

ABSTRACT
Macroautophagy/autophagy has profound implications for aging. However, the true features of autophagy in the progression of aging remain to be clarified. In the present study, we explored the status of autophagic flux during the development of cell senescence induced by oxidative stress. In this system, although autophagic structures increased, the degradation of SQSTM1/p62 protein, the yellow puncta of mRFP-GFP-LC3 fluorescence and the activity of lysosomal proteolytic enzymes all decreased in senescent cells, indicating impaired autophagic flux with lysosomal dysfunction. The influence of autophagy activity on senescence development was confirmed by both positive and negative autophagy modulators; and MTOR-dependent autophagy activators, rapamycin and PP242, efficiently suppressed cellular senescence through a mechanism relevant to restoring autophagic flux. By time-phased treatment of cells with the antioxidant N-acetylcysteine (NAC), the mitochondria uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and ambroxol, a reagent with the effect of enhancing lysosomal enzyme maturation, we found that mitochondrial dysfunction plays an initiating role, while lysosomal dysfunction is more directly responsible for autophagy impairment and senescence. Interestingly, the effect of rapamycin on autophagy flux is linked to its role in functional revitalization of both mitochondrial and lysosomal functions. Together, this study demonstrates that autophagy impairment is crucial for oxidative stress-induced cell senescence, thus restoring autophagy activity could be a promising way to retard senescence.
KEYWORDS: autophagy, lysosomes, mitochondria, oxidative stress, rapamycin, senescence
Introduction
Aging is a complicated process and its mechanism needs to be explored. Cellular senescence happens during aging development and is widely used as an in vitro model for aging research. Stress-induced premature senescence (SIPS) develops faster than replicative senescence and has been established for studying the influence of extracellular or intracellular stress on the aging process.1 Known features of senescent cells include flat and enlarged cellular morphology, increased senescence associated GLB1/β-galactosidase (SA-GLB1/SA-β-gal) activity, the appearance of senescence-associated heterochromatin foci (SAHF), cell growth arrest and senescence-associated secretory phenotype (SASP) such as IL6 and CXCL8/IL8 secretion.2-6
Autophagy plays cytoprotective roles through the turnover of long-lived proteins and scavenging damaged cellular components. Macroautophagy is the most studied type of autophagy and its process comprises 2 stages: the early stage that includes the formation of the phagophore, the precursor to the autophagosome, which captures damaged cellular molecules and organelles, and the late stage that is responsible for the digestion of entrapped components within the autolysosome formed by the fusion of an autophagosome and lysosome.7,8 The updated consensus suggests that the real status of autophagy should be assessed not only by the number of autophagosomes and autolysosomes but also by evaluating the actual autophagic flux, such as monitoring the clearance of cell components in autolysosomes.9,10
The relationship between autophagy and senescence is a basic concern in the field of aging research but is still inconclusive.11 Some studies demonstrate that autophagy is positively correlated with senescence.12,13 Conversely, increasing evidence reveals the negative correlation between autophagy and senescence. For example, Kang et al. reported that autophagy impairment can induce premature senescence in primary human fibroblasts,14 and Harrison et al. verified that rapamycin, an MTOR inhibitor and an autophagy activator, can extend the life span of mice and be a potential anti-aging drug.15
The free radical theory of aging highlights the role of reactive oxygen species (ROS)-induced cellular damage in aging promotion.16 This theory suggests that ROS, when excessively produced in cells under stress conditions, whether naturally or artificially, are detrimental to cell components and homeostasis.17 Mitochondria are a primary source of ROS and the foremost target for ROS damage, and their status has been closely associated with aging phenotypes.18,19 Generally, age-related changes in cells are associated with decreased mitochondrial function, accompanied by increased ROS.20 The mutual influence between mitochondria and autophagy has been suggested,17 providing a new research area to pursue the mechanism and prevention of aging.
To clarify the relationship between autophagy and senescence, 3 levels of work were included in the present study. First, the features of oxidative stress-induced senescent fibroblasts were characterized, with a particular focus on the status of autophagy flux and its role in the development of senescence. Second, the influence of lysosomal and mitochondrial dysfunction on autophagy impairment was analyzed to explore the underlying mechanism. Third, the effect of autophagy modulators on autophagy flux restoration and cellular senescence attenuation was verified. Our results provided new evidence for the negative correlation between autophagy activity and senescence and for the importance of autophagy homeostasis on senescence retardation. This study also provided the first examination of the sequential contribution of mitochondrial and lysosomal dysfunction to the development of oxidative stress-induced senescence.
Results
H2O2 treatment induced cellular senescence with intracellular ROS elevation
First, we established a H2O2-induced SIPS cell model and monitored the progress of cellular senescence for 7 d. As shown, H2O2-treated NIH3T3 cells gradually became enlarged, flattened and were largely positive for SA-GLB1 staining from day 3 (Fig. 1A). The SAHFs in these cells were evident as large and irregularly shaped nuclear puncta (Fig. 1B). The arrest of cell growth was apparent (Fig. 1C), and the levels of TRP53 protein and Cdkn1a, and Il6 mRNA were elevated (Fig. 1D–F). H2O2 treatment also elevated intracellular ROS (Fig. 1G). In addition, this SIPS cell model was successfully established using MRC-5 human lung fibroblast cells (Fig. S1). These results demonstrate that in fibroblasts, the H2O2 treatment protocol we used can reliably induce cellular senescence by 3 to 5 d. Therefore, the following experiments were essentially performed within 5 day after H2O2 treatment.
Figure 1.

Short-term H2O2 treatment is sufficient to induce cellular senescence. NIH3T3 cells were treated with PBS (Ctrl) or with 400 μM H2O2 in PBS as described in the Materials and Methods, and shifted to culture in a complete medium for the indicated days. (A) Images showing the cellular morphology and SA-GLB1 staining. Scale bars: 20 μm. (B) H2O2-treated cells were stained with DAPI to show SAHFs. Circles indicate typical SAHFs. Apoptosis was induced by 1.5 mM H2O2 to show the difference between apoptotic bodies and SAHFs. Scale bars: 20 μm. (C) Cell counting was conducted up to 7 d and growth curves of control, H2O2-treated and serum starved cells are shown. (D) TRP53 was examined by western blot. (E) (F) Relative mRNA levels of Cdkn1a and Il6 were analyzed by qRT-PCR. (G) ROS in NIH3T3 cells was labeled by DCFH-DA probe and quantified by flow cytometry. Data are presented as the means ± SD from 3 independent experiments. *p < 0.05 and **p < 0.01 compared to control.
Autophagic structure increase accompanied with impaired flux of autophagy in senescent cells
The relevance of autophagy and senescence remains elusive.21,22 To elucidate the status of autophagy during SIPS, autophagic structures were first examined using transmission electron microscopy, revealing an apparent increase in the number of vacuole or vesicular-like structures in the cytoplasm of senescent cells (Fig. 2A). To characterize the increased vacuole or vesicular-like structures, LysoTracker Red was used to stain lysosomes and autolysosomes, leading to the observation that lysosomal structures significantly increased in H2O2-treated cells (Fig. 2B). Moreover, in an NIH3T3 cell line stably expressing mRFP-LC3, the punctate LC3 distribution was observed from d 1 after H2O2 exposure, with an apparent increase on d 3 and 5. These results indicate an increase of autophagic structures in H2O2-induced senescent cells.
Figure 2.
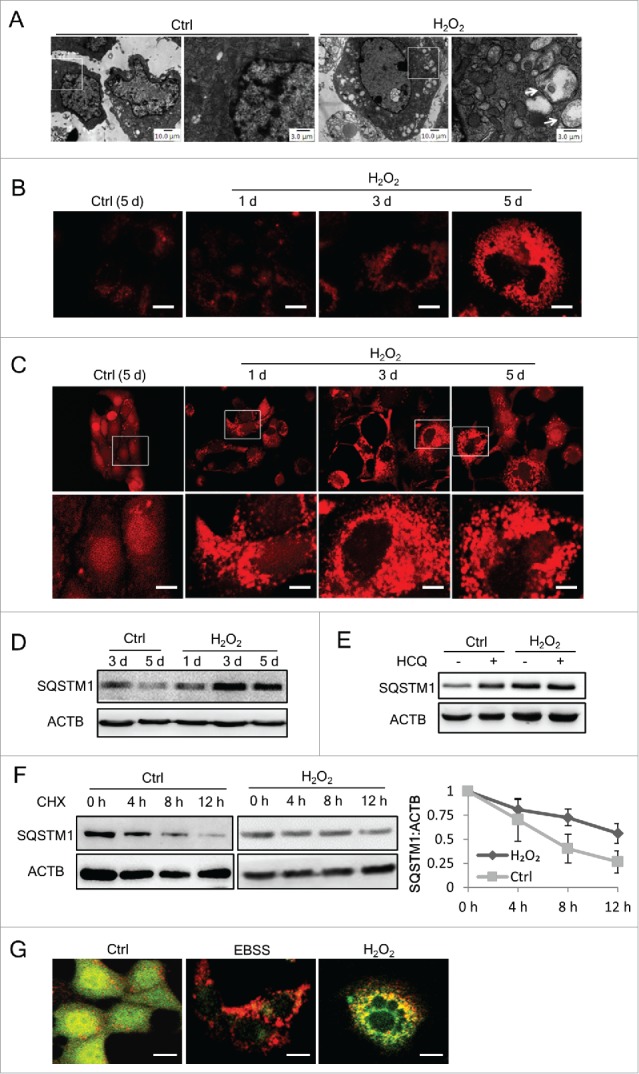
Autophagic structures increase but autophagic flux is impaired in senescent cells. (A) Transmission electronic microscopy of control or H2O2-treated NIH3T3 cells at day 5. The amplified image on the right of each group is selected from the rectangle area of the left image. Arrows show the vesicle-like structures. (B) The lysosome content of NIH3T3 cells was probed with LysoTracker Red DND-99 and images were taken under a fluorescence microscope by the same exposure parameters. Scale bars: 10 μm. (C) Fluorescence images of mRFP-LC3 NIH3T3 cells treated with H2O2 at the indicated time points. Scale bars: 20 μm. Lower panel shows the amplified images of those in rectangles. (D) NIH3T3 cell lysates were collected at the indicated time points and SQSTM1 was examined by western blots. (E) SQSTM1 in NIH3T3 cells was examined by western blots. Both control and H2O2-treated cells were in parallel treated with or without 5 μg/ml HCQ for 12 h before sample collecting. (F) Control or H2O2-treated NIH3T3 cells were cultured for 3 day before adding CHX (100 μg/ml). Samples were collected at the indicated time points after CHX addition, and SQSTM1 was examined by western blot. Representative images are shown at the left, and statistical data calculated from 4 independent experiments are shown at the right. (G) Fluorescence images of mRFP-GFP-LC3 in NIH3T3 cells starved with EBSS for 12 h or at day 3 after H2O2treatment. Scale bars: 10 μm.
To clear whether the accumulation of autophagic structures in these cells was caused by autophagy induction or by blocked autophagic degradation,9,10 we assessed autophagic flux. First, the level of endogenous SQSTM1/p62 protein, which turns over through autophagic degradation, was examined.10 The results showed that SQSTM1 protein in H2O2-treated cells increased on d 3 and 5 (Fig. 2D). The nature of this increase of SQSTM1 was further tested through a blocking experiment using hydroxychloroquine (HCQ), which elevates lysosomal pH and blocks the fusion of autophagosomes with lysosomes, thus inhibiting lysosomal degradation. As shown in Fig. 2E, in contrast with the control cells, HCQ treatment did not cause an increase in the level of SQSTM1 in H2O2-treated cells, suggesting that the degradation capacity of lysosomes H2O2-treated cells was already at a low level similar to that caused by HCQ treatment.
Next, we compared the half-life of SQSTM1 in H2O2-treated and control cells. The results showed that SQSTM1 in H2O2-treated cells was degraded more slowly, as the half-life of SQSTM1 in these cells was approximately 12 h, but that in control cells was 7 h (Fig. 2F). Furthermore, in NIH3T3 cells stably expressing tandem mRFP-GFP-LC3,24 although the puncta-like LC3 protein was predominantly red in EBSS-starved control cells, those in H2O2-treated cells tended to be yellow, reflecting either a lower capacity of lysosomes in the quenching of green fluorescence in lysosomes due to the elevated pH, or a block in the fusion of autophagosomes with lysosomes (Fig. 2G). These results collectively indicate the impairment of autophagic flux in H2O2-induced senescent cells.
Chronic blockage of autophagic flux was sufficient to induce cellular senescence
To get additional evidence, we assessed whether direct impairment of autophagic flux induces cellular senescence. When normal NIH3T3 cells were treated with a sublethal dose of HCQ or leupeptin, 2 lysosome inhibitors capable of blocking autophagic degradation, the cells exhibited increased intracellular ROS (Fig. 3A). In parallel, SA-GLB1-positive cells and Il6 mRNA expression increased after 7 d (Fig. 3A and 3B). Moreover, in an Atg5 knockdown NIH3T3 population with impaired autophagy flux (Fig. 3C and 3D), SA-GLB1-positive cells were apparent on d 15, even without H2O2 treatment (Fig. 3E). An increase in the level of Il6 mRNA was detected in these cells (Fig. 3F). These results indicate that chronic autophagy impairment induces cellular senescence.
Figure 3.
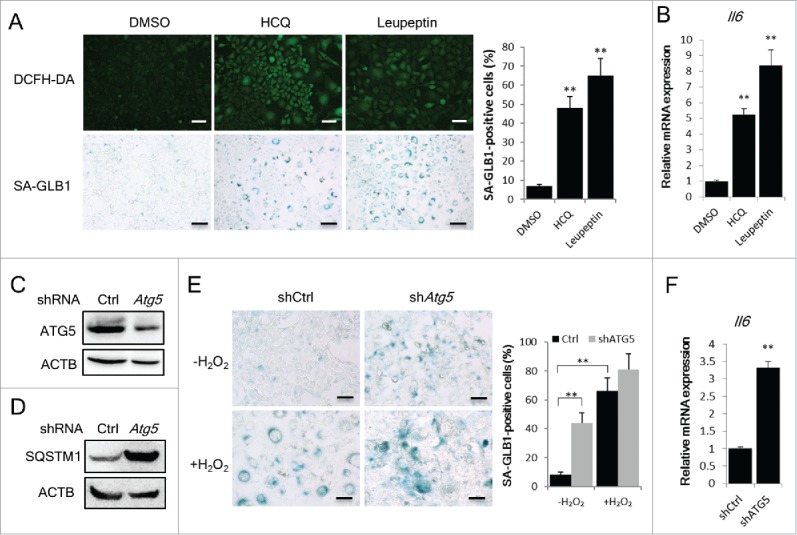
Blockage of autophagy flux is sufficient to induce cellular senescence. (A) NIH3T3 cells were treated with HCQ (3 μg/ml) or leupeptin (5 μg/ml) for 7 d. DCFH-DA fluorescence and SA-GLB1 staining was used to detect intracellular ROS (upper panel) and the senescent state (lower panel) of these cells. The percentages of SA-GLB1-positive cells were quantified. Scale bars: 50 μm. (B) Relative Il6 mRNA expression of the cells treated as described in (A) were quantified by qRT-PCR. (C) and (D) NIH3T3 cells transfected with control shRNA or Atg5 shRNA and positive cells were selected with 2 μg/ml puromycin for 7 d. Images of western blots for ATG5 (C) and SQSTM1 (D) are shown. (E) The cells described in (C) were cultured for another 5 d, and then treated with H2O2 followed by a 3-d incubation. Images of SA-GLB1 staining and the percentages of SA-GLB1-positive cells are shown. Scale bars: 20 μm. (F) Relative Il6 mRNA level in cells cultured for 15 d were quantified by qRT-PCR. The data are presented as means ± SD from 3 independent experiments and **p < 0.01.
MTOR-dependent autophagy activators attenuated senescence through the restoration of autophagy flux
Next, we determined whether autophagy regulators affect cell senescence. The influence of 7 autophagy regulators, 4 activators and 3 inhibitors, on SIPS was examined. The dosages of these chemical compounds were determined in preliminary experiments. As shown in Fig. 4A, rapamycin and PP242, 2 MTOR-dependent autophagy activators, had a potent anti-senescent effect, as these molecules dramatically reduced the percentage of positive cells in SA-GLB1 staining; however, valproic acid (VPA) and LiCl, 2 MTOR-independent autophagy activators, presented only a subtle impact on the senescent state of H2O2-treated cells (Fig. 4A).
Figure 4.
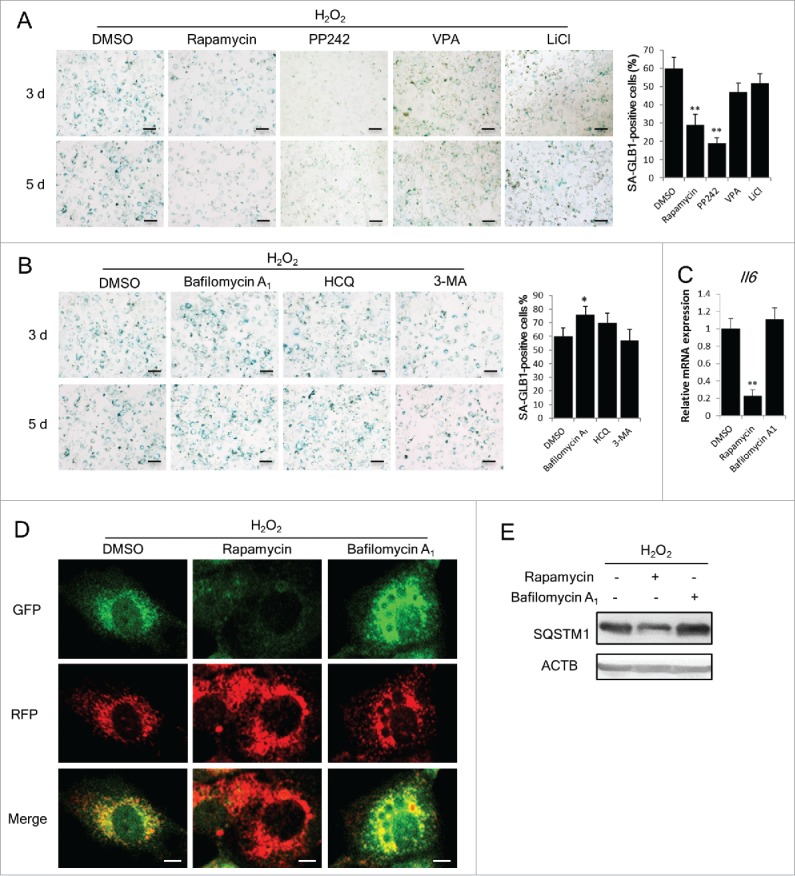
Autophagy restoration attenuates SIPS development. (A) and (B) NIH3T3 cells treated with or without H2O2 were incubated with the indicated reagents for 3 or 5 d. Autophagy activators rapamycin (100 nM), PP242 (500 nM), VPA (1 mM) and LiCl (10 mM) were applied in (A) and autophagy inhibitors bafilomycin A1 (50 nM), HCQ (3 μg/ml) and 3-MA (500 μM), were applied in (B). Images of SA-GLB1 staining are shown in the left, and the percentages of SA-GLB1-positive cells at day 5 are shown in the right. Scale bars: 50 μm. (C) Relative Il6 mRNA level in H2O2-treated NIH3T3 cells was measured by qRT-PCR. (D) Confocal images of cells stably expressing mRFP-GFP-LC3. Scale bars: 5 μm. (E) SQSTM1 in H2O2-treated NIH3T3 cells at d 3 were examined by western blots. The data are presented as means ± SD from 3 independent experiments. *p < 0.05 and **p < 0.01 when compared to DMSO.
As to the autophagy inhibitors, bafilomycin A1 and HCQ, which block lysosomal function, promoted an increase in SA-GLB1-positive cells following H2O2 treatment (Fig. 4B), whereas 3-methyladenine (3-MA), which targets phosphoinositide 3-kinases and phosphatidylinositol 3-kinases and influences autophagy at an early stage,25 did not cause an increase in SA-GLB1 (Fig. 4B). Based on their clear impact on SIPS development, rapamycin and bafilomycin A1 were selected for further investigation. The effects of these 2 reagents on the Il6 mRNA level were consistent with those on SA-GLB1 staining (Fig. 4C). To confirm whether the role of these reagents in SIPS development is relevant to autophagy regulation, we examined autophagy flux using NIH3T3 cells stably expressing tandem mRFP-GFP-LC3 and observed that the ratio of red:yellow puncta in H2O2-treated cells was elevated when rapamycin was applied, but declined in the case when bafilomycin A1 was present (Fig. 4D). In addition, the abundance of SQSTM1 protein decreased in rapamycin-treated cells but not in bafilomycin A1-treated cells (Fig. 4E). The impact of rapamycin and bafilomycin A1 on SIPS was also observed in MRC-5 human fetal lung fibroblast cells (Fig. S1 and Fig. 4A). These results demonstrate that the regulation of autophagic flux affects SIPS development, and the MTOR inhibitor rapamycin is an efficient anti-senescence reagent in this system.
Lysosomal and mitochondrial dysfunction occurs during SIPS development
Considering that the capacity of lysosomal degradation is a rate-limiting factor for autophagic flux,26 we next assessed the function of lysosomes. The results showed that the activity of ACP2 (acid phosphatase 2, lysosomal) decreased in senescent cells (Fig. 5A), concomitant with an increase in the size of lysosomes (Fig. 5B).27 Moreover, the abundance of CTSB (cathepsin B), a representative protease in lysosomes, was reduced in senescent cells (Fig. 5C), concomitant with the decline in activity of this enzyme (Fig. 5D). Taken together, these results reveal that the oxidative stress-induced impairment of autophagic flux in senescent cells is closely associated with the reduced degradation capability of autolysosomes and lysosomes.
Figure 5.
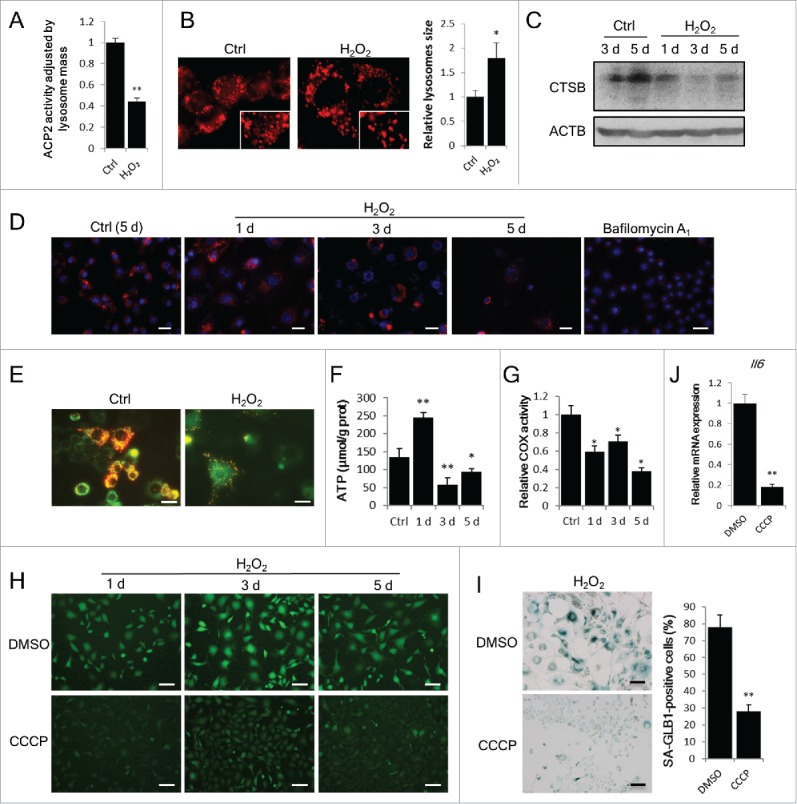
Autophagy impairment couples with lysosomal and mitochondrial dysfunction. NIH3T3 cells were treated with or without H2O2. (A) In vitro ACP2/acid phosphatase activity assay was performed at d 3 after H2O2 treatment. (B) Lysosomes in cells were probed with LysoTracker Red DND-99 at day 3 and images were taken by confocal microscopy. The images in rectangles are 1.5-fold enlarged. Relative lysosome size in control and H2O2-treated cells was quantified. (C) CTSB in control and H2O2-treated cells at the indicated time points were examined by western blot. (D) Cellular CTSB activity was visualized by using the Magic Red Cathepsin B detection kit at the indicated days. Cells treated with bafilomycin A1 (50 nM) for 12 h are shown as a negative control. Scale bars: 50 μm. (E) Mitochondrial membrane potential in cells was measured by JC-1 staining at d 3. Scale bars: 20 μm. (F) Cellular ATP level of control (5 d) and H2O2-treated (1 d, 3 d, 5 d) cells was measured. (G) Mitochondrial COX activity of control (5 d) or H2O2-treated (1 d, 3 d, 5 d) cells was assayed. (H) Cells pretreated with DMSO or 10 μM CCCP for 6 h followed by H2O2 treatment as described in the Materials and Methods. Images of DCFH-DA fluorescence was taken at the indicated days. Scale bars: 50 μm. (I) SA-GLB1 staining was performed at day 5 on CCCP-pretreated cells. Scale bars: 50 μm. (J) Relative Il6 mRNA expression of CCCP-pretreated cells at d 5. The data are presented as means ± SD from 3 independent experiments. *p < 0.05 and **p < 0.01 when compared to control or DMSO.
We also monitored several mitochondrial parameters, as the association between mitochondria and senescence has been suggested in many studies.18,20 Our data showed that mitochondrial depolarization occurred in H2O2-treated cells, indicated by decreased mitochondrial membrane potential (MMP) (Fig. 5E). This result was consistent with the reduced cellular ATP content (Fig. 5F) and decreased activity of mitochondrial COX (cytochrome c oxidase) (Fig. 5G). An uncoupling reagent of oxidative phosphorylation, carbonyl cyanide m-chlorophenyl hydrazone (CCCP),19 was also used. In CCCP preteated cells, the intracellular ROS generation after H2O2 treatment was significantly reduced (Fig. 5H), consistent with attenuated cellular senescence indicated by increased SA-GLB1 staining (Fig. 5I) and elevated Il6 mRNA level (Fig. 5J). Furthermore, we examined the effect of the antioxidant NAC23 on the development of SIPS. Although not attenuated by cotreatment with NAC and H2O2, SIPS was partially rescued when NAC was included in the culture medium after H2O2 treatment (Fig. S2). These data suggest the importance of mitochondria-derived ROS and the dysfunction of mitochondria in SIPS development.
Notably, this mitochondria-derived ROS overproduction did not accompany the decreased mitochondria abundance in senescent cells (data not shown). In contrast, increased mitochondrial DNA (mtDNA) and elevated mtDNA integrity, signs of mitochondrial biogenesis, were detected after H2O2 treatment (Fig. S3).
The role of ROS overproduction in lysosomal dysfunction and autophagy impairment was further determined using another SIPS model established through treatment with pyocyanin, a reagent that induces intracellular ROS production.28 Both ROS overproduction and cellular senescence were induced by pyocyanin in a dose-dependent manner (Fig. S4), and the decrease of ACP2 activity and increase of SQSTM1 protein were also prominent (Fig. S5). Consistently, the treatment of cells with the antioxidant NAC attenuated the senescence induced by pyocyanin (Fig. S6).
Temporally sequential dysfunction of mitochondria and lysosomes during SIPS development
To explore the mechanistic association between lysosomal and mitochondrial dysfunction during SIPS development, a temporally displaced sequence of functional assays for these 2 organelles was conducted. First, we examined the time course of mitochondrial dysfunction and lysosomal dysfunction in H2O2-treated cells based on the increase in ROS generation and the decrease in CTSB activity, respectively. As shown in Fig. 6A, an increase in ROS levels was observed shortly after H2O2 treatment, peaking at 4 to 8 h and showing a nearly 3.5-fold increase. In contrast, a decrease in the activity of CTSB was only observed from day 3 after H2O2 treatment, coinciding with the appearance of another peak of ROS overproduction (Fig. 6B).
Figure 6.
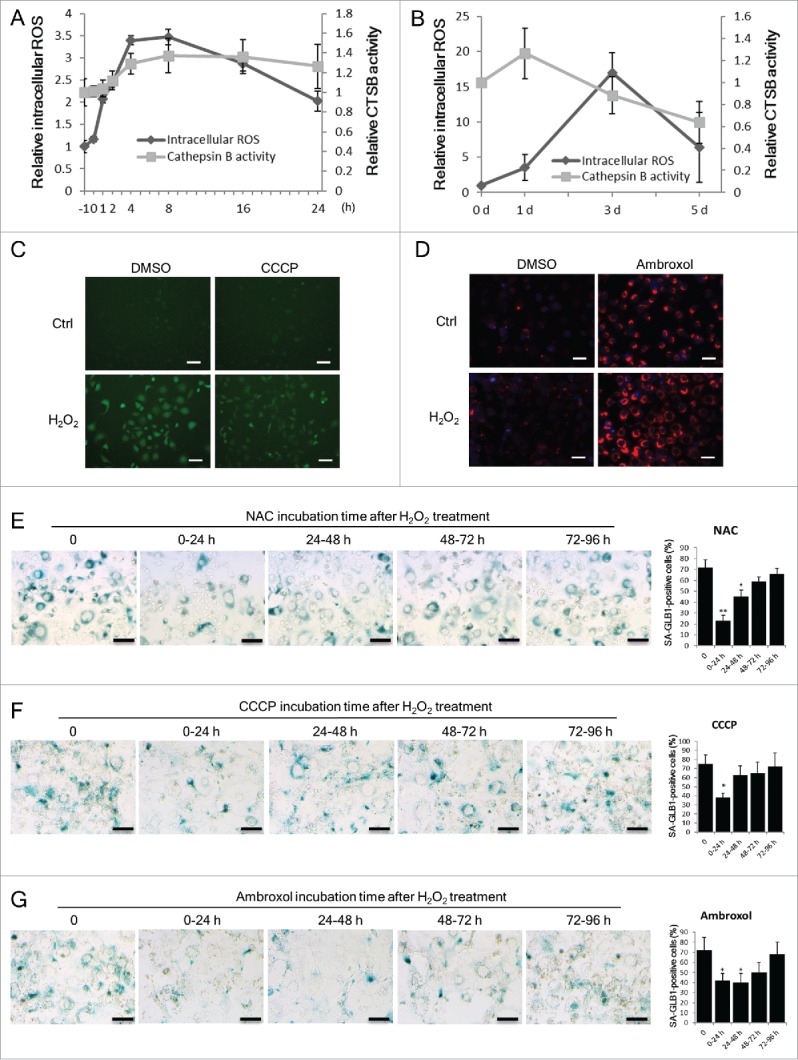
Mitochondria dysfunction precedes lysosomes dysfunction during SIPS development. NIH3T3 cells were treated with or without H2O2. (A) Quantification of intracellular ROS and lysosomal CTSB activity within 24 h after H2O2 treatment. Intracellular ROS was measured by flow cytometry using the DCFH-DA probe. Lysosomal CTSB activity was measured using the fluorescence intensity of the Magic Red Cathepsin B probe. 0 h indicates the end time point of H2O2 treatment. The data of untreated cells (time point −1 h) was set to 1 and other values were normalized. (B) Quantification of intracellular ROS and lysosomal CTSB activity at 0, 1, 3, 5 d after H2O2 treatment. (C) Images of intracellular ROS probe of control or H2O2-treated cells at d 3. DMSO or 10 μM CCCP was added to the culture medium after H2O2treatment. Scale bars: 50 μm. (D) CTSB activity in control or H2O2-treated cells day 3 was visualized with the Magic Red Cathepsin B probe. DMSO or 50 μM ambroxol was added to the culture medium after H2O2 treatment. Scale bars: 50 μm. (E) to (G) H2O2-treated cells were incubated with NAC (2 μM), CCCP (10 μM) or ambroxol (50 μM) at different periods of time. Cells were stained with the SA-GLB1/SA-β-gal assay kit at d 5. Scale bars: 100 μm. The percentages of SA-GLB1-positive cells in each group are quantified. The data are presented as means ± SD from 3 independent experiments. *p < 0.05 and **p < 0.01 when compared to control or samples not treated with NAC, CCCP or ambroxol.
This temporal sequence of mitochondrial and lysosomal dysfunction was further shown through another set of experiments using NAC, CCCP and ambroxol, where the latter acts as an enhancer of lysosome enzyme maturation.29 The suppressive effect of NAC and CCCP on ROS overproduction was obvious (Fig. 6C)23 as was the promoting effect of ambroxol on lysosomal CTSB activity (Fig. 6D); we applied these reagents for a 24-h duration from different time points after H2O2 treatment. As shown in Fig. 6E–6G, the effects of NAC and CCCP on senescence prevention were significant when applied during the first 24 h but blunted when applied later. In contrast, the effect of ambroxol had a relatively wider time window of up to 72 h. In other words, within the first 24 h after H2O2 treatment, defending mitochondria function by NAC and CCCP was more effective on restraining senescence than improving lysosomal function with ambroxol, whereas beyond 24 h, the effect of ambroxol became more prominent. These results suggest that mitochondrial dysfunction precedes lysosomal dysfunction during SIPS development, and both of them, earlier or later, play a role in senescence development.
Autophagy modulators regulated the function of mitochondria and lysosomes in senescent cells
Moreover, we addressed whether the effect of autophagy modulators on cellular senescence is associated with their influence on the functions of mitochondria and lysosomes. With respect to mitochondrial function, MMP was elevated by rapamycin and reduced by bafilomycin A1 (Fig. 7A). Accordingly, rapamycin substantially decreased intracellular ROS levels and increased ATP levels, whereas bafilomycin A1 showed inverse effects (Fig. 7B and 7C). With respect to lysosomal function, rapamycin facilitated an increase in the expression (Fig. 7D) and activity (Fig. 7E) of CTSB in senescent cells. These results provided novel evidence for the effects of rapamycin and bafilomycin A1 on the function of mitochondria and lysosomes.
Figure 7.
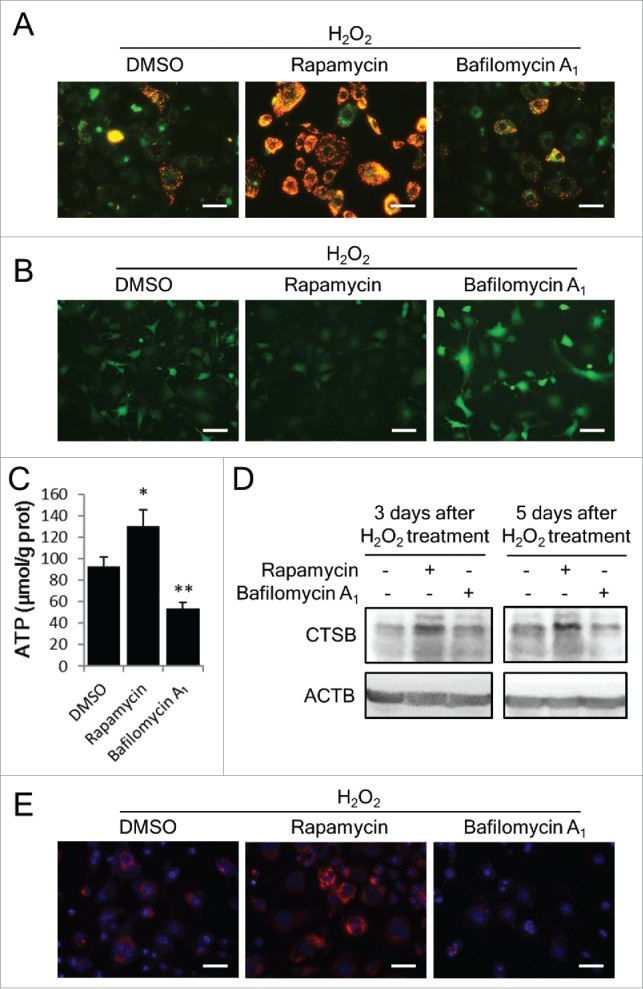
Rapamycin restores mitochondrial and lysosomal function in H2O2-treated cells. NIH3T3 cells were treated with H2O2 and incubated with DMSO, rapamycin or bafilomycin A1 for 3 or 5 d. (A) Images of JC-1 fluorescence from H2O2-treated cells with DMSO, rapamycin or bafilomycin A1 at d 3. Scale bars: 20 μm. (B) DCFH-DA probed intracellular ROS at d 3. Scale bars: 20 μm. (C) Intracellular ATP levels were measured at d 3. (D) CTSB in cells was examined by western blots. (E) Intracellular CTSB activity at d 3 was measured using the Magic Red Cathepsin B kit. Scale bars: 20 μm. The data are presented as means ± SD from 3 independent experiments. *p < 0.05 and **p < 0.01 when compared to DMSO.
Discussion
In the present study, we addressed the functional role of autophagic flux in the development of oxidative stress-induced cellular senescence and attained interesting findings: 1. autophagic flux is impaired in SIPS cells; 2. mitochondrial dysfunction and ROS overproduction temporally occur earlier than lysosomal dysfunction during SIPS development; and 3. the effect of the MTOR-dependent autophagy activator rapamycin on preventing senescence is associated with its role in the restoration of autophagic flux and mitochondrial and lysosomal functions. These findings provide novel evidence for the opinion that autophagy activation is a promising strategy to counteract cellular senescence and suggest that the interplay between mitochondria and lysosomes is crucial for the maintenance of cell health, therefore being a reasonable target for anti-aging intervention.
The relationship between autophagy and senescence seems likely, but the precise correlation between these processes remains unclear, as previous reports have generated contradicting results.11 Although many factors, such as cell type, inducer type, the level of stress intensity, and measuring time point, actually affect the output of autophagy status, increasing evidence shows now that the methods and indices selected for evaluating autophagy activity is of importance. Thus, instead of focusing on the changes of the quantity and size of autophagic structures, which can be caused by the alterations in either autophagic structure formation or autophagic degradation,10 we particularly evaluated the status of autophagic flux during SIPS development. Using a series of functional measurements, particularly concerning the assessment of autolysosomal protein degradation, we ensured that autophagy impairment is positively correlated with senescence development. This result is consistent with the reports of other studies,14,30,31 although the opposite correlation has also been proposed. For example, Young et al. reported that the inhibition of autophagy through the depletion of ATG5 or ATG7 in a tumor cell line suppresses the induction of senescence.12 These inconsistencies might reflect the distinct metabolic characteristics of cells and the variations in senescence induction approaches used in different studies. At any rate, we think that autophagic impairment is a prominent feature of SIPS cells, at least in our system.
The restoration of autophagic flux is considered a potential target for anti-aging therapeutics. Consistent with previous studies,32,33 the results of the present study confirmed the effectiveness of autophagy activation on senescence prevention. Interestingly, we observed that not all autophagy modulators influence SIPS. Only MTOR-dependent autophagy activators, rapamycin and PP242, and lysosomal function-dependent autophagy inhibitors, HCQ and bafilomycin A1, exhibited obvious effects. This parallel comparison of the effects of different autophagy modulators revealed that, for SIPS blockage at least, modulating autophagy at its late stage is more effective than modulation at its early stage. However, this idea requires further study. Nevertheless, the results of the present study confirmed the crucial role of autophagic flux in SIPS development and revealed the effectiveness of MTOR inhibition on SIPS repression.
The association between MTOR and autophagy has been established. Much of the research concerns the inhibitory role of MTOR on ULK activity, which facilitates the early stage of autophagy.34 Other studies have primarily focused on the suppression of MTOR on the activity of TFEB (transcription factor EB), a master transcription factor for autophagic proteins, including those controlling lysosome factors.27,35,38 However, the precise association of MTOR with autophagic flux has not yet been explored in the context of senescent cells. Herein, the results of the present study are novel and encouraging, particularly standing on the fact that rapamycin can activate lysosomal activity in SIPS cells. The effect of MTOR inhibitors on lysosome activation in proliferating cells has been suggested in a previous study.36 However, it shows that only PP242 and Torin, but not rapamycin, worked effectively. As to the inconsistency in the effectiveness of rapamycin, we concerned about the different concentrations of rapamycin used as a 10-fold higher concentration (1 µM) was used in the previous study, than what we used (100 nM). It is known that concentrations of rapamycin higher than 1 µM may elevate the pH of lysosomes.37 Nevertheless, the key molecular mechanism for rapamycin-induced autolysosome activation and autophagic flux restoration in the present study has not been demonstrated. Indeed, although we attempted, we did not obtain evidence that rapamycin could facilitate the nuclear translocation of TFEB.39 This finding implies 2 possibilities: MTOR may regulate the function of autolysosomes via a TFEB-independent mechanism; and/or TFEB may be functionally regulated independent of MTOR. Thus, further assessments are currently underway.
The evidence obtained in the present study also demonstrated the impact of mitochondrial dysfunction on SIPS induction, indicated by the ROS overproduction, the protective role of NAC, the pyocyanin-promoted SIPS development, and the attenuated development of SIPS in CCCP-pretreated or treated cells. Notably, Clara Correia-Melo et al. recently provided evidence of the crucial role of mitochondria in cellular senescence. These authors observed that mitochondria depletion can reduce a spectrum of senescence effectors and phenotypes but preserve ATP production and cell survival via enhanced glycolysis.40 This work supports the concept that mitochondria are a major putative therapeutic target for interventions against aging, owing to their innate role in ROS overproduction. The results of the present study were consistent with this concept. First, the enhanced ROS generation was temporally matched with the increased mtDNA content and enhanced mtDNA integrity during SIPS development (Fig. S3), 2 parameters reflecting the evoked mitochondrial biogenesis. Second, CCCP obviously weakened ROS generation together with SIPS development, in accord with the protective role of uncoupling proteins under stress19 and the “uncoupling to survive theory of aging” proposed by Martin Brand in 2000.42 This theory suggests that the partial uncoupling of mitochondria might be beneficial for longevity. Based on the above concepts and evidence, we think it is better to consider the association of mitochondria with aging not only based on the resultant damage and dysfunction of this organelle but also on their intrinsic and active response upon imbalanced cellular homeostasis. Importantly, this response is not simply or always beneficial for cells, mostly because of the inherent ability of mitochondria on ROS overproduction. Although more evidence and wisdom are needed to refine this consideration, the mitochondria theory of aging itself is now at a new starting line. With respect to the mechanism of stress-induced mitochondrial biogenesis, a recent publication demonstrated the impact of the mitochondrial unfolded protein response (UPRmt)41 It is a promising point to consider.
The interplay between mitochondria and lysosomes in cellular senescence is another attractive aspect. To clarify this issue, time-dependent functional alterations of these 2 organelles were addressed in the present study, with several interesting findings: 1. the dysfunction of mitochondria occurs within hours after H2O2 treatment, much earlier than the dysfunction of lysosomes, likely being the initiating event for senescence development and a trigger of autophagy impairment; 2. although occurring later, the solid association of lysosomal dysfunction with autophagy impairment highlights its responsibility for SIPS development and its position as a characteristic of SIPS; 3. the influence of lysosomes on mitochondria probably exists, as inhibitors of lysosomes induced intracellular ROS generation (Fig. 3A), and rapamycin alleviated the dysfunction of mitochondria (Fig. 7A–C); 4. Our results support the mitochondrial-lysosomal axis theory of aging proposed by Brunk and Terman,43 as the drugs ameliorating the function of each of these organelles exhibit a potent anti-senescence effect.
In conclusion, this study demonstrated the impairment of autophagy in H2O2-induced senescent cells and emphasized the characteristics of autolysosome/lysosome dysfunction for SIPS development. Moreover, together with exploring the initiating role of intracellular ROS and mitochondria dysfunction, the interplay between mitochondria and autophagic flux was defined. Furthermore, this study clarified the efficiency of MTOR inactivation-mediated autophagy restoration on SIPS prevention. These findings increased our understanding of the relationship between autophagy and senescence. Additional studies are needed to explore the underlying mechanism as to how impaired autophagy is associated with senescence and whether a combined treatment targeting both mitochondria and lysosomes is promising for the development of effective strategies against aging and aging-related disorders.
Materials and methods
Reagents and antibodies
Rapamycin (R0395), VPA (P4543), LiCl (203637), bafilomycin A1 (B1793), HCQ (H0915), 3-MA (M9281), NAC (A7250), CHX (C7698) and pyocyanin (P0046) were from Sigma. PP242 was from Cayman Chemical Company (13643). Leupeptin was from Amresco (J580). 2′,7′-dichlor- fluorescein-diacetate (DCFH-DA) was from Applygen (C1300). JC-1 was from Beyotime (C2006). CCCP was from Tocris Bioscience (0452). Ambroxol hydrochloride was from Boehringer Ingelheim (Mucosolvan®, 2 ml; 15 mg). Anti-ACTB (sc-47778), anti-SQSTM1 (sc-28359) and anti-CTSB antibodies (sc-6493) were from Santa Cruz Biotechnologies. Anti-TRP53 antibody was from Cell Signaling Technology (9282).
Cell culture and treatments
The murine fibroblast NIH3T3 and human fetal lung fibroblast MRC-5 cell lines were obtained from Shanghai Institutes for Biological Sciences of Chinese Academy of Sciences and cultured in a complete medium (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) in a humidified incubator at 37°C and 5% CO2.
For monitoring cell proliferation status, normal, H2O2-treated and starved cells were inoculated in 6-well plates in 3 replicates for each time point and harvested by trypsinization at 24-h intervals for 7 d. The numbers of cells were counted under a microscope.
For senescence induction, growing cells at approximately 80% confluence were collected into a microcentrifuge tube after trypsinization, suspended in phosphate-buffered saline (PBS; Sangon Biotech, #BN40100-0500) at 1×106 cells/ml density and exposed to 400 μM H2O2 at 37°C. During H2O2 exposure, the tube was turned upside down gently 5-10 times with a 5 min interval. After 45 min, cells were washed with PBS once and resuspended with complete medium. The control cells were treated with PBS at the same time. Then both control cells and H2O2-treated cells were split into individual wells in culture plate with a density of approximately 2×104 cells/cm2 and cultured with complete medium for various durations as indicated in individual experiments. After treatment, the media of both control and H2O2-treated cells were changed every 3 d.
To evaluate the effect of activators and inhibitors of autophagy activity on SIPS processing, different chemical reagents were added into the culture medium after H2O2 treatment, with their concentrations indicated as in the figure legends. These concentrations were determined by preliminary experiments. The stock solutions of rapamycin and bafilomycin A1 were dissolved in DMSO, those of PP242, VPA, LiCl, HCQ and 3-MA in PBS, respectively.
SA-GLB1/SA-β-gal staining
SA-GLB1 activity was determined using the SA-β-gal staining kit (Beyotime, C0602) according to a standard protocol.3 Senescent cells were identified as bluish green-stained cells by microscopy. More than 500 cells in 6 random fields were counted to determine the percentage of SA-GLB1-positive cells in the total cell population.
SAHF detection
Cells were fixed in situ with 4% paraformaldehyde and washed with PBS. DAPI (Beyotime, C1006) at 300 nM concentration in PBS was added for 5 min incubation. The cells were then washed 3 times by PBS, drained and mounted. DAPI-stained nuclei with blue fluorescence were viewed by fluorescence microscopy.
Western blot analysis
Whole cell lysates were prepared by directly denaturing cell pellets in 2×SDS loading buffer, and then boiling for 10 min. Western blotting assays were done as described previously.44 Anti-TRP53, anti-SQSTM1, and anti-CTSB antibodies were used as primary antibodies. Every experiment was repeated at least 3 times, and representative data are shown.
Real-time quantitative reverse transcription PCR (qRT-PCR)
Total RNA extraction, reverse transcription and real-time PCR amplification were performed as described previously.45 PCR primers for the Cdkn1a gene are 5′-GTGGCCTTGTCGCTGTC TT-3′ (forward) and 5′-GCGCTTGGAGTGA-3′ (reverse), for the Il6 gene are 5′-ACTCACCTCT TCAGAACGAATTG-3′ (forward) and 5′-CCATCTTTGGAAGGTTCAGGTTG-3′ (reverse), and for Rn18s rRNA are 5′-TTGACGGAAGGGCACCACCAG-3′ (forward) and 5′-GCACCACCACCCAC GGAATCG-3′ (reverse). The experiments were performed in triplicate, and the data for the Cdkn1a and Il6 genes were adjusted by the values for Rn18s gene and shown as relative fold changes against control.
Electron microscopy
PBS washed cells were collected into microcentrifuge tubes using cell scrapers. After centrifuging, 4% paraformaldehyde was loaded carefully on cell pellets for an overnight fixation at 4°C. Fixed cells were further treated and sliced, and then the images of their ultrastructure were recorded under a transmission electron microscope.
Lysosome labeling with LysoTracker Red DND-99
Lysosomes were labeled with LysoTracker Red (Life Technologies, L7528) according to the manufacturer's protocol. Briefly, LysoTracker Red was added into cultured cells at a concentration of 1:20,000 dilution from stock solution (1 mM in anhydrous DMSO) and incubated at 37°C for 30 min. Images were taken using a fluorescence microscope and the size of lysosomes was measured by using ImageJ software to analyze the pixels occupied by a single lysosome. We counted at least 50 cells of random area in each group and the average size of control cells was set to one, and those of other groups of cells were normalized.
Stable transfected cells expressing fluorescent LC3 protein
The mRFP-LC3 expression construct (pmRFP-LC3) and mRFP-GFP-LC3 expression construct (ptf-LC3) were provided by Dr. Tamotsu Yoshimori of Osaka University, Japan.24 DNA was transfected into NIH3T3 cells using Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer's protocol. G418 (500 μg/ml; Invitrogen, 11811) was used for selecting stable expression cell clones. More than 50 clones were pooled, expanded and used for experiments. These 2 pooled stable cell populations were named as mRFP-LC3 cells and mRFP-GFP-LC3 cells.
Intracellular ROS detection
Reactive oxygen species production was detected by ROS detecting probe DCFH-DA (Applygen, C1300) combined with flow cytometry detection.46 DCFH-DA turns into a green fluorescent molecule called DCF when oxidized by ROS, so that intracellular ROS levels can be reflected by the fluorescence intensity produced by DCF. Three independent experiments were conducted.
Knockdown of Atg5 by shRNA
An shRNA plasmid targeting mouse Atg5 (AGAACCATACTATTTGCTT) was synthesized by Genechem (25978). NIH3T3 cells were transfected with control or Atg5 shRNA by Lipofectamine 2000 and a polyclonal pool of NIH3T3 cells was selected by adding 2 µg/ml puromycin (Sigma, P8833).
Lysosomal acid phosphatase assay
ACP2 (acid phosphatase 2, lysosomal) activity was assayed by a commercially available kit (Beyotime, P0326) according to the manufacturer's instructions. The activity of ACP2 was normalized by average lysosome content. In brief, LysoTracker Red was used to stain the lysosomes of living cells. After staining, LysoTracker Red-labeled cells were washed with PBS and lysed with 1% Triton X-100 (Sigma-Aldrich, V900502-100ML). The fluorescence intensity was read using a fluorescence microplate reader.
CTSB activity assay
CTSB activity was measured using the Magic Red Cathepsin B detection kit (ImmunoChemistry Technologies, 937). Control or H2O2-treated NIH3T3 cells were cultured in 24-well plates with different treatment times as indicated. Then cells were loaded with Magic Red Cathepsin B reagent for 1 h and washed with PBS twice. More than 10 fluorescent images were taken and representative images are shown. For quantification of CTSB activity, the fluorescence of Magic Red Cathepsin B probe was measured using a fluorescence microplate reader according to the manufacturer's manual.
Mitochondrial membrane potential assay (JC-1)
Mitochondrial membrane potential was monitored by a mitochondrial-specific dual fluorescence probe, JC-1.47 Briefly, JC-1 was added to reach a final concentration of 5 μg/ml and incubated for 20 min, and then the cells were washed twice with medium and imaged under a fluorescence microscope.
Measurement of mitochondrial DNA content and integrity using qPCR
Mitochondrial DNA integrity was measured according to a qPCR method described previously.48 The primers used for qPCR were TGCCCCTCTTCTCGCTCCGG (forward) and GGCGATAACGCATTTGATGGCC (reverse) for amplifying a short fragment in the D-loop; TGGGGGCCAACCAGTAGAACA (forward) and TGCGTCTAGACTGTGTGCTGTCC (reverse) for amplifying a semi-long fragment in the D-loop; GCGCAGTGTAGGCGGAGCTT(forward) and AAAGGAAGCGCAGACCGGCC (reverse) for amplifying a genomic Actb fragment. The mitochondrial DNA content was calculated according to the ratio of short D-loop:genomic Actb fragment. The mitochondrial DNA integrity was calculated according to the ratio of semi-long:short fragment.
Intracellular ATP level
Intracellular ATP level was measured using a commercially available intracellular ATP measurement kit (Nanjing Jiancheng, A095) according to the manufacturer's instructions. The measurement is based on the following principle: ATP and creatine are catalyzed to form creatine phosphate by creatine kinase and colorimetry of phosphomolybdic acid was used to determine the ATP in the samples.
COX (cytochrome c oxidase) activity
Determination of COX activity in protein extracts from the cells was tested using a commercially available COX activity kit (GenMed Scientifics, GMS10014.3.1) according to the manufacturer's instructions. The reduced CYCS (cytochrome c, somatic) can be catalyzed to oxidized CYCS by COX. The change of absorbance at 550 nm is quantified by spectrophotometry to reveal the activity of COX.
Statistical analysis
Data are expressed as means ± SD from at least 3 biological replicates. The difference between control and treated samples was examined using the Student t test. The difference between multiple groups was examined by one-way ANOVA with Bonferroni post-hoc. p < 0.05 is considered to be significant and p < 0.01 was considered highly significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- ATG5
autophagy-related 5
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- CDKN1A/p21
cyclin dependent kinase inhibitor 1A (P21)
- CHX
cycloheximide
- COX
cytochrome c oxidase
- CTSB
cathepsin B
- DAPI
4',6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- EBSS
Earle's balanced salt solution;
- GFP
green fluorescent protein;
- H2O2
hydrogen peroxide
- HCQ
hydroxychloroquine
- IL6
interleukin 6
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MMP
mitochondrial membrane potential
- mRFP
monomeric red fluorescent protein
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTT
methyl thiazolyl tetrazolium
- NAC
N-acetylcysteine
- PBS
phosphate buffered saline
- qRT-PCR
real-time quantitative reverse transcription PCR
- ROS
reactive oxygen species
- SA-GLB1/SA-β-gal
senescence associated GLB1/β-galactosidase
- SAHF
senescence associated heterochromatin foci
- SIPS
stress induced premature senescence
- SQSTM1/p62
sequestosome 1
- TFEB
transcription factor EB
- ULK
unc-51 like kinase
- UPRmt
mitochondrial unfold protein response;
- VPA
valproic acid
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Tamotsu Yoshimori for mRFP-GFP-LC3 plasmid, thank Dr. Canhua Huang and Yuquan Wei for continuous supports and Dr. Ping Lin, Xiujie Wang and Yi Chen for all around convenience.
Funding
This work was supported by National Natural Science Foundation of China (Grant Number 81273224), National Key Research and Development Program (Grant Number 2016YFC1200203), National 973 Basic Research Programs of China (Grant Number 2013CB911300) and also by The Wellcome Trust to Zofia Chrzanowska-Lightowlers (096919/Z/11/Z).
References
- [1].Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol 2000; 35(8):927-45; PMID:11121681; http://dx.doi.org/ 10.1016/S0531-5565(00)00180-7 [DOI] [PubMed] [Google Scholar]
- [2].Chen Q, Amos BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A 1994; 91(10):4130-4; PMID:8183882; http://dx.doi.org/ 10.1073/pnas.91.10.4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al.. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92(20):9363-7; PMID:7568133; http://dx.doi.org/ 10.1073/pnas.92.20.9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113(6):703-16; PMID:12809602; http://dx.doi.org/ 10.1016/S0092-8674(03)00401-X [DOI] [PubMed] [Google Scholar]
- [5].Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep 2003; 4(4):358-62; PMID:12671679; http://dx.doi.org/ 10.1038/sj.embor.embor806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAKSTAT 2013; 2(4):e25763; PMID:24416650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12(9):814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90(4):1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- [9].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140(3):313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12(1):1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gewirtz D. Autophagy and senescence: A partnership in search of definition. Autophagy 2013; 9(5):808-12; PMID:23422284; http://dx.doi.org/ 10.4161/auto.23922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Young A, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al.. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23(7):798-803; PMID:19279323; http://dx.doi.org/ 10.1101/gad.519709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, Rodier F, Davalos AR, Gewirtz DA. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther 2012; 343(3):763-78; PMID:22927544; http://dx.doi.org/ 10.1124/jpet.112.197590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 2011; 6(8):e23367; PMID:21858089; http://dx.doi.org/ 10.1371/journal.pone.0023367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al.. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460(7253):392-5; PMID:19587680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol 2011; 21(10):569-76; PMID:21824781; http://dx.doi.org/ 10.1016/j.tcb.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 2007; 17(9):422-7; PMID:17804237; http://dx.doi.org/ 10.1016/j.tcb.2007.07.009 [DOI] [PubMed] [Google Scholar]
- [18].Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192(7):1001-14; PMID:11015441; http://dx.doi.org/ 10.1084/jem.192.7.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 2011; 51(6):1106-15; PMID:21762777; http://dx.doi.org/ 10.1016/j.freeradbiomed.2011.06.022 [DOI] [PubMed] [Google Scholar]
- [20].Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 2006; 125(7):1241-52; PMID:16814712; http://dx.doi.org/ 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- [21].Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol 2010; 22(2):234-40; PMID:20045302; http://dx.doi.org/ 10.1016/j.ceb.2009.12.005 [DOI] [PubMed] [Google Scholar]
- [22].Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146(5):682-95; PMID:21884931; http://dx.doi.org/ 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- [23].Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta 2013; 1830(8):4117-29; PMID:23618697; http://dx.doi.org/ 10.1016/j.bbagen.2013.04.016 [DOI] [PubMed] [Google Scholar]
- [24].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3(5):452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [25].Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285(14):10850-61; PMID:20123989; http://dx.doi.org/ 10.1074/jbc.M109.080796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1(1):23-36; PMID:16874023; http://dx.doi.org/ 10.4161/auto.1.1.1495 [DOI] [PubMed] [Google Scholar]
- [27].Yu L, McPhee C, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al.. Autophagy termination and lysosome reformation regulated by mTOR. Nature 2010; 465(7300):942-6; PMID:20526321; http://dx.doi.org/ 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Muller M. Pyocyanin induces oxidative stress in human endothelialcells and modulates the glutathione redox cycle. Free Radic Biol Med 2002; 33(11):1527-33; PMID:12446210; http://dx.doi.org/ 10.1016/S0891-5849(02)01087-0 [DOI] [PubMed] [Google Scholar]
- [29].McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, et al.. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014; 137(Pt 5):1481-95; PMID:24574503; http://dx.doi.org/ 10.1093/brain/awu020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bitto A, Lerner C, Torres C, Roell M, Malaguti M, Perez V, Lorenzini A, Hrelia S, Ikeno Y, Matzko ME, et al.. Long-term IGF-I exposure decreases autophagy and cell viability. PLoS One 2010; 5(9):e12592; http://dx.doi.org/ 10.1371/journal.pone.0012592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al.. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012; 1(5):630-41; PMID:22934255; http://dx.doi.org/ 10.4161/onci.20297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4(2):176-84; PMID:18059160; http://dx.doi.org/ 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- [33].Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8(12):1888-95; PMID:19471117; http://dx.doi.org/ 10.4161/cc.8.12.8606 [DOI] [PubMed] [Google Scholar]
- [34].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13(2):132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Settembre C, DiMalta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332(6036):1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhou J, Tan SH, Nicolas V, Bauvy C, Yang ND, Zhang J, Xue Y, Codogno P, Shen HM. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res 2013; 23(4):508-23; PMID:23337583; http://dx.doi.org/ 10.1038/cr.2013.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7(2):188-204; PMID:21107021; http://dx.doi.org/ 10.4161/auto.7.2.14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5(228):ra42; PMID:22692423; http://dx.doi.org/ 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Han X, Tai H, Wang X, Wang Z, Zhou J, Wei X, Ding Y, Gong H, Mo C, Zhang J, et al.. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD+ elevation. Aging Cell 2016; 15(3):416-27; PMID:26890602; http://dx.doi.org/ 10.1111/acel.12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al.. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 2016; 35(7):724-42; PMID:26848154; http://dx.doi.org/ 10.15252/embj.201592862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016; 533(7603):416-9; PMID:27135930; http://dx.doi.org/ 10.1038/nature17989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol 2000; 35(6-7):811-20; PMID:11053672; http://dx.doi.org/ 10.1016/S0531-5565(00)00135-2 [DOI] [PubMed] [Google Scholar]
- [43].Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 2002; 269(8):1996-2002; PMID:11985575; http://dx.doi.org/ 10.1046/j.1432-1033.2002.02869.x [DOI] [PubMed] [Google Scholar]
- [44].Hu JP, Nishishita K, Sakai E, Yoshida H, Kato Y, Tsukuba T, Okamoto K. Berberine inhibits RANKL-induced osteoclast formation and survival through suppressing the NF-κB and Akt pathways. Eur J Pharmacol 2008; 580(1-2):70-9; PMID:18083161; http://dx.doi.org/ 10.1016/j.ejphar.2007.11.013 [DOI] [PubMed] [Google Scholar]
- [45].Mo C, Wang L, Zhang J, Numazawa S, Tang H, Tang X, Han X, Li J, Yang M, Wang Z, et al.. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of Berberine in LPS-Stimulated macrophages and endotoxin-shocked mice. Antioxid Redox Signal 2014; 20(4):574-88; PMID:23875776; http://dx.doi.org/ 10.1089/ars.2012.5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 2010; 594:57-72; PMID:20072909; http://dx.doi.org/ 10.1007/978-1-60761-411-1_4 [DOI] [PubMed] [Google Scholar]
- [47].Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis 2012; 3:e430; PMID:23171850; http://dx.doi.org/ 10.1038/cddis.2012.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal 2012; 17(3):492-504; PMID:22229649; http://dx.doi.org/ 10.1089/ars.2011.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.