introduction of debulking surgery and platinum–taxane combination therapy over 30 years ago, there has been no significant breakthrough impacting the overall survival of ovarian cancer patients. Although over 70% of diagnosed women respond to front‐line standard of care with remission, the disease hides as microscopic or minimal residual within the abdominal cavity for about 18–24 months, recurring thereafter with a phenotype usually not responsive to current chemotherapeutic agents (Bast, 2011; Bast et al., 2009; Coleman et al., 2013; Kurman and Shih Ie, 2011; Romero and Bast, 2012; Vaughan et al., 2011). Because patients are left without treatment between remission and recurrence, it is imperative to develop consolidation therapies that can be used chronically after standard of care. We previously reported that mifepristone and other antiprogestins are potential candidates to achieve such a goal (Goyeneche and Telleria, 2015; Telleria and Goyeneche, 2012) as they: (i) block growth of ovarian cancer cells in vitro and in vivo (Goyeneche et al., 2007); (ii) arrest ovarian cancer cells at the G1 phase of the cell cycle by increasing the abundance of Cdk inhibitors p21cip1 and p27kip1, and inhibiting the activity of cyclin dependent kinase 2 (Cdk2) (Goyeneche et al., 2012); (iii) do not require progesterone receptors to block cell growth (Tieszen et al., 2011); (iv) prevent the repopulation of ovarian cancer cells following cytotoxic chemotherapy with cisplatin (Freeburg et al., 2009b) or cisplatin plus paclitaxel (Gamarra‐Luques et al., 2012); (v) synergize with inhibitors of the PI3K/Akt survival pathway killing ovarian cancer cells (Wempe et al., 2013); and (vi) growth‐arrest ovarian cancer cells that are sensitive or resistant to cisplatin and/or paclitaxel (Freeburg et al., 2009a; Gamarra‐Luques et al., 2014).
Using unbiased genomic and proteomic approaches to shed light on the signaling evoked by mifepristone as an anti‐ovarian cancer agent, we discovered in the present work that mifepristone induces the unfolded protein response (UPR) and increases autophagic flux in ovarian cancer cells. The UPR represents a series of signaling transduction events that ameliorate the accumulation of unfolded/misfolded proteins caused by loss of homeostasis in the endoplasmic reticulum (ER) (a.k.a. ER stress) (Gardner et al., 2013; Hetz, 2012; Luo and Lee, 2013; Rutkowski and Kaufman, 2007; Sano and Reed, 2013; Schonthal, 2013; Verfaillie et al., 2013). The UPR promotes protein folding by increasing expression of resident ER chaperones, transiently blocking global protein synthesis, and enhancing ER assisted degradation (ERAD) (Sano and Reed, 2013). During ERAD, misfolded proteins are recognized and retro‐translocated to the cytoplasm where they are ubiquitinated and targeted for proteasome degradation (Brodsky, 2012; Lederkremer, 2009; Needham and Brodsky, 2013; Vembar and Brodsky, 2008). When ER stress is acute, the corrective measures induced by the UPR allows for cell survival; conversely, when the stress is chronic and the load of unfolded/misfolded proteins critically exceeds the homeostatic capacity of the ER, the UPR triggers cell death (Hetz, 2012; Logue et al., 2013; Sano and Reed, 2013; Urra et al., 2013; Verfaillie et al., 2013). In cases where excess accumulation of unfolded proteins leads to protein aggregation, the amelioration of ER stress and restoration of the protein folding environment of the ER is further achieved by the activation of autophagy (Nagelkerke et al., 2014; Wang and Terpstra, 2013). Herein, we prove that mifepristone triggers the UPR as a consequence of increased protein synthesis. Secondly, we show that mifepristone increases autophagic flux and kills ovarian cancer cells in combination with a lysosome inhibitor. Lastly, we demonstrate that if mifepristone is associated with a proteasome inhibitor that interferes with ERAD, thus not allowing the cells to degrade misfolded/unfolded proteins via the ubiquitin proteasome system (UPS), the cells die.
2. Materials and methods
2.1. Cell lines and in vitro exposure of the cells to drugs
The human carcinoma cell line OV2008 and its platinum‐resistant sibling OV2008/C13 were obtained from Dr. Stephen Howell (University of California, San Diego) and were maintained in RPMI‐1640 (Mediatech, Herndon, VA) supplemented with 5% heat inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 10 mM HEPES (Mediatech), 4 mM l‐glutamine (Mediatech), 1 mM sodium pyruvate (Mediatech), 100 IU penicillin (Mediatech), and 100 μg/ml streptomycin (Mediatech). The SKOV‐3 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured in RPMI‐1640 (Mediatech) with 5% fetal bovine serum (Atlanta Biologicals), 10 mM HEPES (Mediatech), 4 mM l‐glutamine (Mediatech), 0.45% D (+) glucose (Sigma Chemical Co., St. Louis, MO), 1 mM sodium pyruvate (Mediatech), 1× non‐essential amino acids (Mediatech), 100 IU penicillin (Mediatech), 100 μg/ml streptomycin (Mediatech), and 0.01 mg/ml human insulin (Roche, Indianapolis, IN). All cells were maintained at 37 °C in a humidified atmosphere containing 95% air/5% CO2. The drugs used to treat the cells were vehiculized in DMSO, and the maximal concentration of DMSO in the culture media was ≤0.2% (v/v). The providers of the drugs utilized were as follows: mifepristone (Sigma); MG‐132 (Enzo Life Sciences, Farmingdale, NY); bortezomib (Millennium Pharmaceuticals, Cambridge, MA); chloroquine (Sigma); salubrinal (EMD Millipore, Billerica, MA); cycloheximide (Sigma).
2.2. Cell proliferation and viability
Following different treatment approaches, the number of cells as well as their viability was assessed via microcapillary fluorescence cytometry as we previously reported in detail (Freeburg et al., 2009b).
2.3. Deoxyribonucleic acid microarray
Total RNA was extracted in TRI reagent with a Polytron homogenizer. After centrifugation, the aqueous layer containing total RNA was removed, mixed with RLT buffer (Qiagen, Valencia, CA) and ethanol, and centrifuged through a Qiagen RNeasy column. The column was washed, and the sample was treated with ribonuclease‐free deoxyribonuclease (Qiagen) to remove residual DNA. Total RNA was eluted from the column using nuclease free water. The quality and quantity of RNA were assessed using the RNA 6000 Nano LabChip in an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA).
The Codelink Whole Human Genome Bioarray (Applied Microarrays, Tempe, AZ) was used for the DNA microarray analysis of gene expression. These microarrays contain ∼53,000 single stranded 30‐mer oligonucleotide probes for human genes/transcribed sequences. Sample processing and hybridization into DNA microarrays was carried out as previously described (Eyster and Brannian, 2009) using 1 μg total RNA as starting material. The slides were scanned with a GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA) and analyzed with GenePix Pro (Molecular Devices), CodeLink (Applied Microarrays), Acuity (Molecular Devices), and GeneSpring 7.0 (Agilent) software. The GenePix Pro software aligned and acquired the microarray image. Codelink software 5.0 was used to apply the background correction. Fold expression values were obtained from analysis by using the Acuity software. GeneSpring software was used to normalize the expression of each gene to the median gene expression and each slide to the 50th percentile of gene expression, and for statistical analysis of the microarray data. The complete data for these microarrays is available at the NCBI Gene Expression Omnibus (www.ncbi:nlm.nih.gov/geo) and can be accessed through GEO accession number GSE29763.
2.4. Class comparison and pathway analyses
The microarray data generated using the Codelink Whole Human Genome Bioarray using OV2008 and OV2008/C13 cells were imported into BRB Arraytools, log2‐transformed, and quantile normalized. Probe intensities <10 were thresholded, and exclusion filters (<20% of probe values have at least a 1.5‐fold change in either direction from the probe's median value or >50% missing data) was applied. Out of 53,079 probe values, 1927 passed the filter. The resulting probe values were subjected to class comparison analysis limited to each cell line. The significantly affected genes were subjected to Ingenuity Pathway Analysis (IPA) as previously described (Roy et al., 2014).
A building pathway flow searching database was additionally performed using Pathway Studio® (Elsevier Inc.) and a mammalian dataset (ResNet 2.5 database). The hypothetical pathways affected by mifepristone in ovarian cancer cells were built based on genes we previously discovered to be regulated by the synthetic steroid following hypothesis‐driven experiments (Goyeneche et al., 2007; Tieszen et al., 2011; Wempe et al., 2013) in combination with the most representative genes significantly regulated by mifepristone and discovered in the present study via unbiased genomic and proteomic platforms.
2.5. Real time RT‐PCR
Real time RT‐PCR was performed as previously described (Eyster et al., 2007). GAPDH was used as the endogenous control (housekeeping) gene. Predesigned primers and probes were obtained from Life Technologies (Grand Island, NY) (Hs00607129_gH for HSPA5, Hs00355782_m1 for CDKN1A, Hs01597588_m1 for CDKN1B, and Hs00358796_g1 for CHOP). Data were analyzed by GraphPad Prism (GraphPad Software, San Diego, CA).
2.6. Protein identification by mass spectrometry analysis
The proteins were obtained upon cellular fractionation to increase the efficiency in the number of proteins to be detected. The cellular pellet was resuspended in low salt buffer (10 mM HEPES pH7.9, 10 mM KCl, 1.5 mM MgCl2) for 15 min on ice and then homogenized using a motorized pestle. The lysate was centrifuged at 4 °C at 800 g. The supernatant constituted a fraction enriched in cytosolic components. The pellet, enriched mainly in membranes and organelles, was lysed using NP40 buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% NP40, and 50 mM NaF). The lysate was centrifuged at 10,000 g for 20 min, and the supernatant was transferred to a new tube. The proteins obtained in both fractions in‐solution were reduced with 50 mM DTT (Sigma–Aldrich, Saint Louis, MO) at 65 °C for 5 min, alkylated with 100 mM iodoacetamide (Sigma–Aldrich), and digested using sequencing grade trypsin (Promega, Madison, WI) overnight at 37 °C. The digestion was stopped by the addition of 0.5% acetic acid, frozen in dry ice, and concentrated using a Savant SpeedVac centrifuge (Thermo Scientific, Hudson, NH). The tryptic‐digested peptides were dissolved in 100 mM ammonium formate pH10 and separated through 2D‐nanoLC with dilution using a 2D‐nanoAcquity UPLC (Waters Corporation, Milford, MA). The first dimension was performed in XBridge BEH130 C18, 5 μm, 300 μm × 50 mm NanoEase Column (Waters Corporation, Milford, MA) using as solvent A1 20 mM ammonium formate pH10 and B1, 100% acetonitrile (Optima LC/MS, Fisher Scientific, Pittsburgh, PA) LC‐MS grade. The flow at 1st dimension was 2 μl/min, and 11 different step gradients (dilution method) were performed for 20 min each. The second dimension included trapping and desalting online through 180 μm × 20 mm, 5 μm symmetry C18 nanoAcquity UPLC trap column (Waters) at a flow 20 μl/min, 99% A2 (water, 0.1% formic Acid), and 1% B2 (100% acetonitrile, 0.1% formic acid) for 20 min. After the peptides were desalted and concentrated, they were separated online in the second dimension through BEH130 C18 1.7 μm, 100 μm × 100 mm nanoAcquity UPLC column. The standard solvent gradient used was: 0–2 min, 3% B2 isocratic; 2–40 min, 3–85% B2 linear, at a flow rate of 400 nl/min for 60 min. The eluted ions were analyzed by one full precursor MS scan (400–1500 m/z) followed by four MS/MS scans of the most abundant ions detected in the precursor MS scan while operating under dynamic exclusion or direct data acquisition system (DDAS). Spectra obtained in the positive ion mode with nano ESI‐Q‐Tof Synapt G1 mass spectrometer (Waters) were deconvoluted and analyzed using the MassLynx software 4.1 (Waters). A peak list (PKL format) was generated to identify +1 or multiple charged precursor ions from the mass spectrometry data file. The instrument was calibrated in MS/MS mode using 100 fmol of (Glu1)‐Fibrinopeptide B human (Sigma) with a RMS residual of 3.857 e−4 amu or 6.9413 e−1 ppm or 7.722 e0 ppm. Parent mass (MS) and fragment mass (MS/MS) peak ranges were 400–1500 Da and 65–1500 Da, respectively. Mascot server v2.5.0 and Mascot Daemon Toolbox v2.5.1 (www.matrix‐science.com, UK) in MS/MS ion search mode (local licenses) were applied to conduct peptide matches (peptide masses and sequence tags) and protein searches against NCBInr v20150531 (67337701 sequences, 24122812982 residues) taxonomy filter (Homo sapiens) (311623 sequences) and IPI Human v 3.80 (86719 sequences, 34928216 residues). The following parameters were set for the search: carbamidomethyl (C) on cysteine was set as fixed; variable modifications included asparagine and glutamine deamidation and methionine oxidation. One missed cleavage was allowed; monoisotopic masses were counted; the precursor peptide mass tolerance was set at 50 ppm; fragment mass tolerance was 0.3 Da, and the ion score or expected cut‐off was set at 5. The MS/MS spectra were searched with MASCOT using a 95% confidence interval (C.I. %) threshold (p < 0.05). A minimum score of 46 was used for peptide identification. The protein redundancy that appeared at the database under different gi (protein accession number) was limited to human with the first priority assigned to that specie.
2.7. Western blot analysis
Following treatment, cells were harvested, washed with PBS, pelleted, and maintained at −80 °C until whole cell extracts were prepared. Cell lysates were obtained, protein quantitated, and subjected to gel electrophoresis and western blotting as previously described (Gamarra‐Luques et al., 2012). The list of antibodies and the dilutions utilized are described in Table S1.
2.8. Xbp1 mRNA splicing
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) per manufacturer's instructions. Reverse transcription (RT) reaction and polymerase chain reaction (PCR) were done using a SuperScript® first‐strand synthesis system for RT‐PCR (Invitrogen). The forward primer for PCR amplification of spliced and total human Xbp1 mRNA was 5′‐CCTGGTTGCTGAAGAGGAGG‐3′ and the reverse primer was 5′‐CCATGGGGAGTTCTGGAG‐3′. For ACTB (β‐actin), the primers were 5′‐CCTGTACGCCAACACAGTGC‐3′ (forward) and 5′‐ATACTCCTGCTTGCTGATCC‐3′ (reverse). PCR was carried out with an initial cycle at 94 °C for 3 min, followed by 35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s with a final extension at 72 °C for 5 min. The size of amplified unspliced Xbp1 mRNA is 145 base pairs (bp), the size of amplified spliced Xbp1 mRNA is 119 bp, and the size of amplified ACTB (β‐actin) mRNA is 211 bp. The PCR products were run on a 2% agarose gel, stained with ethidium bromide (Molecular Probes), and photographed under UV light. A DNA marker (Bionexus Lo™ DNA marker) was used to determine the size of the PCR products (Bionexus, Oakland, CA).
2.9. Puromycin incorporation assay
Puromycin is an aminoacyl‐tRNA mimetic that enters the site A of the ribosome and terminates translation prematurely. However, when exposure to puromycin is done short before ending an experiment, conjugation of puromycin with nascent polypeptide chains generate short‐lived puromycylated peptides that are released from the ribosome and can be detected with an anti‐puromycin antibody by western blot. Incorporation of puromycin into nascent amino acid chains is a sensitive indicator of instant or ongoing mRNA translation rate as one molecule of puromycin is incorporated per nascent peptide (Liu et al., 2012; Schmidt et al., 2009). For each experiment, puromycin (Sigma) was pulsed to the culture media to a final concentration of 1 μM at 37 °C for 30 min. Thereafter, cells were harvested and processed for western blot analysis of puromycylated peptides.
2.10. Assessment of the activity of the proteasome
We transiently transfected ovarian cancer cells with an expression plasmid termed GFPu using Effectene transfection reagent (Qiagen). GFPu (Li et al., 2011) is a modified, enhanced, green fluorescence protein (EFGP) driven by a cytomegalovirus promoter; it carries an ubiquitination signal sequence (CL1 degron, ACKNWFSSLSHFVIHL) that is used as surrogate substrate for the proteasome (Bence et al., 2001). In the absence of changes in synthesis, GFPu protein abundance, as assessed by fluorescence microscopy or western blot, inversely reflects proteasome proteolytic function (Dong et al., 2004). The caspase‐like (B1) peptidase activity in vitro was done as previously described (Li et al., 2011). We used the synthetic fluorogenic peptide Suc‐LLE‐AMC with or without ATP and in the presence or absence of MG‐132. The fraction of the activity that was inhibited by MG‐132 was attributed to the proteasome.
2.11. Drug interaction analysis
To characterize the pharmacological interaction between mifepristone and chloroquine, and mifepristone and bortezomib, we used the CalcuSyn software (Biosoft, Cambridge, UK), which utilizes the combination index (CI) as a method for quantifying drug interaction. We previously described in detail the calculation of the CI (Gamarra‐Luques et al., 2012). Briefly, for a specific drug association, a CI < 1 indicates synergism (defined as a more than expected additive effect), CI = 1 indicates additivism, whereas CI > 1 indicates antagonism.
3. Results
3.1. Mifepristone inhibits the growth of ovarian cancer cells of different genetic backgrounds and platinum sensitivities
We first confirmed the anti‐growth capacity of mifepristone on three ovarian cancer cell lines, one sensitive to platinum and carrying wild‐type p53 (OV2008), its sibling cell line resistant to platinum yet maintaining wild‐type p53 (OV2008/C13), and the platinum semi‐resistant and p53 null, SKOV‐3 cell line (Freeburg et al., 2009a). The growth inhibition curves are shown in Figure 1. The concentration needed to inhibit 50% of growth (GI50) ranged from 8 to 14 μM, confirming our previous assessment with these cell lines in which mifepristone at concentrations up to 20 μM was cytostatic, whereas at higher concentrations it had lethal effects (Freeburg et al., 2009a; Goyeneche et al., 2012).
Figure 1.
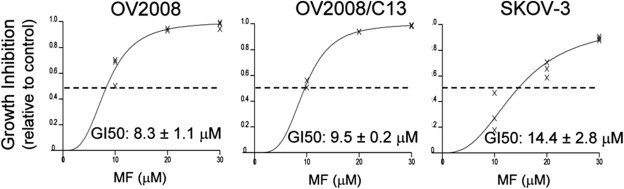
OV2008 (p53 wt/platinum sensitive), OV2008/C13 (p53 wt/platinum resistant), and SKOV‐3 (p53 null/platinum semi‐resistant) cells were cultured in the presence of DMSO (vehicle), 10, 20, or 30 μM mifepristone for 7 days. The values of concentrations of the drug causing 50% inhibition of growth (GI50) were calculated using software (Calcusyn, Biosoft) designed to study drug interaction that analyzes the median effective dose or Dm, which is analogous to the GI50. The experiments were run 3 times in triplicates for each cell line.
3.2. Functional genomic and proteomic analyses of mifepristone‐treated ovarian cancer cells
To identify genes and proteins differentially regulated by mifepristone, we exposed the ovarian cancer cells to a cytostatic 20 μM concentration of mifepristone for 24 h. A DNA microarray analysis identified six genes (ZNF488, S100P, DDIT3, UNC5B, TRIB3, and CHAC1) differentially expressed in the three cell lines, being then unrelated to platinum sensitivity or p53 status (Table 1). There were also several genes significantly downregulated and upregulated regardless of platinum sensitivity (Figure S1 and Tables S2 and S3). Ingenuity Pathway Analysis (IPA) of genes differentially regulated by mifepristone in two cell lines (OV2008 and OV2008/C13) identified the ER stress and the unfolded protein response (UPR) pathways as the top candidates significantly affected by mifepristone (Tables S4 and S5).
Table 1.
Genes differentially expressed upon exposure to 20 μM mifepristone for 24 h regardless of the sensitivity of the cells to cisplatin and p53 status as detected by DNA microarray analysis and expressed as fold changes when compared to vehicle‐treated controls.
| Symbol | Gene description | OV2008 | OV2008/C13 | SKOV‐3 | Protein class |
|---|---|---|---|---|---|
| ZNF488 | Zinc finger protein 488 | −2.99 | −2.92 | −1.95 | Transcription factor |
| S100P | S100 calcium binding protein P | 9.49 | 3.41 | 2.02 | Signaling molecule |
| DDIT3 | DNA‐Damage‐Inducible Transcript 3/CHOP | 6.06 | 5.41 | 2.08 | Transcription factor |
| UNC5B | Unc‐5 homolog B (C. elegans) | 2.81 | 2.94 | 3.90 | Receptor |
| TRIB3 | Tribbles homolog 3 (Drosophila) | 6.47 | 4.63 | 2.21 | Protein kinase |
| CHAC1 | Cation transport regulator homolog 1 (E. coli) | 4.34 | 4.78 | 2.01 | Cation transporter |
We also found differentially expressed proteins in response to 20 μM mifepristone for 24 h using a 2D nanoLC‐MS/MS proteomic approach (Figure 2 and Tables S6 and S7). For this initial screening we found notably increased, in all ovarian cancer cells exposed to cytostatic doses of mifepristone, a mRNA encoding for DNA‐damage inducible transcript 3 (DDIT3), also known as CCAAT‐enhancer‐binding protein homolog protein (CHOP) or GADD153 (Growth arrest and DNA damage inducible gene 153) (Oyadomari and Mori, 2004) (Table 1). Using the proteomic platform, it was clear that the main protein consistently and significantly upregulated by mifepristone in the three cell lines was GRP78 (glucose‐regulated protein, 78 kDa), a member of the family of heat shock proteins of 70 kDa, also termed BIP (binding immunoglobulin protein) or heat shock 70 kDa protein 5 (HSPA5) (Figure 2). GRP78 is the master chaperone of the ER (Lee, 2014). Together, CHOP and GRP78 were proposed to balance the stress of the ER in a ‘yin’ (CHOP; pro‐cell death factor) ‘yang’ (GRP78; pro‐survival factor) manner (Schonthal, 2013). We further confirmed the expression of these differentially regulated proteins by western blot. Figure 3A shows that GRP78 and CHOP were upregulated by 20 μM mifepristone in a time‐dependent manner in all cell lines studied. As control for mifepristone‐mediated cytostatic activity, we measured the increased expression of the cyclin‐dependent kinase inhibitors p27kip1 and p21cip1, both previously shown to mediate cell cycle arrest induced by mifepristone (Goyeneche et al., 2007, 2012). Additionally, we validated the DNA microarray data for four genes in OV2008 cells: p21cip1, p27kip1, GRP78, and CHOP. We confirmed that p21cip1, GRP78, and CHOP mRNA levels increased in response to mifepristone by using a real‐time RT‐PCR approach, whereas p27kip1, which increased in response to mifepristone at the level of protein abundance, did not significantly change at the level of mRNA (Figure 3B). This latter finding was in agreement with the fact that p27kip1 is known to be regulated post‐transcriptionally (Slingerland and Pagano, 2000).
Figure 2.
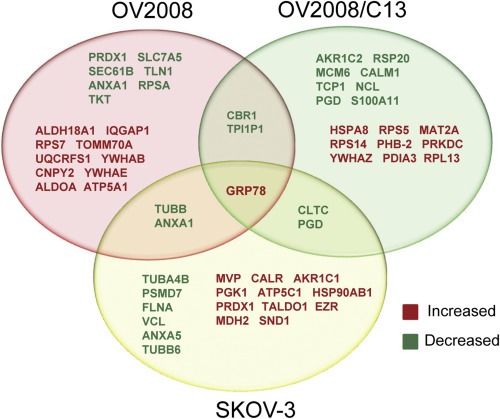
Venn diagram displaying differentially expressed proteins in response to 24 h exposure to 20 μM mifepristone as detected by 2D‐nanoLC‐mass spectrometry (MS/MS). The experiment was conducted 3 times in triplicate for each cell line.
Figure 3.
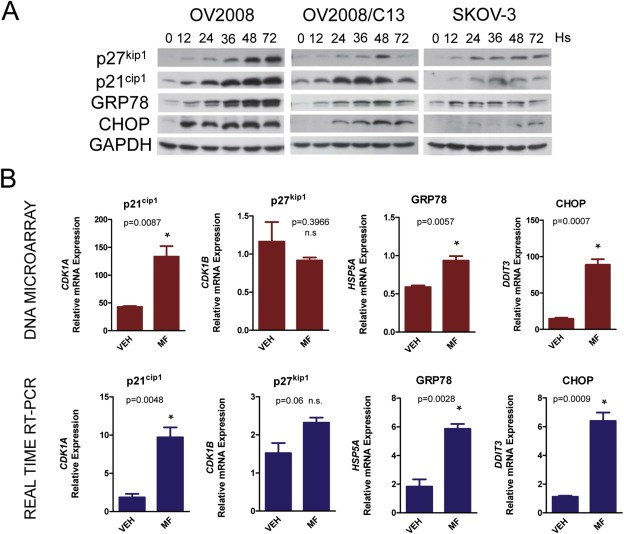
OV2008, OV2008/C13, and SKOV‐3 cells were exposed to 20 μM mifepristone for 12, 24, 36, 48, or 72 h (Hs). At the end of the experiments, cells were subjected to RNA and protein isolation. (A) Western blot analysis of cell cycle inhibitors p21cip1 and p27kip1, and of ER stress proteins GRP78 and CHOP. Results shown are representative of 3 separate experiments. (B) Comparison of the expression of the mRNA encoding for the indicated proteins as detected by DNA microarray and real‐time RT‐PCR in OV2008 cells. This experiment was done 3 times in triplicate. Statistical analysis was done using Student's t‐test.
3.3. Mifepristone triggers the unfolded protein response
Because mifepristone upregulated two key proteins involved in the ER stress response (CHOP and GRP78), we studied the expression of other proteins involved in ER stress‐related homeostasis, as well as compared the effect of mifepristone against those of two well‐known ER stressors, thapsigargin and tunicamycin. Thapsigargin causes ER stress by blocking calcium channels in the ER membrane (Chen et al., 2000), whereas tunicamycin is a glycosidase inhibitor, causing accumulation of non‐glycosylated proteins that are incapable of being exported from the ER (Miyake et al., 2000; Noda et al., 1999). Figure 4A shows that thapsigargin and tunicamycin caused remarkable increase in the abundance of GRP78, which was more marked than that caused by mifepristone. In addition, the three drugs were similarly potent in increasing the expression of the ER chaperone protein disulfide isomerase (PDI) and the sensor of the unfolded protein response (UPR), inositol‐requiring protein 1 (IRE1). None of the compounds, however, significantly changed the levels of other ER chaperones such as endoplasmic oxidoreductin‐1‐like protein (ERO1α) and calcium‐binding protein, calnexin (Gutierrez and Simmen, 2014). We further observed that the downstream transcription factor involved in the UPR, ATF4, was increased by the three drugs, but more potently by thapsigargin when compared to tunicamycin or mifepristone.
Figure 4.
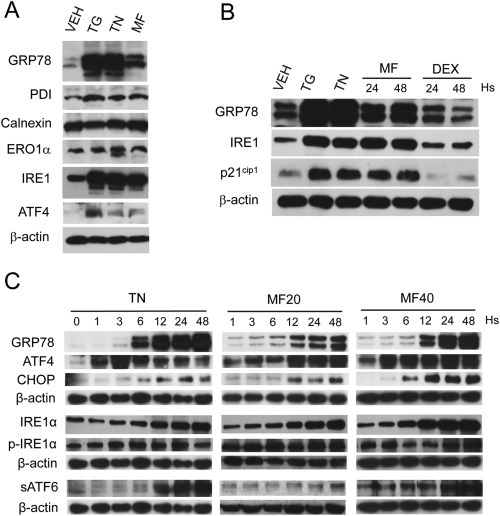
(A) OV2008 cells were exposed to 20 μM mifepristone (MF) for 48 h, or to 300 nM thapsigargin (TG) or 2 μg/ml tunicamycin (TN) for 24 h. (B) The induction of GRP78 by 20 μM MF for 24 h or 48 h followed that of ER stressors TG (300 nM) or TN (2 μg/ml) used for 24 h; the effect of MF was not mimicked by an equimolar concentration of a related steroid, dexamethasone (DEX). (C) Time‐course induction of UPR‐related proteins by 20 μM MF (MF20) or 40 μM MF (MF40) in comparison to the induction achieved by the known ER stressor TN (2 μg/ml). Results are representative of at least 2 independent experiments with a similar outcome.
To demonstrate that the effect of mifepristone was not steroid‐structure related, we compared the expression of the ER stress‐related proteins GRP78 and IRE1 and that of the cell cycle inhibitor p21cip1 in response to mifepristone, against their responses to the related synthetic steroid and glucocorticoid agonist, dexamethasone. Figure 4B shows that cell cycle arrest, anticipated by the increase in p21cip1, and ER stress response, marked by the upregulation of GRP78 and IRE1, were induced by mifepristone but not by an equimolar concentration of dexamethasone, indicating the specificity of the mifepristone‐induced ER stress response.
To maintain homeostasis when the load of unfolded proteins exceeds the folding capacity of the ER, GRP78 detaches from the ER membrane sensors PERK, IRE1, and ATF6, and unleashes the UPR (Clarke et al., 2014; Gardner et al., 2013; Nagelkerke et al., 2014). We performed a time‐course and dose–response experiment to determine the induction of various components of the UPR pathway by mifepristone. We assessed the activation of the pathway downstream of PERK by measuring the increases in transcription factors ATF4 and CHOP, which occurred following a same trend in mifepristone‐treated cells when compared to tunicamycin‐treated cells (Figure 4C, upper panels). The UPR sensor IRE1 was upregulated and phosphorylated upon 48 h of cellular exposure to tunicamycin or mifepristone (Figure 4C, middle panels). Finally, a third branch of the UPR pathway driven by the activation of ATF6 was activated by tunicamycin and mifepristone as demonstrated by the increase in the cleaved product of ATF6 formed upon trafficking of ATF6 from the ER to the Golgi apparatus, where it is cleaved to release the soluble transcription factor (sATF6) (Figure 4C, lower panels). The induction of the ER stress response by mifepristone was dose‐dependent (Figure 4C); for instance, cells treated with 20 μM mifepristone show increases in the expression of UPR related proteins that were smaller in magnitude (e.g. GRP78, CHOP, and IRE1α), or occurred later in time (e.g. ATF4, CHOP, and sATF6), when compared to cells treated with 40 μM mifepristone.
3.4. Mifepristone increases mRNA translation rate involving the eIF2α/ATF4 pathway
One homeostatic mechanism whereby the UPR controls ER stress as a consequence of accumulation of misfolded proteins is the transient decline in the rate of mRNA translation (Hetz, 2012; Nagelkerke et al., 2014). However, when we studied mRNA translation rate by labeling peptides with puromycin, we found that mifepristone caused a time‐related increase in protein synthesis lasting approximately 36 h (Figure 5A). Such increase in global protein puromycylation was dose‐dependent up to 5 μM mifepristone, reaching a plateau at doses of 10 and 20 μM. The accumulation of GRP78, however, kept increasing with the dose of mifepristone (Figure 5B).
Figure 5.
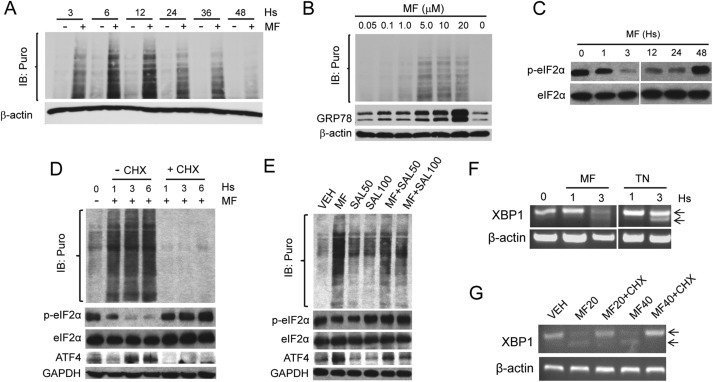
Mifepristone (MF) increases the incorporation of puromycin (Puro) in newly synthesized peptides. (A) Time‐course upon treatment with 20 μM MF. (B) Dose‐response after treatment with MF for 24 h. (C) Effect of MF on the phosphorylation of eIF2α on Serine 51 (p‐eIF2α). (D) Effect of cycloheximide (CHX) on MF‐induced puromycylation and ATF4 induction; CHX (10 μg/ml) was added to the culture concurrently with 20 μM MF for the indicated times. (E) Salubrinal 50 μM (SAL50) or 100 μM (SAL100) and MF (20 μM) were added either individually or concurrently for 3 h. Cultures in A,B, D and E were pulsed with 1 μM puromycin for 30 min at 37 °C before stopping the experiments, and puromycylated proteins were detected with the mouse antibody clone 12D10. IB; immunoblot. (F) Effect of 20 μM MF on the splicing of XPB1 mRNA assessed by RT‐PCR; TN, tunicamycin (2 μg/ml). Arrows indicate total and spliced XPB1 mRNA variants. (G) Splicing of XPB1 mRNA in ovarian cancer cells co‐incubated for 3 h with MF and CHX (10 μg/ml); MF20, 20 μM; MF40, 40 μM. All results presented were performed in OV2008 cells and are representative of 2 or 3 independent experiments that had a similar outcome.
Polypeptide chain translation initiator factor eIF2α is a limiting factor for protein synthesis under conditions of cellular stress based on the capacity of its alpha subunit to become phosphorylated on serine 51 (Ser‐51) (Koromilas, 2015), thus limiting the availability of eIF2α needed for initiation of translation (Bhat et al., 2015). We show that ovarian cancer cells have elevated phosphorylation of eIF2α on Ser‐51 (p‐eIF2α), which was rapidly yet temporarily diminished by mifepristone, without affecting total levels of eIF2α, and concurrently with an increase in puromycin incorporation (Figure 5C and D). Mifepristone also increased the expression of ATF4 (Figure 5D and E), which is known to be induced more efficiently under conditions of ER stress (Koromilas, 2015; Nagelkerke et al., 2014). Increased puromycylation, dephosphorylation of eIF2α on Ser‐51, and induction of ATF4 by mifepristone, were all prevented by the protein elongation inhibitor cycloheximide (Schneider‐Poetsch et al., 2010) (Figure 5D). To study whether mifepristone augments puromycylation by increasing eIF2α availability for translation initiation, we co‐incubated ovarian cancer cells with mifepristone and salubrinal, a small molecule inhibitor of the serine/threonine protein phosphatase that removes Ser‐51 phosphorylation from eIF2α (Boyce et al., 2005). Figure 5E shows that salubrinal prevented mifepristone‐induced Ser‐51 dephosphorylation of eIF2α while diminishing mifepristone‐induced puromycylation and ATF4 induction. Altogether, results shown in Figure 5 (panels A–E) suggest that mifepristone increases mRNA translation rate by targeting the eIF2α/ATF4 signaling pathway.
3.5. The increase in protein synthesis induced by mifepristone contributes to the unfolded protein response
Because protein synthesis was induced by mifepristone in association with induction of the UPR, we examined whether such increase in mRNA translation rate was related to the causation of ER stress and the unleashing of the UPR. We exposed ovarian cancer cells to mifepristone and measured the splicing of XBP1 mRNA as a non‐translatable readout of the UPR; this splicing occurs as a consequence of the RNAse activity of activated IRE1α, and leads to the formation of a potent transcriptional activator (XPB1s) that induces the expression of ER chaperones and ERAD proteins (Ron and Walter, 2007). Figure 5F shows that mifepristone triggered a time‐course dependent splicing of XPB1 mRNA similar to that caused by the well‐known ER stressor tunicamycin. Such splicing was, however, abrogated when the cells were co‐incubated with cycloheximide (Figure 5G), hence indicating that protein synthesis is required for MF‐induced UPR.
3.6. Mifepristone increases autophagic flux in ovarian cancer cells
It has been shown that induction of autophagy is a mechanism of homeostasis that facilitates the elimination of long‐lived proteins (Wang and Terpstra, 2013). Autophagy is particularly active in response to ER stress and involves the catabolism of proteins within double‐membrane vesicles, the autophagosomes, which fuse with lysosomes forming the autolysosomes. In the short term, this mechanism allows for cell survival, whereas in the long term, it leads to cell death (Platini et al., 2010). We studied the dynamics of the process of autophagy by assessing LC3II levels and autophagic flux. LC3 proteins are cleaved by Atg4 generating LC3‐I, which conjugates with phosphatidylethanolamine giving rise to a lipidated product termed LC3‐II. As LC3‐II is degraded in autolysosomes, its level is widely used as a marker of autophagy (Barth et al., 2010). However, increase in LC3‐II levels detected by western blot may indicate either increased autophagy induction or impaired autophagosome removal. Hence, to assess if mifepristone‐associated ER stress involved an increase in autophagic flux, we studied LC3‐II turnover in the presence or absence of the lysosome degradation inhibitor bafilomycin A1. If autophagy processing is increased by mifepristone, then LC3‐II levels should increase further in the presence of the lysosome degradation inhibitor, as the transit of LC‐3II through the autophagy pathway will be inhibited (Zhang et al., 2013). Mifepristone was capable of increasing the levels of LC3‐II in a dose‐dependent manner. Yet, such increase was enhanced remarkably by bafilomycin A1 (Figure 6A). Furthermore, when mifepristone was combined with the selective eIF2α dephosphorylation inhibitor salubrinal (Boyce et al., 2005), the autophagic flux was substantially abrogated (Figure 6A). GRP78 accumulated in response to mifepristone, but not in response to bafilomycin A1 (Figure 6A). Salubrinal ameliorated the increase in GRP78 induced by mifepristone. Moreover, salubrinal prevented the loss of viability induced by a lethal concentration of mifepristone (Figure 6B).
Figure 6.
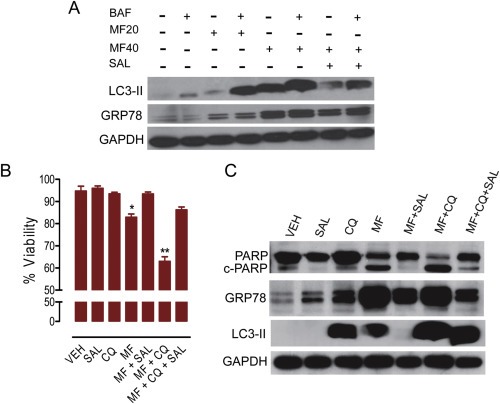
OV2008 cells were incubated for 48 h in the presence of salubrinal (SAL, 100 μM), chloroquine (CQ, 40 μM), mifepristone (MF, 40 μM), or their combinations. Viability (B) was assayed using microcytometry as previously described in detail (Freeburg et al., 2009b), whereas protein expression (A, C) was studied by western blot using the indicated antibodies. BAF = bafilomycin A1 (100 nM, added 1 h before ending the experiment). *p < 0.05 vs. VEH; **p < 0.01 vs. MF (one‐way ANOVA followed by Newman–Keuls Multiple Comparison Test). Results are representative of at least 2 independent experiments with a similar outcome.
3.7. Chloroquine increases mifepristone‐induced cytotoxicity
Chloroquine inhibits lysosomes by increasing the pH of the lysosomal compartment, thus preventing the activity of lysosomal acid proteases and causing autophagosomes to accumulate; it is widely used in the clinic to treat malaria and other medical conditions (Solomon and Lee, 2009). When we exposed ovarian cancer cells to lethal doses of mifepristone, usage of chloroquine increased further the number of dying cells (Figure 6B) in association with total cleavage of the marker of cell death, cleaved PARP (Galluzzi et al., 2012) (Figure 6C). The interaction amongst mifepristone and chloroquine was synergistic for the combinations of 10, 20 or 40 μM mifepristone with 40 μM chloroquine (CI = 0.613, 0.565, and 0.424, respectively). The lethality caused by the combination of mifepristone with chloroquine—and assessed by measuring viability and cleavage of PARP—was reversed by the presence of salubrinal, which also reduced MF‐induced GRP78 levels (Figure 6B and C).
3.8. Mifepristone potentiates the toxicity of proteasome inhibitors
One canonical mechanism whereby the stress of the ER is relieved is by enhancing ER‐associated degradation (ERAD) via the ubiquitin proteasome system (UPS), in which misfolded proteins are recognized and retro‐translocated to the cytoplasm where they are ubiquitinated and targeted for proteasome degradation (Brodsky, 2012; Vembar and Brodsky, 2008). We first studied whether the ER stress induced by mifepristone associated with a compensatory increase in the activity of the UPS. We assessed the accumulation of poly‐ubiquitinated proteins as a surrogate marker of proteasome activity (Bedford et al., 2011; Wang and Terpstra, 2013). The level of poly‐ubiquitinated proteins did not change much when comparing cells treated with mifepristone against cells treated with tunicamycin, whereas the overall ubiquitination of drug‐treated cells was not different from that depicted in vehicle‐treated cells, suggesting that neither tunicamycin nor mifepristone affect the activity of the proteasome (Figure 7A).
Figure 7.
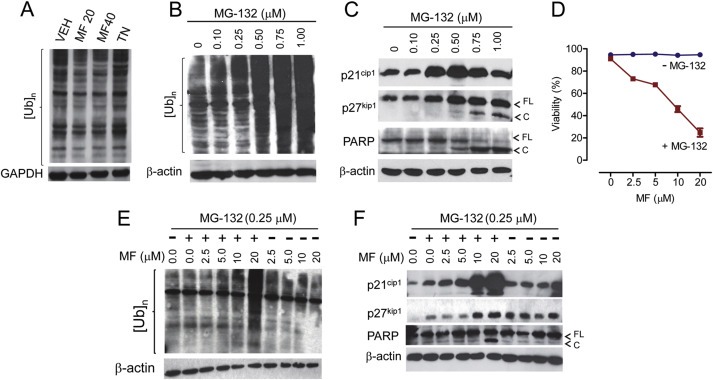
(A) Neither mifepristone (MF) (MF20, 20 μM; MF40, 40 μM) nor tunicamycin (TN; 2 μg/ml) impact the processing of poly‐ubiquitinated proteins ([Ub]n) in OV2008 cells. Concentrations of MG‐132—an inhibitor of the proteasome—0.5 μM or higher cause accumulation of [Ub]n (B) and lethality (C). (D) Reduced viability of OV2008 ovarian cancer cells exposed for 72 h to a range of cytostatic concentrations of MF and a fixed (0.25 μM) cytostatic dose of MG‐132 as determined using microcapillary cytometry. Lethal interaction between MG‐132 and MF for 72 h involves accumulation of [Ub]n (E), accumulation of p21cip1, and cleavage of PARP (FL = full length; C = cleaved) (F). GAPDH or β‐actin were used as loading controls. Results are representative of at least 2 independent experiments with a similar outcome.
We next rationalized that by causing ER stress, mifepristone may interact with proteasome inhibitors blocking ovarian cancer cell growth; this is because lack of ERAD/26S proteasome function would eliminate one critical relief mechanism for an ER overwhelmed by mifepristone‐induced accumulation of misfolded/unfolded proteins. We exposed cells to various concentrations of the proteasome inhibitor MG‐132 and found that doses up to 0.25 μM did not kill the cells and did not accumulate poly‐ubiquitinated proteins whereas doses of 0.50 μM and higher were sufficient to block the proteasome and to kill the cells as demonstrated by the accumulation of poly‐ubiquitinated proteins (Figure 7B) and cleavage of PARP (Figure 7C). We then exposed ovarian cancer cells to various concentrations of mifepristone in the presence or absence of a non‐killing concentration of MG‐132. Results in Figure 7D show that, while in a range of 2.5–20 μM mifepristone did not kill the cells, when combined with a non‐lethal dose of MG‐132 (0.25 μM), it triggered cell death in a dose‐dependent manner. At the 0.25 μM dose used, MG‐132 alone did not accumulate poly‐ubiquitinated proteins, but it did so when it was combined with 20 μM mifepristone (Figure 7E). This result suggests that the ER stress induced by mifepristone allowed a sub‐optimal dose of MG‐132 to cause proteasome functional insufficiency. The toxicity of this drug combination was further evidenced by the cleavage of PARP and p21cip1 (Galluzzi et al., 2012) (Figure 7F).
MG‐132 is useful for laboratory investigation but has not reached the clinic; hence, we tested the previous concept using bortezomib, approved for the treatment of multiple myeloma and mantle cell lymphoma (Buac et al., 2013). We found that 5 nM bortezomib was sufficient to kill ∼60% ovarian cancer cells when combined with a cytostatic dose of mifepristone (Figure 8A). The interaction among mifepristone and bortezomib was synergistic for the combinations of 5 and 10 nM bortezomib with 20 μM mifepristone (CI = 0.389 and 0.685, respectively).
Figure 8.
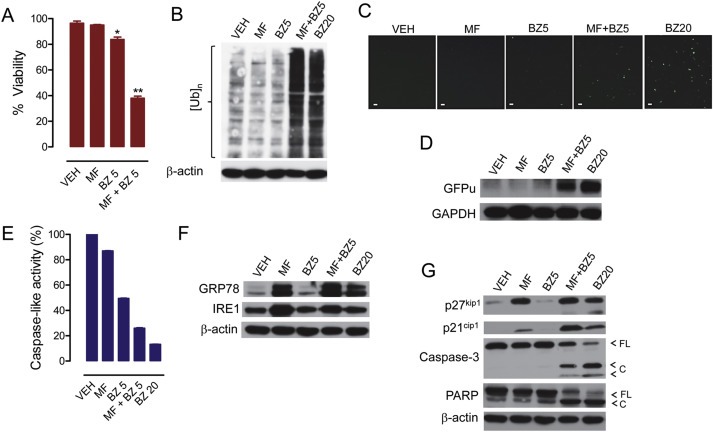
OV2008 cells were exposed for 48 h to 20 μM mifepristone (MF), 5 nM bortezomib (BZ5), 20 nM BZ (BZ20), or the combination of 20 μM MF with 5 nM BZ, and viability was assessed (A). Proteins were isolated, electrophoresed, and subjected to western blot for poly‐ubiquitination (B), ER stress‐related proteins (F), and cyclin dependent kinase inhibitors p21cip1 and p27kip1, caspase‐3, and PARP (G). In a separate experiment, the peptidergic activity of the proteasome was measured in response to similar treatments (E). To further assess the activation of the proteasome in response to cytostatic doses of BZ (5 nM) and MF (20 μM), the cells were transfected with a plasmid expressing GFPu, a green fluorescence protein with carboxyl fusion of an ubiquitination signal sequence (degron CL1); accumulation of GFPu correlates with a reduction in the activity of the proteasome. Panel (C) shows the accumulation of GFPu via fluorescence microscopy (scale bar = 50 μm), whereas panel (D) shows GFPu accumulation by western blot analysis. Results are representative of at least 2 independent experiments with a similar outcome.
Five nM bortezomib or 20 μM mifepristone alone did not accumulate poly‐ubiquitinated proteins. Such accumulation, however, occurred when the drugs were combined (Figure 8B). The inhibition of the activity of the proteasome by the combination of 5 nM bortezomib with 20 μM mifepristone was further confirmed by the accumulation of the surrogate substrate GFPu (Figure 8C and D) and the inhibition of the caspase‐like peptidase activity of the proteasome (Figure 8E). Such effects observed with the drug combination (i.e. 5 nM bortezomib/20 μM mifepristone) were similar to those observed when bortezomib was used alone but at an effective proteasome inhibitory concentration of 20 nM, as demonstrated by the accumulation of poly‐ubiquitinated proteins (Figure 8B) and of GFPu (Figure 8C and D), and by the decline in the caspase‐like activity of the 20S proteasome (Figure 8E). Also at the 20 nM concentration, bortezomib monotherapy induced ER stress judged by the accumulation of GRP78 (Figure 8F) and triggered cell death (Figure 8G).
Mifepristone alone, as previously described, caused accumulation of GRP78 and IRE1, whereas bortezomib, at the sub‐optimal dose of 5 nM did not (Figure 8F). Cells exposed to the combination of 5 nM bortezomib with 20 μM mifepristone depicted as much GRP78 and IRE1 as cells receiving mifepristone alone (Figure 8F). The combination of 5 nM bortezomib with 20 μM mifepristone induced accumulation of p21cip1 beyond that caused by mifepristone alone, while inducing cleavage of caspase‐3 and of downstream substrate PARP (Figure 8G). These results indicate that when mifepristone and bortezomib are combined, the cells undergo caspase‐associated cell death.
Salubrinal—the selective eIF2α dephosphorylation inhibitor and cytoprotector against ER stressors (Boyce et al., 2005; Gao et al., 2013; Gong et al., 2012)—rescued the cells from the toxicity of high‐dose mifepristone as shown by cell survival (Figure 9A) and cleavages of caspase‐3 and PARP (Figure 9C), and prevented mifepristone‐induced accumulation of GRP78, CHOP and LC3II (Figure 9C and D). However, in cells subjected to the toxicity of the combination mifepristone/bortezomib, salubrinal only in part ameliorated lethality (Figure 9A), slightly reduced the levels of GRP78 (Figure 9C) and CHOP (Figure 9D), but did not prevent the accumulation of poly‐ubiquitinated proteins (Figure 9B).
Figure 9.
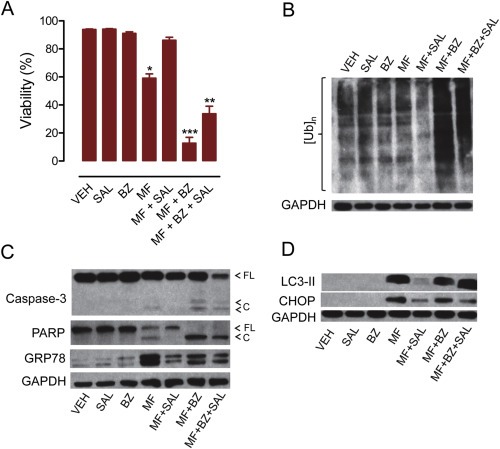
OV2008 cells were incubated with mifepristone (MF; 40 μM), salubrinal (100 μM), bortezomib (5 nM), or a combination of the two for 48 h. (A) Viability was detected by cytometry of cells treated with the depicted drugs. *p < 0.05 vs. vehicle; **p < 0.01 vs. MF + BZ; ***p < 0.001 vs. vehicle (one‐way ANOVA followed by Tukey's Multiple Comparison Test). (B) Poly‐ubiquitination ([Ub]n) of proteins as detected by western blot. (C) Expression of caspase‐3 and its cleavage forms, the caspase‐3 downstream substrate PARP, and ER master chaperone GRP78. (D) Accumulation of LC3‐II caused by MF and its prevention by the small molecule salubrinal (SAL). Also shown is the transcription factor CHOP upregulated by MF. Results are representative of at least 2 independent experiments with a similar outcome.
4. Discussion
Cancer cells, when compared to non‐cancer cells, operate with increased expression of ER stress‐related proteins and a prevalent UPR, a phenomenon coined as ER aggravation (Schonthal, 2013) or UPR addiction (Nagelkerke et al., 2014); this allows the cancer cells to heavily rely on the UPR for survival in the environment within which they usually proliferate: reduced nutrients, acidosis, energy deficiency, and low oxygen tension (hypoxia) (Giampietri et al., 2015; Healy et al., 2009; Nagelkerke et al., 2014; Schonthal, 2013). Two approaches are under exploration to target the ER in cancer cells: (i) inhibition of the compensatory UPR components usually hyperactive in malignant cells (e.g. developing GRP78 inhibitors); and (ii) further increasing ER stress using ER stress aggravators. In this work we provide evidence that mifepristone is a potent stressor of the ER in ovarian cancer cells—hence, an ER stress aggravator—thus supporting the second therapeutic approach.
For the first time in ovarian cancer cells, we show that mifepristone induces ER stress as suggested by the activation of all arms of the UPR. We demonstrate that mifepristone, despite not affecting the activity of the proteasome directly, potentiates the effect of proteasome inhibitors in killing ovarian cancer cells. We showed this in ovarian cancer cells with functional p53 (7, 8), and in p53 deficient ovarian cancer cells (Figure S2). Likewise, other compounds have been shown to potentiate the action of proteasome inhibitors when increasing ER stress. For instance, the toxicity of bortezomib was enhanced by ER stressors and dysregulators of the UPR such as TNF in colon cancer cells (Nowis et al., 2007), the multi‐kinase inhibitor sorafenib in hepatic cancer cells (Honma and Harada, 2013), eayarestatin—an inhibitor of the ER associated ATPase p97 that retro‐translocates proteins from the ER to the proteasome—in cervical cancer cells (Brem et al., 2013), Bcl‐2 antagonists in diffuse lymphocytic B‐cell lymphoma (Dasmahapatra et al., 2009), tunicamycin and thapsigargin in pancreatic cancer cells (Nawrocki et al., 2005), and photodynamic therapy in cervical cancer cells (Szokalska et al., 2009). In ovarian cancer cells, bortezomib increased the toxicity of TRAIL (Saulle et al., 2007), histone deacetylase inhibitors (Bazzaro et al., 2008; Fang et al., 2011), carboplatin (Al‐Eisawi et al., 2013), and the natural compound indole‐3‐carbinol (Taylor‐Harding et al., 2012). Here we show that mifepristone‐induced ER stress aggravation, combined with proteasome inhibition, provides preclinical therapeutic advantage towards ovarian cancer cells—i.e. lethality—when compared to either compound used individually.
We also report for the first time that mifepristone increases autophagic flux in ovarian cancer cells. Increase in autophagy has been shown to be a homeostatic mechanism that balances the accumulation of unfolded or misfolded proteins in the ER (Nagelkerke et al., 2014). For instance, ER stress and autophagy were observed in human endothelial cells in response of oxidized low‐density lipoproteins (Muller et al., 2011) and in mouse embryo fibroblasts and human lung cancer cells in response to the protease inhibitor nelfinavir (Johnson et al., 2015). The increase in autophagic flux by mifepristone may be a compensatory mechanism to diminish ER workload; this is because mifepristone‐induced LC‐3II accumulation was completely abrogated by salubrinal, which is an inhibitor of eIF2α dephosphorylation that protects cells from the toxicity caused by several ER stressors (Boyce et al., 2005; Gong et al., 2012; Matsuoka and Komoike, 2015). Furthermore, we show that induction of autophagy after mifepristone may have a survival purpose as the cells die when induction of autophagy is prevented by adding the lysosome inhibitor chloroquine. Such toxicity, however, seems to involve the eIF2α/ATF4 branch of the UPR, as it was reversed by salubrinal. Thus, we can conclude that ovarian cancer cells respond to mifepristone with ER stress, and, downstream of it, autophagy as a manner to limiting the toxicity of the synthetic steroid.
Another novel finding in this work was the acute increase in mRNA translation rate induced by mifepristone. Usually, cells that are under ER stress temporarily down‐regulate global translation, while unleashing the translation of a subset of mRNAs with upstream open reading frames (uORFs) in their 5′ untranslated region (5′UTR) and resistant to eIF2α repression (Andreev et al., 2015); such are the cases of transcripts encoding for ATF4, CHOP, and TRIB3 (Barbosa et al., 2013), or transcripts with intra‐ribosomal entry sites (IRES) in their 5′UTR that are translated via a cap‐independent mechanism, such as the transcript encoding for the Cdk inhibitor p27kip1 (Gopfert et al., 2003). This phenomenon has the purpose of alleviating the ER from accumulated misfolded and/or unfolded proteins, thus preventing ER‐mediated cell death due to proteotoxicity (Hetz, 2012). In our studies, however, the acute increase in protein synthesis by mifepristone in ovarian cancer cells was accountable for the UPR triggered by the steroid, since mifepristone‐induced XPB1 mRNA cleavage was prevented by inhibiting protein synthesis. Thus, the increase in protein synthesis rate upon mifepristone exposure may cause ER protein overload leading to ER stress, which it is then ameliorated by a compensatory UPR.
The increase in mRNA translation rate by mifepristone involves the temporary reduction of eIF2α phosphorylation on Ser‐51 (p‐eIF2α). This result is relevant as the pharmacological inhibition of this particular phosphorylation site was proposed to be key to impair tumor growth using combination anti‐cancer agents, particularly in cancer cells having increased basal eIF2α Ser‐51 phosphorylation (Koromilas, 2015; Koromilas and Mounir, 2013), as it is the case for the ovarian cancer cells studied in the present work. Supporting this concept, the small molecule GSK2656157, which is an ATP‐competitive inhibitor of the activity of PERK (Axten et al., 2012)—the kinase that phosphorylates eIF2α on Ser‐51 under conditions of ER stress—significantly reduced the growth of tumors induced by human pancreatic and multiple myeloma cancer cells in immunosuppressed mice (Atkins et al., 2013).
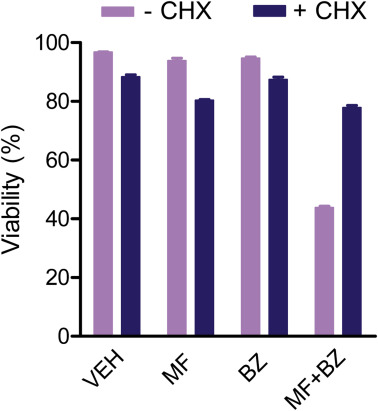
Investigations support the idea that increased protein synthesis in response to ER stress may induce cell death as a consequence of the simultaneous transcriptional induction of ATF4 and CHOP, ATP depletion, increase in oxidative stress, and expression of genes that promote further load of proteins within the ER (Han et al., 2013; Marciniak et al., 2004). Here we show that ATF4 is induced in response to mifepristone, and it is followed by the up‐regulation of CHOP, a target of ATF4. Another evidence for a positive contribution of increased protein synthesis to mifepristone‐induced toxicity is the fact that cycloheximide significantly mitigated the lethality induced by the combination mifepristone/bortezomib (Figure S3). Our results provide further support to the chemotherapeutic utility of emerging compounds that de‐repress protein translation in the face of increased levels of ER stress (Sidrauski et al., 2013), which is a condition under which most cancer cells operate.
The activation of the ER stress response by mifepristone is further evidenced by the significant upregulation of CHAC1 in all cell lines studied. This gene was defined as a cation transport regulator‐like protein 1, acting in ER‐stress mediated cell death downstream of ATF‐4 and CHOP (Mungrue et al., 2009). Supporting an active role of ATF4 and CHOP in the killing of ovarian cancer cells when mifepristone was used at lethal doses, or when it was used at cytostatic doses but combined with either a proteasome inhibitor or an autophagy inhibitor, is the remarkable capacity of mifepristone to induce expression of TRIB3, which was described as an ER stress gene induced by ATF4‐CHOP, and demonstrated to be involved in cell death (Ohoka et al., 2005). Another piece of evidence supporting the effect of mifepristone‐induced eIF2α‐ATF4‐CHOP pathway in the phenotype of the cells, is the induction of autophagy as ATF4‐CHOP are required for the transcription of several genes involved in the formation and functioning of the autophagosome upon amino acid starvation, ER stress, or hypoxia (B'Chir et al., 2013; Rzymski et al., 2010).
In addition to the genes related to the ER stress response, we also discovered genes differentially expressed by mifepristone and encoding for structural proteins as well as proteins involved in various functions within the cells, including metabolic and cell communication processes. Using the known proteins affected by mifepristone and involved in cell cycle arrest which we previously discovered (Cdk2 and its regulators), and adding the genes differentially expressed by mifepristone in the three ovarian cancer cell lines regardless of sensitivity to platinum and p53 background (see Table 1), we performed a pathway building analysis that led to the development of several predicted regulatory pathways that should be the subject of further hypothesis‐driven studies (Figure S4 and Table S8). Based on published literature, the pathway building analysis predicted the interconnection among all genes utilized for the analysis. For instance, the analysis projected that upregulation of the calcium binding protein S100P leads to downregulation of Bcl‐2 (Shimamoto et al., 2014), which we found to be true when mifepristone induced cell death in combination with PI3K inhibitors (Wempe et al., 2013). S100P is predicted to increase expression of ezrin, which is confirmed in our results in SKOV‐3 cells (see Table S6). Ezrin links the plasma membrane and the actin cytoskeleton (Algrain et al., 1993), which we previously demonstrated becomes dysregulated by mifepristone (Brandhagen et al., 2013). It is likely that ezrin also plays a role in mediating mifepristone‐induced dysregulation of cellular adhesion, migration, and actin cytoskeleton organization. This is because ezrin inhibits E‐cadherin (CDH1) (Hunter, 2004; Li et al., 2008) and, in this manner, can mediate the morphological alterations caused by mifepristone we previously described (Brandhagen et al., 2013). The pathway analysis also anticipated that upregulation of HSPA5 (GRP78) should negatively affect caveolin‐1 (CAV1) (Moon et al., 2015); indeed, CAV1 was shown to be downregulated by mifepristone (see Table S6). Caveolin‐1 may link mifepristone activity with the augmented autophagic flux as predicted in the pathway analysis by the regulation of proteins involved in autophagy (Figure S3). This is relevant considering that caveolin‐1 facilitates the interaction between Fas and LC3B (Tanaka et al., 2012), two proteins predicted to be affected by mifepristone. Two more predictions were the regulations between CHOP (DDIT3) and the pseudokinase TRIB3 (Ohoka et al., 2007), and the upregulation of CHAC1 by CHOP (Mungrue et al., 2009) we show in this study. Finally, another evidence relevant to our studies is that ZNF488, shown to enhance migration and invasion of nasopharyngeal cancer cells (Zong et al., 2016), was significantly downregulated by mifepristone in all cell lines studied (see Table 1). Thus, it is possible that ZNF488 mediates the inhibition of migration and invasion induced by mifepristone in ovarian cancer cells (our unpublished observations). The predictive pathways set in motion by mifepristone and developed when combined hypothesis‐driven and discovery‐based approaches (depicted in Figure S4) will need validation in future investigations to unleash their roles in mifepristone‐mediated effects towards ovarian cancer cells, such as cell growth inhibition, activation of the UPR, modification in morphology, cytoskeleton dynamics, adhesion and migration of the cells, and activation of autophagy.
Mifepristone has been shown to be an antiprogestin and an antiglucocorticoid agent; thus, it can be assumed that it drives its anti‐ovarian cancer effect impacting either progesterone receptors (PR) or glucocorticoid receptors (GR). With respect to PR, evidence shows that presence of ‘classical’ (nuclear) PR is not required for mifepristone to block the growth of various types of cancer cells, including ovarian cancer cells (Tieszen et al., 2011). The same concentrations of mifepristone we used to induce growth arrest in our previous work were used in the current study to induce ER stress in ovarian cancer cells expressing or not expressing PR. Thus, we rationalize that the presence of ‘classical’ PR is not required for the induction of the ER stress response and would not impact the usage of this drug for anti‐ovarian cancer therapy.
It is possible, however, that GR may drive the anti‐growth effect and ER stress induction by mifepristone. There are two isoforms of GR, GRα and GRβ. Evidence supports GRα as driving GR‐mediated transactivation activity, whereas GRβ functions as a dominant negative inhibitor of GRα (Taniguchi et al., 2010). Our laboratory has shown that mifepristone blocked growth of cancer cells that have negligible expression of GRα, including OVCAR‐3 ovarian cancer cells (Tieszen et al., 2011), suggesting that the presence of GRα may not be required for the anti‐proliferative action of mifepristone. It remains to be assessed if GRβ, which is ubiquitously present in all cancer cell lines we so far studied (Tieszen et al., 2011), and is able to bind mifepristone and transactivate genes in the absence of GRα (Kadmiel and Cidlowski, 2013; Lewis‐Tuffin et al., 2007), plays a role in the anti‐growth effect and ER stress response induced by the synthetic steroid.
The three drugs utilized in this work, mifepristone, chloroquine, and bortezomib, are already used in the clinic. Mifepristone is utilized to terminate early pregnancies (working as an antiprogestin) (Newhall and Winikoff, 2000) and to ameliorate hyperglycemia in patients with endogenous Cushing's syndrome (working as an antiglucocorticoid) (Fleseriu et al., 2012). Chloroquine is used to treat malaria (Solomon and Lee, 2009), and bortezomib is prescribed for the treatment of multiple myeloma and mantle cell lymphoma (Buac et al., 2013). Hence, the systemic toxicity and tolerability studies for these compounds have already been performed, making them valuable candidates for rapid repurposing for cancer therapy.
In summary, in this work we combined unbiased genomic and proteomic tools with hypothesis‐driven approaches to discover and validate the effect of mifepristone as a potent stressor of the ER leading to the induction of the UPR. We provide evidence that the UPR induced by mifepristone is mediated by an early surge in the rate of protein synthesis as a consequence of the increased availability of translation initiator factor eIF2α. We also show for the first time that mifepristone increases autophagic flux. Lastly, we demonstrate that blocking the UPS or the autophagy pathway in combination with mifepristone leads to cell death. The translational value of this study is that using mifepristone in combination with drug/s that dysregulate the protein quality control machinery of the cell, may cause sufficient cellular stress to tip cell fate toward proteotoxic cell death in otherwise non‐dividing ovarian cancer cells (Figure 10). The clinical relevance of this approach is that it can be used as a consolidation therapy for ovarian cancer patients while in remission, following frontline chemotherapy aiming to delay or to prevent recurrence.
Figure 10.
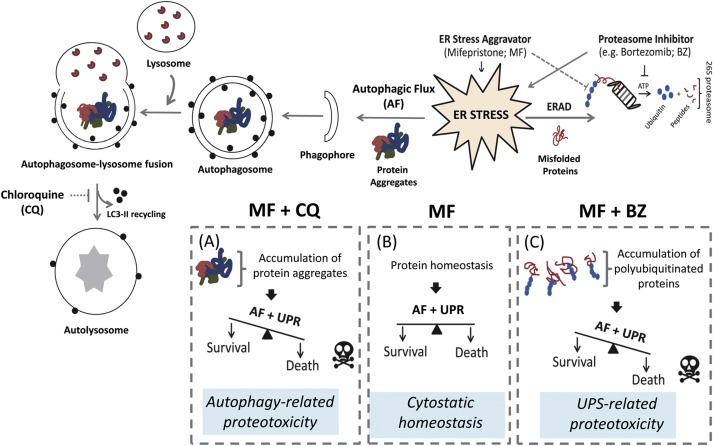
Schematic model whereby ER stress aggravation triggered by mifepristone (MF) (B) is potentiated by either proteasome inhibition (C) or lysosomal inhibition (A) leading to cell death. AF: autophagic flux; ERAD: ER associated degradation; UPR: unfolded protein response; UPS: ubiquitin proteasome system; CQ: chloroquine; BZ: bortezomib.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Figure S1 (A) Venn diagram displaying the number of transcripts up‐ or downregulated (>2 fold) after 24 h exposure to 20 μM mifepristone. (B) Number of transcripts induced (maroon) or repressed (green) by mifepristone in each cell line.
{kind=link}
Figure S2 Exposure of SKOV‐3 cells to various doses of bortezomib for 72 h in the absence (−) or presence (+) of 20 μM mifepristone (MF). Viability was assessed by microcapillary fluorescence cytometry.
{kind=link}
Figure S3 Protein synthesis inhibitor cycloheximide (CHX) mitigated OV2008 cell death induced by the combination treatment of bortezomib (BZ) and mifepristone (MF). Cells where cultured for 72 h in the presence of DMSO (VEH), 20 μM MF, 5 nM BZ, or the combination of 20 μM MF with 5 nM BZ. All groups were done in the continuous presence or absence of 10 μg/ml CHX. This experiment was repeated twice with a similar outcome.
{kind=link}
Figure S4 Pathway building analysis of hypothetical molecular networks regulated by mifepristone based on previously known molecules affected by the steroid, as well as those discovered using genomic and proteomic analysis in the present study. The analysis was performed using Pathway Studio®. The name description of each gene and gene products depicted in the diagram are described in Table S8. Symbols highlighted in green represent proteins previously discovered to be regulated by mifepristone. Symbols highlighted in yellow represent proteins discovered to be regulated by mifepristone in the current genomic and proteomic analysis (see Table 1). The meaning of the different symbols and types of interactions among proteins is also displayed.
{kind=link}
Acknowledgments
This research was supported by K22CA121991, ARRA Supplement K22CA121991‐S1, and R15CA164622 from the National Cancer Institute, NIH, 2 P20 RR016479 from NIH/NCRR SD‐BRIN, and by start‐up research funds from the Department of Pathology, Faculty of Medicine, McGill University. We are indebted to Millennium Pharmaceuticals for providing bortezomib (Velcade®) for preclinical research purposes.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.05.001.
Zhang Lei, Hapon Maria B., Goyeneche Alicia A., Srinivasan Rekha, Gamarra-Luques Carlos D., Callegari Eduardo A., Drappeau Donis D., Terpstra Erin J., Pan Bo, Knapp Jennifer R., Chien Jeremy, Wang Xuejun, Eyster Kathleen M., Telleria Carlos M., (2016), Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.05.001.
References
- Al-Eisawi, Z. , Beale, P. , Chan, C. , Yu, J.Q. , Huq, F. , 2013. Carboplatin and oxaliplatin in sequenced combination with bortezomib in ovarian tumour models. J. Ovarian Res. 6, 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algrain, M. , Turunen, O. , Vaheri, A. , Louvard, D. , Arpin, M. , 1993. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J. Cell Biol. 120, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev, D.E. , O'Connor, P.B. , Fahey, C. , Kenny, E.M. , Terenin, I.M. , Dmitriev, S.E. , Cormican, P. , Morris, D.W. , Shatsky, I.N. , Baranov, P.V. , 2015. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. Elife. 4, e03971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins, C. , Liu, Q. , Minthorn, E. , Zhang, S.Y. , Figueroa, D.J. , Moss, K. , Stanley, T.B. , Sanders, B. , Goetz, A. , Gaul, N. , Choudhry, A.E. , Alsaid, H. , Jucker, B.M. , Axten, J.M. , Kumar, R. , 2013. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Axten, J.M. , Medina, J.R. , Feng, Y. , Shu, A. , Romeril, S.P. , Grant, S.W. , Li, W.H. , Heerding, D.A. , Minthorn, E. , Mencken, T. , Atkins, C. , Liu, Q. , Rabindran, S. , Kumar, R. , Hong, X. , Goetz, A. , Stanley, T. , Taylor, J.D. , Sigethy, S.D. , Tomberlin, G.H. , Hassell, A.M. , Kahler, K.M. , Shewchuk, L.M. , Gampe, R.T. , 2012. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. [DOI] [PubMed] [Google Scholar]
- B'Chir, W. , Maurin, A.C. , Carraro, V. , Averous, J. , Jousse, C. , Muranishi, Y. , Parry, L. , Stepien, G. , Fafournoux, P. , Bruhat, A. , 2013. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa, C. , Peixeiro, I. , Romão, L. , 2013. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 9, e1003529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth, S. , Glick, D. , Macleod, K.F. , 2010. Autophagy: assays and artifacts. J. Pathol. 221, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast, R.C. , 2011. Molecular approaches to personalizing management of ovarian cancer. Ann. Oncol. 22, (Suppl. 8) viii5–viii15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast, R.C. , Hennessy, B. , Mills, G.B. , 2009. The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer. 9, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzaro, M. , Lin, Z. , Santillan, A. , Lee, M.K. , Wang, M.C. , Chan, K.C. , Bristow, R.E. , Mazitschek, R. , Bradner, J. , Roden, R.B. , 2008. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 14, 7340–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford, L. , Lowe, J. , Dick, L.R. , Mayer, R.J. , Brownell, J.E. , 2011. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence, N.F. , Sampat, R.M. , Kopito, R.R. , 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bhat, M. , Robichaud, N. , Hulea, L. , Sonenberg, N. , Pelletier, J. , Topisirovic, I. , 2015. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 14, 261–278. [DOI] [PubMed] [Google Scholar]
- Boyce, M. , Bryant, K.F. , Jousse, C. , Long, K. , Harding, H.P. , Scheuner, D. , Kaufman, R.J. , Ma, D. , Coen, D.M. , Ron, D. , Yuan, J. , 2005. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 307, 935–939. [DOI] [PubMed] [Google Scholar]
- Brandhagen, B.N. , Tieszen, C.R. , Ulmer, T.M. , Tracy, M.S. , Goyeneche, A.A. , Telleria, C.M. , 2013. Cytostasis and morphological changes induced by mifepristone in human metastatic cancer cells involve cytoskeletal filamentous actin reorganization and impairment of cell adhesion dynamics. BMC Cancer. 13, 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem, G.J. , Mylonas, I. , Bruning, A. , 2013. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol. Oncol. 128, 383–390. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L. , 2012. Cleaning up: ER-associated degradation to the rescue. Cell. 151, 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buac, D. , Shen, M. , Schmitt, S. , Kona, F.R. , Deshmukh, R. , Zhang, Z. , Neslund-Dudas, C. , Mitra, B. , Dou, Q.P. , 2013. From bortezomib to other inhibitors of the proteasome and beyond. Curr. Pharm. Des. 19, 4025–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L.Y. , Chiang, A.S. , Hung, J.J. , Hung, H.I. , Lai, Y.K. , 2000. Thapsigargin-induced grp78 expression is mediated by the increase of cytosolic free calcium in 9L rat brain tumor cells. J. Cell. Biochem. 78, 404–416. [DOI] [PubMed] [Google Scholar]
- Clarke, H.J. , Chambers, J.E. , Liniker, E. , Marciniak, S.J. , 2014. Endoplasmic reticulum stress in malignancy. Cancer Cell. 25, 563–573. [DOI] [PubMed] [Google Scholar]
- Coleman, R.L. , Monk, B.J. , Sood, A.K. , Herzog, T.J. , 2013. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 10, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasmahapatra, G. , Lembersky, D. , Rahmani, M. , Kramer, L. , Friedberg, J. , Fisher, R.I. , Dent, P. , Grant, S. , 2009. Bcl-2 antagonists interact synergistically with bortezomib in DLBCL cells in association with JNK activation and induction of ER stress. Cancer Biol. Ther. 8, 808–819. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Dong, X. , Liu, J. , Zheng, H. , Glasford, J.W. , Huang, W. , Chen, Q.H. , Harden, N.R. , Li, F. , Gerdes, A.M. , Wang, X. , 2004. In situ dynamically monitoring the proteolytic function of the ubiquitin-proteasome system in cultured cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 287, H1417–H1425. [DOI] [PubMed] [Google Scholar]
- Eyster, K.M. , Brannian, J.D. , 2009. Gene expression profiling in the aging ovary. Methods Mol. Biol. 590, 71–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyster, K.M. , Klinkova, O. , Kennedy, V. , Hansen, K.A. , 2007. Whole genome deoxyribonucleic acid microarray analysis of gene expression in ectopic versus eutopic endometrium. Fertil. Steril. 88, 1505–1533. [DOI] [PubMed] [Google Scholar]
- Fang, Y. , Hu, Y. , Wu, P. , Wang, B. , Tian, Y. , Xia, X. , Zhang, Q. , Chen, T. , Jiang, X. , Ma, Q. , Xu, G. , Wang, S. , Zhou, J. , Ma, D. , Meng, L. , 2011. Synergistic efficacy in human ovarian cancer cells by histone deacetylase inhibitor TSA and proteasome inhibitor PS-341. Cancer Invest. 29, 247–252. [DOI] [PubMed] [Google Scholar]
- Fleseriu, M. , Biller, B.M. , Findling, J.W. , Molitch, M.E. , Schteingart, D.E. , Gross, C. , Investigators, S.S. , 2012. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J. Clin. Endocrinol. Metabol. 97, 2039–2049. [DOI] [PubMed] [Google Scholar]
- Freeburg, E.M. , Goyeneche, A.A. , Seidel, E.E. , Telleria, C.M. , 2009. Resistance to cisplatin does not affect sensitivity of human ovarian cancer cell lines to mifepristone cytotoxicity. Cancer Cell Int. 9, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeburg, E.M. , Goyeneche, A.A. , Telleria, C.M. , 2009. Mifepristone abrogates repopulation of ovarian cancer cells in between courses of cisplatin treatment. Int. J. Oncol. 34, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi, L. , Vitale, I. , Abrams, J.M. , Alnemri, E.S. , Baehrecke, E.H. , Blagosklonny, M.V. , Dawson, T.M. , Dawson, V.L. , El-Deiry, W.S. , Fulda, S. , Gottlieb, E. , Green, D.R. , Hengartner, M.O. , Kepp, O. , Knight, R.A. , Kumar, S. , Lipton, S.A. , Lu, X. , Madeo, F. , Malorni, W. , Mehlen, P. , Nunez, G. , Peter, M.E. , Piacentini, M. , Rubinsztein, D.C. , Shi, Y. , Simon, H.U. , Vandenabeele, P. , White, E. , Yuan, J. , Zhivotovsky, B. , Melino, G. , Kroemer, G. , 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarra-Luques, C. , Hapon, M.B. , Goyeneche, A.A. , Telleria, C.M. , 2014. Resistance to cisplatin and paclitaxel does not affect the sensitivity of human ovarian cancer cells to antiprogestin-induced cytotoxicity. J. Ovarian Res. 7, 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarra-Luques, C.D. , Goyeneche, A.A. , Hapon, M.B. , Telleria, C.M. , 2012. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer. 12, 200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, B. , Zhang, X.Y. , Han, R. , Zhang, T.T. , Chen, C. , Qin, Z.H. , Sheng, R. , 2013. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol. Sin. 34, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, B.M. , Pincus, D. , Gotthardt, K. , Gallagher, C.M. , Walter, P. , 2013. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampietri, C. , Petrungaro, S. , Conti, S. , Facchiano, A. , Filippini, A. , Ziparo, E. , 2015. Cancer microenvironment and endoplasmic reticulum stress response. Mediators Inflamm. 417281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, T. , Wang, Q. , Lin, Z. , Chen, M.L. , Sun, G.Z. , 2012. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochem. Biophys. Res. Commun. 427, 461–465. [DOI] [PubMed] [Google Scholar]
- Gopfert, U. , Kullmann, M. , Hengst, L. , 2003. Cell cycle-dependent translation of p27 involves a responsive element in its 5′-UTR that overlaps with a uORF. Hum. Mol. Genet. 12, 1767–1779. [DOI] [PubMed] [Google Scholar]
- Goyeneche, A.A. , Caron, R.W. , Telleria, C.M. , 2007. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin. Cancer Res. 13, 3370–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyeneche, A.A. , Seidel, E.E. , Telleria, C.M. , 2012. Growth inhibition induced by antiprogestins RU-38486, ORG-31710, and CDB-2914 in ovarian cancer cells involves inhibition of cyclin dependent kinase 2. Invest. New Drugs. 30, 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyeneche, A.A. , Telleria, C.M. , 2015. Antiprogestins in gynecological diseases. Reproduction. 149, R15–R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez, T. , Simmen, T. , 2014. Endoplasmic reticulum chaperones and oxidoreductases: critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 4, 291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Back, S.H. , Hur, J. , Lin, Y.H. , Gildersleeve, R. , Shan, J. , Yuan, C.L. , Krokowski, D. , Wang, S. , Hatzoglou, M. , Kilberg, M.S. , Sartor, M.A. , Kaufman, R.J. , 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy, S.J. , Gorman, A.M. , Mousavi-Shafaei, P. , Gupta, S. , Samali, A. , 2009. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharmacol. 625, 234–246. [DOI] [PubMed] [Google Scholar]
- Hetz, C. , 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Honma, Y. , Harada, M. , 2013. Sorafenib enhances proteasome inhibitor-mediated cytotoxicity via inhibition of unfolded protein response and keratin phosphorylation. Exp. Cell Res. 319, 2166–2178. [DOI] [PubMed] [Google Scholar]
- Hunter, K.W. , 2004. Ezrin, a key component in tumor metastasis. Trends Mol. Med. 10, 201–204. [DOI] [PubMed] [Google Scholar]
- Johnson, C.E. , Hunt, D.K. , Wiltshire, M. , Herbert, T.P. , Sampson, J.R. , Errington, R.J. , Davies, D.M. , Tee, A.R. , 2015. Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol. Oncol. 9, 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel, M. , Cidlowski, J.A. , 2013. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 34, 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koromilas, A.E. , 2015. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta. 1849, 871–880. [DOI] [PubMed] [Google Scholar]
- Koromilas, A.E. , Mounir, Z. , 2013. Control of oncogenesis by eIF2alpha phosphorylation: implications in PTEN and PI3K-Akt signaling and tumor treatment. Future Oncol. 9, 1005–1015. [DOI] [PubMed] [Google Scholar]
- Kurman, R.J. , Shih Ie, M. , 2011. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum. Pathol. 42, 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederkremer, G.Z. , 2009. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 19, 515–523. [DOI] [PubMed] [Google Scholar]
- Lee, A.S. , 2014. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer. 14, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Tuffin, L.J. , Jewell, C.M. , Bienstock, R.J. , Collins, J.B. , Cidlowski, J.A. , 2007. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol. Cell. Biol. 27, 2266–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Powell, S.R. , Wang, X. , 2011. Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J. 25, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Wu, M. , Wang, H. , Xu, G. , Zhu, T. , Zhang, Y. , Liu, P. , Song, A. , Gang, C. , Han, Z. , Zhou, J. , Meng, L. , Lu, Y. , Wang, S. , Ma, D. , 2008. Ezrin silencing by small hairpin RNA reverses metastatic behaviors of human breast cancer cells. Cancer Lett. 261, 55–63. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Xu, Y. , Stoleru, D. , Salic, A. , 2012. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. U. S. A. 109, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue, S.E. , Cleary, P. , Saveljeva, S. , Samali, A. , 2013. New directions in ER stress-induced cell death. Apoptosis. 18, 537–546. [DOI] [PubMed] [Google Scholar]
- Luo, B. , Lee, A.S. , 2013. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 32, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak, S.J. , Yun, C.Y. , Oyadomari, S. , Novoa, I. , Zhang, Y. , Jungreis, R. , Nagata, K. , Harding, H.P. , Ron, D. , 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, M. , Komoike, Y. , 2015. Experimental evidence shows salubrinal, an eIF2alpha dephosphorylation inhibitor, reduces xenotoxicant-induced cellular damage. Int. J. Mol. Sci. 16, 16275–16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake, H. , Hara, I. , Arakawa, S. , Kamidono, S. , 2000. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J. Cell. Biochem. 77, 396–408. [DOI] [PubMed] [Google Scholar]
- Moon, H.G. , Qin, Z. , Quan, T. , Xie, L. , Dela Cruz, C.S. , Jin, Y. , 2015. Matrix protein CCN1 induced by bacterial DNA and CpG ODN limits lung inflammation and contributes to innate immune homeostasis. Mucosal Immunol. 8, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, C. , Salvayre, R. , Negre-Salvayre, A. , Vindis, C. , 2011. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 18, 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungrue, I.N. , Pagnon, J. , Kohannim, O. , Gargalovic, P.S. , Lusis, A.J. , 2009. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J. Immunol. 182, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelkerke, A. , Bussink, J. , Sweep, F.C. , Span, P.N. , 2014. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta. 1846, 277–284. [DOI] [PubMed] [Google Scholar]
- Nawrocki, S.T. , Carew, J.S. , Pino, M.S. , Highshaw, R.A. , Dunner, K. , Huang, P. , Abbruzzese, J.L. , McConkey, D.J. , 2005. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 65, 11658–11666. [DOI] [PubMed] [Google Scholar]
- Needham, P.G. , Brodsky, J.L. , 2013. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim. Biophys. Acta. 1833, 2447–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhall, E.P. , Winikoff, B. , 2000. Abortion with mifepristone and misoprostol: regimens, efficacy, acceptability and future directions. Am. J. Obstet. Gynecol. 183, S44–S53. [DOI] [PubMed] [Google Scholar]
- Noda, I. , Fujieda, S. , Seki, M. , Tanaka, N. , Sunaga, H. , Ohtsubo, T. , Tsuzuki, H. , Fan, G.K. , Saito, H. , 1999. Inhibition of N-linked glycosylation by tunicamycin enhances sensitivity to cisplatin in human head-and-neck carcinoma cells. Int. J. Cancer. 80, 279–284. [DOI] [PubMed] [Google Scholar]
- Nowis, D. , McConnell, E.J. , Dierlam, L. , Palamarchuk, A. , Lass, A. , Wojcik, C. , 2007. TNF potentiates anticancer activity of bortezomib (Velcade) through reduced expression of proteasome subunits and dysregulation of unfolded protein response. Int. J. Cancer. 121, 431–441. [DOI] [PubMed] [Google Scholar]
- Ohoka, N. , Hattori, T. , Kitagawa, M. , Onozaki, K. , Hayashi, H. , 2007. Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J. Biol. Chem. 282, 35687–35694. [DOI] [PubMed] [Google Scholar]
- Ohoka, N. , Yoshii, S. , Hattori, T. , Onozaki, K. , Hayashi, H. , 2005. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S. , Mori, M. , 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. [DOI] [PubMed] [Google Scholar]
- Platini, F. , Perez-Tomas, R. , Ambrosio, S. , Tessitore, L. , 2010. Understanding autophagy in cell death control. Curr. Pharm. Des. 16, 101–113. [DOI] [PubMed] [Google Scholar]
- Romero, I. , Bast, R.C. , 2012. Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 153, 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D. , Walter, P. , 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Roy, D. , Mondal, S. , Wang, C. , He, X. , Khurana, A. , Giri, S. , Hoffmann, R. , Jung, D.B. , Kim, S.H. , Chini, E.N. , Periera, J.C. , Folmes, C.D. , Mariani, A. , Dowdy, S.C. , Bakkum-Gamez, J.N. , Riska, S.M. , Oberg, A.L. , Karoly, E.D. , Bell, L.N. , Chien, J. , Shridhar, V. , 2014. Loss of HSulf-1 promotes altered lipid metabolism in ovarian cancer. Cancer Metab. 2, 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski, D.T. , Kaufman, R.J. , 2007. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32, 469–476. [DOI] [PubMed] [Google Scholar]
- Rzymski, T. , Milani, M. , Pike, L. , Buffa, F. , Mellor, H.R. , Winchester, L. , Pires, I. , Hammond, E. , Ragoussis, I. , Harris, A.L. , 2010. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 29, 4424–4435. [DOI] [PubMed] [Google Scholar]
- Sano, R. , Reed, J.C. , 2013. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 1833, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulle, E. , Petronelli, A. , Pasquini, L. , Petrucci, E. , Mariani, G. , Biffoni, M. , Ferretti, G. , Scambia, G. , Benedetti-Panici, P. , Cognetti, F. , Humphreys, R. , Peschle, C. , Testa, U. , 2007. Proteasome inhibitors sensitize ovarian cancer cells to TRAIL induced apoptosis. Apoptosis. 12, 635–655. [DOI] [PubMed] [Google Scholar]
- Schmidt, E.K. , Clavarino, G. , Ceppi, M. , Pierre, P. , 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Schneider-Poetsch, T. , Ju, J. , Eyler, D.E. , Dang, Y. , Bhat, S. , Merrick, W.C. , Green, R. , Shen, B. , Liu, J.O. , 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonthal, A.H. , 2013. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 85, 653–666. [DOI] [PubMed] [Google Scholar]
- Shimamoto, S. , Tsuchiya, M. , Yamaguchi, F. , Kubota, Y. , Tokumitsu, H. , Kobayashi, R. , 2014. Ca2+/S100 proteins inhibit the interaction of FKBP38 with Bcl-2 and Hsp90. Biochem. J. 458, 141–152. [DOI] [PubMed] [Google Scholar]
- Sidrauski, C. , Acosta-Alvear, D. , Khoutorsky, A. , Vedantham, P. , Hearn, B.R. , Li, H. , Gamache, K. , Gallagher, C.M. , Ang, K.K. , Wilson, C. , Okreglak, V. , Ashkenazi, A. , Hann, B. , Nader, K. , Arkin, M.R. , Renslo, A.R. , Sonenberg, N. , Walter, P. , 2013. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2, e00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingerland, J. , Pagano, M. , 2000. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17. [DOI] [PubMed] [Google Scholar]
- Solomon, V.R. , Lee, H. , 2009. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 625, 220–233. [DOI] [PubMed] [Google Scholar]
- Szokalska, A. , Makowski, M. , Nowis, D. , Wilczynski, G.M. , Kujawa, M. , Wojcik, C. , Mlynarczuk-Bialy, I. , Salwa, P. , Bil, J. , Janowska, S. , Agostinis, P. , Verfaillie, T. , Bugajski, M. , Gietka, J. , Issat, T. , Glodkowska, E. , Mrowka, P. , Stoklosa, T. , Hamblin, M.R. , Mroz, P. , Jakobisiak, M. , Golab, J. , 2009. Proteasome inhibition potentiates antitumor effects of photodynamic therapy in mice through induction of endoplasmic reticulum stress and unfolded protein response. Cancer Res. 69, 4235–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, A. , Jin, Y. , Lee, S.J. , Zhang, M. , Kim, H.P. , Stolz, D.B. , Ryter, S.W. , Choi, A.M. , 2012. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am. J. Respir. Cell Mol. Biol. 46, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi, Y. , Iwasaki, Y. , Tsugita, M. , Nishiyama, M. , Taguchi, T. , Okazaki, M. , Nakayama, S. , Kambayashi, M. , Hashimoto, K. , Terada, Y. , 2010. Glucocorticoid receptor-beta and receptor-gamma exert dominant negative effect on gene repression but not on gene induction. Endocrinology. 151, 3204–3213. [DOI] [PubMed] [Google Scholar]
- Taylor-Harding, B. , Agadjanian, H. , Nassanian, H. , Kwon, S. , Guo, X. , Miller, C. , Karlan, B.Y. , Orsulic, S. , Walsh, C.S. , 2012. Indole-3-carbinol synergistically sensitises ovarian cancer cells to bortezomib treatment. Br. J. Cancer. 106, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telleria, C.M. , Goyeneche, A.A. , 2012. Antiprogestins in ovarian Cancer. In Farghaly S., Ovarian Cancer – Clinical and Therapeutic Perspectives. InTechopen; [Google Scholar]
- Tieszen, C.R. , Goyeneche, A.A. , Brandhagen, B.N. , Ortbahn, C.T. , Telleria, C.M. , 2011. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. 11, 207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra, H. , Dufey, E. , Lisbona, F. , Rojas-Rivera, D. , Hetz, C. , 2013. When ER stress reaches a dead end. Biochim. Biophys. Acta. 1833, 3507–3517. [DOI] [PubMed] [Google Scholar]
- Vaughan, S. , Coward, J.I. , Bast, R.C. , Berchuck, A. , Berek, J.S. , Brenton, J.D. , Coukos, G. , Crum, C.C. , Drapkin, R. , Etemadmoghadam, D. , Friedlander, M. , Gabra, H. , Kaye, S.B. , Lord, C.J. , Lengyel, E. , Levine, D.A. , McNeish, I.A. , Menon, U. , Mills, G.B. , Nephew, K.P. , Oza, A.M. , Sood, A.K. , Stronach, E.A. , Walczak, H. , Bowtell, D.D. , Balkwill, F.R. , 2011. Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer. 11, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar, S.S. , Brodsky, J.L. , 2008. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie, T. , Garg, A.D. , Agostinis, P. , 2013. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 332, 249–264. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Terpstra, E.J. , 2013. Ubiquitin receptors and protein quality control. J. Mol. Cell. Cardiol. 55, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wempe, S.L. , Gamarra-Luques, C.D. , Telleria, C.M. , 2013. Synergistic lethality of mifepristone and LY294002 in ovarian cancer cells. Cancer Growth Metastasis. 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.J. , Chen, S. , Huang, K.X. , Le, W.D. , 2013. Why should autophagic flux be assessed?. Acta Pharmacol. Sin. 34, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, D. , Yin, L. , Zhong, Q. , Guo, W.-j. , Xu, J.-h. , Jiang, N. , Lin, Z.-r. , Li, M.-z. , Han, P. , Xu, L. , He, X. , Zeng, M.-s. , 2016. ZNF488 enhances the invasion and tumorigenesis in nasopharyngeal carcinoma via the Wnt signaling pathway involving epithelial mesenchymal transition. Cancer Res. Treat. 48, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Figure S1 (A) Venn diagram displaying the number of transcripts up‐ or downregulated (>2 fold) after 24 h exposure to 20 μM mifepristone. (B) Number of transcripts induced (maroon) or repressed (green) by mifepristone in each cell line.
Figure S2 Exposure of SKOV‐3 cells to various doses of bortezomib for 72 h in the absence (−) or presence (+) of 20 μM mifepristone (MF). Viability was assessed by microcapillary fluorescence cytometry.
Figure S3 Protein synthesis inhibitor cycloheximide (CHX) mitigated OV2008 cell death induced by the combination treatment of bortezomib (BZ) and mifepristone (MF). Cells where cultured for 72 h in the presence of DMSO (VEH), 20 μM MF, 5 nM BZ, or the combination of 20 μM MF with 5 nM BZ. All groups were done in the continuous presence or absence of 10 μg/ml CHX. This experiment was repeated twice with a similar outcome.
Figure S4 Pathway building analysis of hypothetical molecular networks regulated by mifepristone based on previously known molecules affected by the steroid, as well as those discovered using genomic and proteomic analysis in the present study. The analysis was performed using Pathway Studio®. The name description of each gene and gene products depicted in the diagram are described in Table S8. Symbols highlighted in green represent proteins previously discovered to be regulated by mifepristone. Symbols highlighted in yellow represent proteins discovered to be regulated by mifepristone in the current genomic and proteomic analysis (see Table 1). The meaning of the different symbols and types of interactions among proteins is also displayed.