

ABSTRACT
Improving biological functions of endothelial progenitor cells (EPCs) is beneficial to maintaining endothelium homeostasis and promoting vascular re-endothelialization. Because macroautophagy/autophagy has been documented as a double-edged sword in cell functions, its effects on EPCs remain to be elucidated. This study was designed to explore the role and molecular mechanisms of store-operated calcium entry (SOCE)-activated autophagy in proliferation of EPCs under hypercholesterolemia. We employed oxidized low-density lipoprotein (ox-LDL) to mimic hypercholesterolemia in bone marrow-derived EPCs from rat. Ox-LDL dose-dependently activated autophagy flux, while inhibiting EPC proliferation. Importantly, inhibition of autophagy either by silencing Atg7 or by 3-methyladenine treatment, further aggravated proliferative inhibition by ox-LDL, suggesting the protective effects of autophagy against ox-LDL. Interestingly, ox-LDL increased STIM1 expression and intracellular Ca2+ concentration. Either Ca2+ chelators or deficiency in STIM1 attenuated ox-LDL-induced autophagy activation, confirming the involvement of SOCE in the process. Furthermore, CAMKK2 (calcium/calmodulin-dependent protein kinase kinase 2, β) activation and MTOR (mechanistic target of rapamycin [serine/threonine kinase]) deactivation were associated with autophagy modulation. Together, our results reveal a novel signaling pathway of SOCE-CAMKK2 in the regulation of autophagy and offer new insights into the important roles of autophagy in maintaining proliferation and promoting the survival capability of EPCs. This may be beneficial to improving EPC transplantation efficacy and enhancing vascular re-endothelialization in patients with hypercholesterolemia.
KEYWORDS: autophagy, CAMKK2, cell-based therapy, endothelial progenitor cells, MTOR, oxidized low-density lipoprotein, proliferation, store-operated calcium entry
Introduction
Atherosclerosis, the leading cause of cardiovascular diseases, is initiated by the hypercholesterolemia-induced disruption of vascular endothelium integrity. Endothelial progenitor cells (EPCs) play a major role in the maintenance of endothelial integrity and contribute to re-endothelialization of injured vessels as well as revascularization of ischemic tissues. Originated from bone marrow and spleen, EPCs were first described and isolated by Asahara and his colleagues nearly 2 decades ago.1 The number of EPCs is in correlation with many cardiovascular diseases such as acute myocardial infarction, hypercholesterolemia and hypertension.2-4 Recently, EPC-based therapy has demonstrated that increasing the number or improving function of circulating EPCs may be promising in the treatment of atherosclerotic diseases. However, the poor viability of EPCs after transplantation limits the large-scale use of cell-based therapy. Therefore, exploring new strategies in improving the survival post-transplantation is crucial to enhancing the success of EPC therapy.5
Autophagy may serve as a potential target for the cell therapy improvement. Recently, Wang et al. have demonstrated that hypoxia-activated autophagy promotes EPCs survival and differentiation, suggesting that autophagy in hypoxic preconditioning may be beneficial to the adaptation of transplanted EPCs in cell-based therapy.6 Autophagy involves an evolutionarily conserved process that turns over long-lived proteins or impaired organelles through a lysosomal-mediated pathway and acts as a survival mechanism under stress conditions to maintain cellular homeostasis. Autophagic dysregulation is associated with a majority of cardiovascular pathological processes, such as atherosclerosis, cardiac hypertrophy and cardiomyopathies.7,8 A decline in autophagy flux contributes to aging-elicited cardiac hypertrophy and contractile dysfunction.9 In cardiac myocytes, ischemia and reperfusion (I/R) impair the autophagy flux, whereas enhancing autophagy constitutes a protective effect against I/R injury.10 Moderate autophagy impedes foam cell formation in macrophages and vascular smooth muscle cells (VSMCs) to stabilize atherosclerotic plaques by preventing apoptosis and plaque necrosis.11-13 However, it has not been explored whether autophagy has any effects on EPC proliferation during the repairing process of injured vessels and ischemic tissues.
The role of calcium signaling in autophagy regulation has met with a great deal of controversy in the past decade. Although various signal messengers and downstream pathways are involved in autophagy regulation, recently calcium has been recognized as an important player in this process in different cell types.14-17 Calcium is mostly considered as an activator of autophagy because calcium-mobilizing agents and calcium ionophores can promote autophagy by elevating intracellular calcium concentrations ([Ca2+]i).17-19 On the contrary, some laboratories report that [Ca2+]i suppresses autophagy.20-22 This may be attributed to the binding of BECN1 with ITPR3 in the endoplasmic reticulum membrane and/or the presence of endoplasmic reticulum-mitochondria microdomains, where calcium would be taken up by mitochondria, promoting ATP production and then inhibiting autophagy via AMPK pathway. Store-operated calcium entry (SOCE) is a major mechanism for calcium entry in EPCs in nonexcited cells. Since our laboratory has previously demonstrated SOCE promoted proliferation and migration of EPCs,23-25 we proposed that SOCE may also be involved in the regulation of autophagy in EPCs.
In the present study, we utilized ox-LDL to mimic a hypercholesterolemia environment in EPCs in vitro. As a major risk factor in the pathogenesis of atherogenesis, ox-LDL has been known to damage vascular endothelial cells, leading to an alteration in the structural integrity and the function of the endothelial barrier. In EPCs, ox-LDL decreases cell proliferation capacity as well as impairs other cell functions including migration, adhesion and vasculogenesis.26-28 It has been reported that ox-LDL changes electrophysiological properties and increases [Ca2+]i,29-31 but the detailed mechanism especially in EPCs has not been elucidated.
We hypothesized that autophagy may protect against inhibition of EPC proliferation by ox-LDL. Furthermore, [Ca2+]i increase via SOCE and CAMKK2 activation contributed to the mechanisms of autophagy induced by ox-LDL in EPCs.
Results
Ox-LDL decreases EPC proliferative activity
A new approach of real-time cell analyzer (RTCA) and a traditional method of cell counting kit-8 (CCK-8) were used to measure the proliferation of EPCs after exposure to 0, 10, 30, 60 or 100 μg/ml ox-LDL. After ox-LDL treatment, RTCA results showed that the normalized cell index of EPCs significantly decreased with the increase of ox-LDL concentration, indicating that ox-LDL decreased proliferative ability in a dose-dependent manner (Fig. 1A). In accordance with RTCA results, CCK-8 assay revealed similar results after different time intervals and doses of ox-LDL treatments (Fig. 1B).
Figure 1.
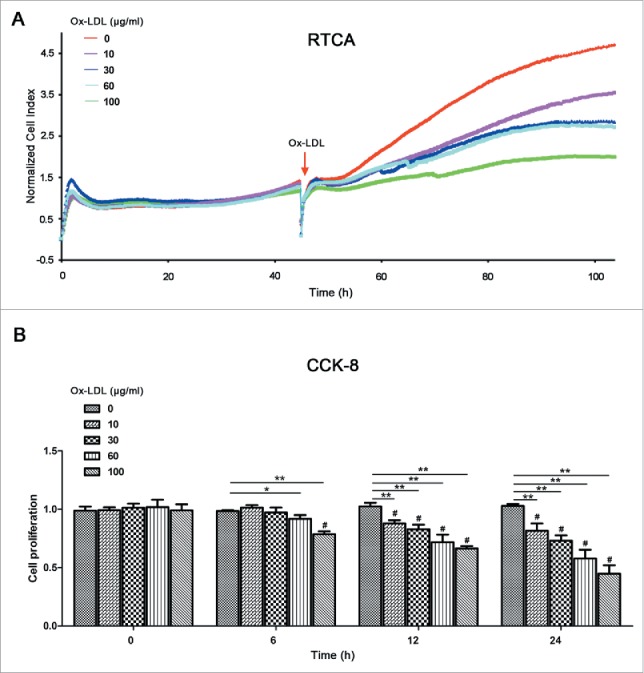
Ox-LDL treatment decreases EPC proliferative activity. (A) After seeding on E-plates for 48 h, EPCs were treated with different concentrations of ox-LDL (0, 10, 30, 60 or 100 μg/ml) respectively (red arrow) and monitored by RTCA. The normalized cell index indicated ox-LDL dose-dependently decreased EPC proliferation. Representative graphs were shown from 3 independent experiments. (B) CCK-8 results showed ox-LDL reduced EPC proliferative activity in dose- and time-dependent manners after 0, 6, 12 or 24 h ox-LDL exposure at a series of concentrations (0, 10, 30, 60 or 100 μg/ml). (Cells were isolated from 3 rats for 1 experiment and 3 independent experiments were performed; mean + SD; #P < 0.01, compared with 0 h within the same concentration group; *P < 0.05, **P < 0.01, compared with 0 μg/ml `ox-LDL in the same time of exposure).
Ox-LDL activates autophagy flux in EPCs
To confirm autophagy activity after ox-LDL exposure, we approached western blots to detect MAP1LC3B (microtubule-associated protein 1 light chain 3 β) and SQSTM1/p62, which are general biomarkers for autophagy. Results showed that at constant 12 h, 60 or 100 μg/ml ox-LDL significantly increased the MAP1LC3B-II:MAP1LC3B-I ratio compared with control (Fig. 2A), whereas SQSTM1 level significantly decreased in EPCs (Fig. 2B). Furthermore, at constant 60 μg/ml ox-LDL, the ratio of MAP1LC3B-II:MAP1LC3B-I increased significantly after 12 h (Fig. 2C) and SQSTM1 showed an opposite tendency with time, compared with control (Fig. 2D). Pretreatment with autophagy inhibitors, bafilomycin A1 (Baf) or chloroquine (CQ) before ox-LDL further enhanced MAP1LC3B-II accumulation (Fig. 2E), indicating that ox-LDL stimulated the autophagy flux. All data above suggested that ox-LDL increased autophagy in EPCs.
Figure 2.

Ox-LDL activates autophagy flux in EPCs. (A and B) EPCs were incubated with ox-LDL (0, 10, 30, 60 or 100 μg/ml) for 12 h. Western blots revealed that ox-LDL markedly increased the ratio of MAP1LC3B-II:MAP1LC3B-I (A) and decreased SQSTM1 (B) in a dose-dependent manner. (C and D) EPCs were continuously exposed to ox-LDL (60 μg/ml) for either 0, 6, 12,18 or 24 h. Western blots showed that the ratio of MAP1LC3B-II:MAP1LC3B-I increased (C) and SQSTM1 decreased (D) with time. (E) EPCs were pretreated with Baf (50 nM) for 3 h or CQ (20 μM) for 12 h before treated with ox-LDL (60 μg/ml) for 12 h. MAP1LC3B-II:MAP1LC3B-I ratio increased significantly in Baf+ox-LDL or CQ+ox-LDL group compared with ox-LDL, Baf or CQ alone, showing that autophagy flux was activated. (Cells were isolated from 3 rats for 1 experiment and at least 3 independent experiments were performed, western blot results were normalized to the controls (given as one-fold), mean + SD, **P < 0.01).
To further corroborate these findings, we utilized a pH-sensitive tandem GFP-mRFP-LC3 adenoviral construct to monitor puncta formation induced by autophagy under a laser confocal scanning microscope (LCSM). EPCs were infected with tandem GFP-mRFP-LC3 adenovirus for 24 h and continuously incubated 12 h with 60 μg/ml ox-LDL before being observed under an LCSM. Yellow puncta, reflective of RFP and GFP fluorescence combination, marks autophagosomes, whereas free red puncta (RFP only) marks autolysosomes where acidic pH quenches GFP fluorescence.32 Results showed that both free red and yellow puncta in merged images increased significantly in the ox-LDL treated group compared with control (Fig. 3A and 3B), suggesting increases of both autophagosomes and autolysosomes. Pretreatment with Baf followed by ox-LDL increased more yellow puncta but decreased free red puncta in merged images (Fig. 3A and 3B), further indicating the activation of autophagy by ox-LDL and the successful blocking of autophagy flux by Baf. In addition, green puncta in ox-LDL increased significantly compared with control, a further increase was detected with Baf pretreatment (Fig. 3C). Ox-LDL also increased red puncta in mRFP, but red puncta remained stable when cells were pretreated with Baf (Fig. 3D).
Figure 3.

Ox-LDL induces autophagosome and autolysosome formation in EPCs. (A) EPCs were infected by tandem GFP-mRFP-LC3 adenovirus for 24 h before exposure to ox-LDL (60 μg/ml) 12 h, baf (10 nM) 6 h plus ox-LDL (60 μg/ml) 12 h or baf (10 nM) 6 h alone. All the groups were observed under an LCSM. Representative images showed puncta formation in different groups. Scale bar: 25 μM. (B) Quantitative analysis of yellow and free red puncta in merged images. Ox-LDL increased the number of yellow and free red puncta in merged images compared with control. Baf plus ox-LDL further increased the number of yellow puncta but decreased free red puncta compared with ox-LDL alone. (C) Quantitative analysis of green puncta. (D) Quantitative analysis of red puncta. (n = 10 cells per group, cells were isolated from 3 rats for 1 experiment and 3 independent experiments were performed, mean + SD, *P < 0.05, **P < 0.01).
Results from western blots and immunofluorescence confirmed the induction of autolysosome formation, demonstrating that ox-LDL activated autophagy flux in EPCs.
Autophagy alleviates ox-LDL-induced inhibition of proliferation
To address the impacts of autophagy in ox-LDL-induced EPC proliferation reduction, we utilized gene-silencing as well as pharmacological techniques to inhibit autophagy activity. As shown in Fig. 4B, in the Atg7 silencing group, Atg7 was knocked down in EPCs after a 72-h lentiviral infection, while MAP1LC3B-II formation and ATG12–ATG5 conjugation decreased but SQSTM1 accumulation increased compared with control, suggesting autophagy was effectively inhibited. Under the same condition, no significant difference of proliferative activity was observed in control or Atg7 silencing groups after 96-h infection (Fig. 4A). However, 60 μg/ml ox-LDL application after Atg7 silencing significantly reduced proliferation compared with ox-LDL alone (Fig. 4C). A similar pattern was shown in the CCK-8 experiment (Fig. 4D). When applying the autophagy pharmacological inhibitor 3-methyladenine (3-MA) before ox-LDL, EPC proliferation was reduced further, compared with ox-LDL alone in both RTCA and CCK-8 assays (Fig. 4E and 4F). The selected 3-MA concentration (2 mM) has been previously confirmed to inhibit autophagy.6
Figure 4.

Autophagy was a protective response in EPC proliferation. (A) EPCs were seeded on E-plates for 24 h before lentiviral vector containing shRNA targeting Atg7 (shAtg7) infection (black arrow). RTCA indicated no significant difference among control, vector-control (VC) and shAtg7 groups. (B) Representative western blots for the detection of ATG7, ATG12–ATG5 conjugate, MAP1LC3B-II and SQSTM1 after infection showed that Atg7 was successfully knocked down and autophagy was effectively inhibited in Atg7 silencing group. (C) Cells were exposed to ox-LDL (60 μg/ml) (red arrow) after shAtg7 infection. The normalized cell index indicated that EPC proliferative activity fell down more quickly than ox-LDL alone and control groups after silencing Atg7. (D) CCK-8 assay showed a similar result that silencing Atg7 before ox-LDL treatment significantly reduced proliferative activity compared with ox-LDL alone and control groups. (E) 3-MA (2 mM) was added to inhibit autophagy (black arrow) before 60 μg/ml ox-LDL (red arrow). The normalized cell index indicated that after 3-MA inhibition, EPC proliferative activity fell down more quickly than ox-LDL alone and control groups. (F) CCK-8 assay showed similar results that the proliferative activity in 3-MA+ox-LDL group reduced significantly compared with ox-LDL alone and control groups. (Cells were isolated from 3 rats for one experiment and 3 independent experiments were performed, mean + SD, **P < 0.01).
As a consequence, autophagy inhibition by either gene silencing or inhibitor exacerbated the reduction of proliferative activity elicited by ox-LDL. These results suggested that autophagy acted as a protective response in EPC proliferation activity.
Ox-LDL evokes EPC calcium release and activates the store-operated calcium entry
Our previous studies confirm that [Ca2+]i especially from store-operated calcium channels (SOCCs) is an important factor in regulating EPC functions.24,25 However, in EPCs, it is still unclear whether ox-LDL is capable of increasing [Ca2+]i and if the change of [Ca2+]i is associated with autophagy. To confirm this, we treated EPCs with various concentrations of ox-LDL for 12 h and then loaded with the calcium probe fluo3-AM. Ox-LDL increased fluorescence intensity in EPCs under an LCSM. The higher the added ox-LDL concentration, the stronger the fluorescence intensity that was detected (Fig. S1A and B). In each experiment, Fmin was obtained by measuring fluorescence intensity in the presence of BAPTA-AM (cell-permeable intracellular Ca2+ chelator) and EGTA (extracellular Ca2+ chelator). Fmax was detected with saturating intracellular Ca2+ (ionomycin plus Ca2+). Calculation of [Ca2+]i (as described in the Materials and Methods) also revealed ox-LDL significantly increased [Ca2+]i in a dose-dependent manner (Fig. S1C).
To further elucidate whether SOCE was involved in the [Ca2+]i increase after ox-LDL exposure, we measured the changes of [Ca2+]i sensor protein, STIM1 of SOCCs, after ox-LDL treatment for 12 h. Significantly increased protein level of STIM1 was observed in 60 and 100 μg/ml ox-LDL groups over control (Fig. 5A), suggesting that ox-LDL treatment upregulated STIM1 expression. However, no significant changes of TRPC1 and ORAI1 of SOCCs were detected with ox-LDL (data not shown). In order to analyze whether STIM1 was involved in the [Ca2+]i increase induced by ox-LDL, we approached shRNA-Stim1 to silence Stim1 effectively (Fig. 5B). Stim1 knockdown followed by ox-LDL decreased both fluo3 calcium fluorescence intensity (Fig. 5C and 5D) and [Ca2+]i (Fig. 5E) compared with either ox-LDL alone or empty lentiviral vector plus ox-LDL. The data suggested that ox-LDL increased [Ca2+]i through STIM1 mediated SOCE.
Figure 5.

Ox-LDL upregulates STIM1 and increases [Ca2+]i. (A) Representative western blots for detection of STIM1 after treated with ox-LDL (0, 10, 30, 60 or 100 μg/ml) respectively for 12 h and quantitative analysis demonstrated that ox-LDL significantly increased the protein level of STIM1 at the concentration of 60 or 100 μg/ml. (B) Representative western blots and quantitative analysis showed shRNA targeting Stim1 (shStim1) effectively silenced STIM1 protein expression after 72 h infection. (C) EPCs were infected by shStim1 followed by ox-LDL (60 μg/ml) for 12 h, then examined with the fluorescent dye fluo3 under an LCSM. The representative images showed the fluorescence intensity in different groups. Scale bar: 100 μm. (D) Quantitative analysis showed that the fluorescence intensity in ox-LDL and VC+ox-LDL groups were significantly stronger than control and shStim1+ox-LDL groups. (Control: n = 97 cells, Ox-LDL: n = 91 cells, ShStim1+ox-LDL: n = 94 cells, VC+ox-LDL: n = 97 cells). (E) The exact [Ca2+]i was calculated by the equation mentioned above, indicating that ox-LDL increased [Ca2+]i, silencing Stim1 reversed the increase elicited by ox-LDL. (Cells were isolated from 3 rats for 1 experiment and 3 independent experiments were performed, western blot results were normalized to the controls (given as 1-fold), mean + SD, *P < 0.05, **P < 0.01).
To further confirm this, we incubated EPCs with fluo3-AM, and then added different doses of ox-LDL in Ca2+ free medium. Ox-LDL induced a [Ca2+]i transient increase in calcium-free solution (Fig. 6A and 6B, first wave), due to the release from calcium stores in EPCs. The subsequent reapplication of extracellular Ca2+ triggered a sharp increase of [Ca2+]i (Fig. 6A and 6C, second wave) from the effects of SOCCs. The increase of [Ca2+]i after reapplying extracellular Ca2+ was partly inhibited by either silencing Stim1 (Fig. 6D and 6E) or the SOCCs pharmacological inhibitor 2-aminoethyl diphenylborinate (2-APB) (Fig. 6F), which further implicated ox-LDL-induced [Ca2+]i increase via SOCCs.
Figure 6.

Ox-LDL increases [Ca2+]i through SOCE. (A) EPCs stained with fluorescent dye fluo3, were used to measure [Ca2+]i under an LCSM. Different concentrations of ox-LDL (0, 10, 30, 60 or 100 μg/ml ) were added to the medium without Ca2+, followed by Ca2+ (2 mM) retrieving. Ox-LDL elicited calcium stores release in EPCs (first wave) and dose-dependently activated extracellular Ca2+ influx (second wave). (B and C) Quantitative analysis of Ca2+ release and entry amplitudes. Concentrations of 30, 60 or 100 μg/ml ox-LDL elicited intracellular Ca2+ store release (B) and extracellular Ca2+ influx (C) significantly (ox-LDL 0 μg/ml: n = 40 cells, 10 μg/ml: n = 23 cells, 30 μg/ml: n = 21 cells, 60 μg/ml: n = 34 cells and 100 μg/ml: n = 25 cells). (D) Stim1 in EPCs was silenced by shRNA for 72 h before performing the [Ca2+]i measurement. Ox-LDL (60 μg/ml) was added to the medium without calcium, followed by adding back 2 mM Ca2+. The purple (shStim1), green (VC) and red (control) traces depicted representative time-course of [Ca2+]i changes in EPCs. (E) Quantitative analysis of Ca2+ entry amplitudes indicated that silencing Stim1 reduced Ca2+ influx elicited by ox-LDL (shStim1: n = 14 cells, VC: n = 14 cells and control: n = 25 cells). (F) Representative time-courses of [Ca2+]i changes showed that specific SOCCs inhibitor 2-APB inhibited extracellular Ca2+ influx elicited by ox-LDL (n = 12 cells). (Cells were isolated from 3 rats for one experiment and 3 independent experiments were performed, mean + SD, *P < 0.05, **P < 0.01).
SOCE contributes to ox-LDL-induced autophagy
To observe the effects of [Ca2+]i on ox-LDL-induced autophagy, we pretreated EPCs with EGTA or BAPTA-AM for 20 min before ox-LDL application. Although incubation with either EGTA or BAPTA-AM alone had no significant effect on the basal level of autophagy, ox-LDL-induced autophagy was reversed by incubating the cells with EGTA or BAPTA-AM (Fig. 7A), suggesting that calcium was required in ox-LDL-induced autophagy. Besides, Stim1 knockdown alleviated the autophagy level activated by ox-LDL (Fig. 7B), indicating the involvement of SOCCs in the autophagy activation process under ox-LDL exposure.
Figure 7.

SOCE contributes to autophagy induction. (A) EPCs were pretreated with EGTA (10 mM) or BAPTA-AM (20 μM) for 20 min followed by ox-LDL (60 μg/ml) for 12 h. Western blots showed that EGTA or BAPTA-AM significantly decreased the ratio of MAP1LC3B-II:MAP1LC3B-I increased by ox-LDL. (B) Stim1 was silenced by shRNA for 72 h before ox-LDL (60 μg/ml) exposure, autophagy level in Stim1-knocked down group decreased significantly compared with ox-LDL alone group. (C) Treated EPCs (as in A and B) were infected by GFP-mRFP-LC3 adenovirus for 24 h and observed under an LCSM. Representative images showed puncta forming in different groups. Scale bar: 25 μM. (D) Quantitative analysis of the yellow puncta in EGTA or BAPTA-AM pretreatment groups, indicating pretreatment with EGTA or BAPTA-AM decreased yellow puncta (n = 8 cells per group). (E) Quantitative analysis of the yellow puncta in Stim1-knockdown groups, showing silencing Stim1 decreased yellow puncta (n = 8 cells per group). (Cells were isolated from 3 rats for one experiment and 3 independent experiments were performed, western blot results were normalized to the controls (given as one-fold), mean + SD, *P < 0.05, **P < 0.01).
GFP-mRFP-LC3 marked EPCs under an LCSM also revealed that ox-LDL increased the number of autophagosomes (yellow puncta) in EPCs, while pretreatment with either EGTA or BAPTA-AM respectively partly inhibited this effect (Fig. 7C and 7D). Moreover, the autophagosome increase by ox-LDL was also partly reversed by silencing Stim1, implicating the participation of STIM1 in the ox-LDL-induced autophagy activation (Fig. 7C and 7E).
Both the results from western blots and autophagosome calculations manifested that the [Ca2+]i increase by SOCE contributed to ox-LDL-induced autophagy in EPCs.
The SOCE-CAMKK2-MTOR pathway is related to the ox-LDL-induced autophagy activation
In our study, we demonstrated that ox-LDL-activated autophagy required SOCCs-mediated Ca2+ influx in EPCs, but the downstream Ca2+ signaling was unclear. In other cell types, CAMKK2 signaling shows a relationship with autophagy.33 To test whether this is also the case in our ox-LDL-induced autophagy process, we applied western blots to confirm that ox-LDL dose-dependently increased the Ser511 phosphorylation of CAMKK2 with no change of CAMKK2 protein expression, suggesting the activation of CAMKK2 (Fig. 8A). Pretreated EPCs with STO-609 (10 μM), a CAMKK2 inhibitor,34 not only inhibited CAMKK2 phosphorylation (Fig. 8B) but also reduced AMPK phosphorylation at Thr172 (Fig. 8C), the downstream target protein of CAMKK2, implicating inhibition of CAMKK2 activity by STO-609. In addition, STO-609 reversed the ratio of MAP1LC3B-II:MAP1LC3B-I increased by ox-LDL as well (Fig. 8D), indicating the association of CAMKK2 phosphorylation with autophagy induction by ox-LDL. CAMKK2 activation was also related to SOCE because phosphorylation of CAMKK2 was inhibited by shStim1 (Fig. 8E). The data above suggested that ox-LDL-induced autophagy was associated with CAMKK2 activation and at least in part through SOCCs. In addition, ox-LDL dose-dependently decreased the phosphorylation levels of MTOR at Ser2448 and its substrate, RPS6KB/p70S6K (ribosomal protein S6 kinase 1) at Thr389 (Fig. 8F and 8H). STO-609 pretreatment before ox-LDL significantly inhibited phosphorylation of MTOR (Fig. 8G) and RPS6KB (Fig. 8I) compared with ox-LDL alone, suggesting the association of ox-LDL-induced autophagy with CAMKK2 and MTOR kinase.
Figure 8.

Phosphorylation of CAMKK2 and dephosphorylation of MTOR are related to ox-LDL-induced autophagy. (A) EPCs were treated with different concentrations of ox-LDL (0, 10, 30, 60 or 100 μg/ml) for 12 h. Western blot analysis showed that ox-LDL dose-dependently increased phosphorylation of CAMKK2. (B and C) EPCs were pretreated with the CAMKK2 inhibitor STO-609 (10 μM) before ox-LDL (60 μg/ml) exposure, the phosphorylation of CAMKK2 and AMPK were significantly decreased compared with ox-LDL alone. (D) Western blots indicated that STO-609 reduced the ratio of MAP1LC3B-II:MAP1LC3B-I increased by ox-LDL. (E) Stim1 was silenced with shRNA for 72 h before ox-LDL (60 μg/ml) exposure 12 h, the phosphorylation of CAMKK2 was significantly decreased in Stim1-knocked down group compared with ox-LDL alone. (F) EPCs were treated with different concentrations of ox-LDL (0, 10, 30, 60 or 100 μg/ml) for 12 h. Western blots analysis showed that phosphorylation of MTOR decreased significantly at the concentration of 30, 60 or 100 μg/ml of ox-LDL. (G) STO-609 (10 μM) were pretreated with EPCs before ox-LDL (60 μg/ml) exposure, STO-609 reversed phosphorylation of MTOR compared with ox-LDL alone groups. (H) EPCs were treated with different concentrations of ox-LDL (0, 10, 30, 60 or 100 μg/ml) for 12 h. Western blots analysis showed that phosphorylation of RPS6KB decreased significantly at the concentration of 30, 60 or 100 μg/ml of ox-LDL. (I) EPCs were pretreated with STO-609 (10 μM) before ox-LDL (60 μg/ml), STO-609 reversed phosphorylation of RPS6KB compared with ox-LDL alone groups. (Cells were isolated from 3 rats for 1 experiment and 3 independent experiments were performed, western blot results were normalized to the controls (given as 1-fold), mean + SD, *P < 0.05, **P < 0.01).
As a consequence, our results revealed that ox-LDL upregulated CAMKK2 phosphorylation in EPCs partly through SOCE. Moreover, phosphorylated CAMKK2 may contribute to MTOR inhibition and autophagy induction.
Discussion
In the present study, we found that ox-LDL inhibited proliferation as well as activated autophagy flux in dose- and time-dependent manners in EPCs. Autophagy activation may act as an endogenous protective response against ox-LDL-induced proliferative inhibition, as inhibition of autophagy further reduced proliferation level. In addition, ox-LDL elevated [Ca2+]i partly through SOCCs whereas SOCE elevation was associated with autophagy increase. Furthermore, we revealed that ox-LDL-activated autophagy was associated with CAMKK2 phosphorylation and MTOR dephosphorylation. We conclude that ox-LDL inhibits EPC proliferation but also activates the endogenous protective response by autophagy as well, and the autophagy activation is associated with SOCE and CAMKK2 signaling pathway. For the first time, we confirm that SOCE contributes to the autophagy induction under ox-LDL exposure in EPCs.
Previous studies have demonstrated that ox-LDL impairs EPCs proliferation and inhibits tube formation.26-28,35 Consistently, we found ox-LDL dose-dependently inhibited EPCs proliferation (Fig. 1). Although not fully elucidated, the mechanisms may involve increasing expression of OLR1/LOX-1 (oxidized low-density lipoprotein [lectin-like] receptor 1),26 activation of MAPK1436 and downregulation of SELE/E-selectin or the ITGAV-ITGB5 heterodimeric protein (integrin αvβ5).37 In addition, recent reports suggest that reduction of integrin-linked kinase expression and phosphorylation of downstream protein kinase are related to ox-LDL-induced impairment of EPC biological function.27 Thus, reducing the negative effects of ox-LDL may be beneficial to EPCs in re-endothelialization. In our study, we found that ox-LDL not only inhibited proliferation but also activated self-protective response by activating autophagy flux.
Ox-LDL is considered as atherogenic and apoptotic as well. Previous studies have focused on ox-LDL-induced apoptosis in VSMCs,38-40 endothelial cells41-44 and EPCs.45 The differences in oxidative status, concentration and the treatment time of ox-LDL as well as cell species might account for the different effects of our study compared with previous studies. In our study, 60 μg/ml ox-LDL at 12 h had no significant influence on EPC apoptosis rate (Fig. S2A) and mitochondrion membrane potential (MMP, Fig. S2B), but showed significant changes at 24 h. These results implicated that at the ox-LDL concentration and time we used, there might be hardly any influence from apoptosis. Furthermore, at our condition, ox-LDL may activate autophagy before apoptosis induction in EPCs.
Moderate autophagy exerted cardio-protective effect, but may be deleterious when activated to excess.7,8,46 In advanced atherosclerosis, autophagy stabilizes atherosclerotic plaques by preventing macrophage apoptosis and plaque necrosis, as well as by impeding foam-cell formation in VSMCs. When inhibiting autophagy by silencing Atg7 or 3-MA in EPCs, the proliferation of EPCs decreased further under ox-LDL exposure (Fig. 4C to 4F). These data suggested that the activation of autophagy promoted the resistance of EPCs to ox-LDL for survival. However, the mechanism of ox-LDL in autophagy regulation remains controversial. Some studies indicate that ox-LDL induces autophagosome and autolysosome formation by upregulating expression of OLR147,48 or peroxisome proliferator-activated receptor gamma (PPARG)49 and their downstream production of reactive oxygen species (ROS). Excessive generated ROS in response to damaged mitochondria which escape from autophagy, causes cellular dysfunction and cell death.50 Conversely, recent studies show that ox-LDL increases expression of OLR1 and subsequently attenuates a protective autophagy response in bovine aortic endothelial cells51 and impairs autophagy, partly through activation of the MTOR signaling pathway in VSMCs.11,52 Various concentrations and oxidative status of ox-LDL as well as the corresponding signal pathways might be related to the disputed effects of ox-LDL-induced autophagy. In our study, we applied physiological and pathological serum concentrations of ox-LDL (10-100 μg/ml) to treat EPCs,26,53,54 and detected a dose-dependent increase of autophagy flux (Fig. 2) and (Fig. 3).
SOCE-mediated intracellular calcium increase seems essential but also controversial in ox-LDL-autophagy regulation. Previous study have revealed that mitochondrial ion channels are involved in angiotensin-II-induced autophagy in VSMCs.55 Ox-LDL is reported to induce a prompt and transient Ca2+ increase in a dose-dependent manner,31 Our study further confirmed that Ca2+ rise after ox-LDL stimulation was primarily due to Ca2+ influx through SOCCs in EPCs. More importantly, SOCE contributed to autophagy induction by ox-LDL. However, the role of Ca2+ in autophagy regulation is still on debate. Many reports suggest that Ca2+ and protein-driven modulation of calcium, enhances autophagy.17,18,56-58 On the contrary, other studies consider Ca2+ as an autophagy inhibitor.20,59 They demonstrate that BECN1 bound with ITPR3, which locates on the endoplasmic reticulum membrane and/or the presence of endoplasmic reticulum-mitochondria microdomains. Ca2+ could be taken up by the complex of BECN1 and ITPR3 to promote production of ATP then inhibit autophagy.20-22 In colorectal cancer cells, the SOCE inhibitor SKF-96365 induces cytoprotective autophagy,60 but suppresses autophagy in both epithelial and neuronal cells.61 Different cell status and complex downstream calcium signaling might account for the controversial results.
We showed that ox-LDL activated autophagy by promoting SOCE. However, how ox-LDL is internalized then regulating SOCE remains to be elucidated. Previous findings in leukocytes consider that cell surface receptors, which interact with phospholipase C, are involved in the calcium-mobilizing effect of ox-LDL.30 In endothelial cells, OLR1 has been confirmed as a cell surface receptor to regulate autophagy under ox-LDL exposure or low shear stress.62,63 As the precursor of endothelial cells, it is likely that OLR1 is also responsible for ox-LDL internalization so that ox-LDL may have the effect of releasing calcium stores, upregulating STIM1 expression and then activating SOCE in EPCs. Of course this needs to be elucidated in future studies.
The SOCE-CAMKK2-MTOR signal pathway may be important in Ca2+-induced autophagy regulation. In our study, ox-LDL phosphorylated CAMKK2 and inhibited MTOR activity to activate autophagy in EPCs (Fig. 8A, 8F, and 8H), whereas Ca2+ influx through SOCE phosphorylated CAMKK2 to activate autophagy in EPCs (Fig. 8E), which verified the involvement of SOCE-CAMKK2-MTOR signal pathway in ox-LDL-autophagy induction. Although different in cell types and research purposes, evidence shows that the CAMKK2-MTOR signal pathway is involved in Ca2+-activated autophagy in Hela, MCF-717 and PC 12 cells33 as well as with other pharmacological agents that increase [Ca2+]i-induced autophagy.18 In vascular endothelial cells, the canonical MTOR axis is involved in autophagy regulation with ox-LDL treatment.64 In addition, the MAPK1/ERK2-MAPK3/ERK1 pathway is also proposed to be essential for Ca2+-activated autophagy.19 It is reasonable to speculate that ox-LDL elevates SOCE, increases the activity of CAMKK2, and then activates the autophagy response, which contributes to the protection of the EPC proliferation. However, our data suggested that the SOCE-CAMKK2 pathway may not be the only one to activate autophagy in ox-LDL stimulation. Mitochondrial damage and ROS generation or endoplasmic reticulum stress may also be associated with autophagy induction under ox-LDL exposure in other cell lines.44,50,65 Whether this is also the case in EPCs needs to be clarified.
In conclusion, as shown in the schematic drawing (Fig. 9), we indicated that ox-LDL inhibited EPC proliferation but also activated the protective autophagy response. The increased autophagy was regulated at least in part by the novel signaling pathway of SOCE-CAMKK2-MTOR.
Figure 9.
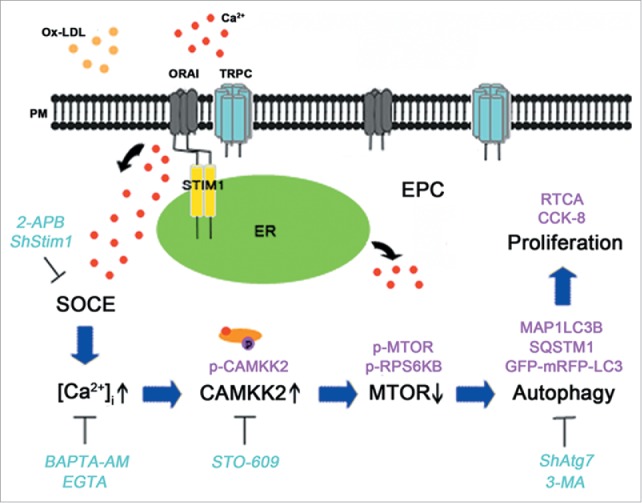
A schematic drawing of the calcium signal and autophagy pathway in EPCs after ox-LDL exposure. Ox-LDL activates SOCE and elevates [Ca2+]i in EPCs, furthermore the [Ca2+]i increases phosphorylation of CAMKK2 and decreases the downstream protein MTOR phosphorylation to activate autophagy. Autophagy serves as a protective role in ox-LDL-induced inhibition of proliferation. (Lines with arrowheads indicate induction or activation, and lines with bars at the end indicate blockade or inhibition).
Our results provided novel fundamental evidence in regulating EPC proliferation through protective autophagy. In patients with hypercholesterolemia, enhancing protective autophagy will be beneficial to improving EPC survival rate and cell transplantation efficacy, thus promoting vascular re-endothelialization and slowing down the progress of atherosclerosis. Furthermore, modification of SOCCs might be developed into a potential therapeutic breakthrough for the protective autophagy regulation. Our findings may provide a survival-promoting mechanism and a potential therapeutic target of EPCs as well as the optimization of stem cell transplantation strategies in the context of hypercholesterolemia.
Materials and methods
Reagents
EGM-2MV BulletKit medium was purchased from Lonza, containing endothelial basal medium (EBM-2, CC-3156) and 10% fetal bovine serum (CC-4101A) and supplemented with recombinant Homo sapiens (rHs) FGF2/FGF-B (CC-4101A), rHsIGF1 (CC-4115A), rHsEGF (CC-4317A), rHsVEGF (CC-4414A), ascorbic acid (CC-4116A), heparin (CC-4396A). Antibodies against rat MAP1LC3B (2775S), ATG7 (8558S), SQSTM1 (5114S), STIM1 (5668), ORAI1 (3280), p-CAMKK2 (12818), MTOR (2972), p-MTOR (2971), RPS6KB (2708) and p-RPS6KB (9205) were obtained from Cell Signal Technology. Antibodies against CAMKK2 (ab96531), AMPK (ab32047) and p-AMPK (ab133448) were obtained from Abcam. Antibody against ATG5 (AP1812a) was from Abgent and this antibody could detect both free ATG5 (molecular mass 32 kDa) and ATG12–ATG5 conjugate (molecular mass 55 kDa). Lymphoprep (1.083, 10831), chloroquine (CQ, C6628), bafilomycin A1 (Baf, B1793), FITC-UEA-I (L9006), 3-methyladenine (3-MA, M9281), 2-aminoethyl diphenylborinate (2-APB, D9745) and STO-609 (S1318) were purchased from Sigma. One,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labeled acetylated LDL (DiI-acLDL, L3484) was acquired from Molecular Probes. Antibodies against CD34 (SC-7324) and TRPC1 (SC-15055) were purchased from Santa Cruz Biotechnology. FITC-conjugated anti-rat PROM1/CD133 (BS-0395R), PECAM1/CD31 (BS-0468R), KDR/VEGFR2 (ER-1542) antibodies and the corresponding isotype control IgGs were obtained from Bioss.
Ox-LDL preparation
Ox-LDL (YB-002) was obtained from Yiyuan Biotechnologies. This lot of ox-LDL derived from human LDL. LDL is oxidized using 2 μM Cu2SO4 at 37°C overnight. Oxidation is terminated by adding excess EDTA-Na2 and analyzed on agarose gel electrophoresis for migration versus LDL. This lot of ox-LDL migrates 1.2-fold further than the native LDL. TBARS (thiobarbituric acid reactive substances) is determined colorimetrically by using malondialdehyde (MDA) as a standard: 12.0 nM MDA per mg of protein in ox-LDL. The best response we used was seen with 60 μg/ml ox-LDL which was based on mimicking a high level of ox-LDL in hypercholesterolemia as described in published studies.51,53
Isolation and characterization of EPCs
All animal procedures were approved by the Experimental Animal Ethics Committee of the Third Military Medical University before performing the study and conformed to the regulations of Guide for the Care and Use of Laboratory Animals (8th edition, National Research Council, USA, 2011). In our study, male Sprague-Dawley rats (150 to 180g, Chongqing, China) were anesthetized with an intramuscular injection of 100 mg/kg ketamine and 5 mg/kg xylazine, then sacrificed by cervical dislocation. Bone marrow was harvested by flushing the femurs and tibias. Bone marrow-derived mononuclear cells (BMNCs) were isolated using density-gradient centrifugation followed by washing 3 times in phosphate-buffered saline (PBS; Boster Biological Technology, AR0032). At last BMNCs were resuspended in EGM-2MV BulletKit medium and seeded on common culture plates (Corning, 430639, USA) or E-plates (ACEA Biosciences, L8).
To confirm the phenotype of EPCs, we incubated the cells with 10 mg/ml DiI-acLDL for 4 h and 10 mg/ml FITC-UEA-I for 1 h. At last the cells were incubated with 1 μg/ml DAPI. Triple-stained cells positive for Dil-acLDL, lectin and DAPI were identified as EPCs (Fig. S3A). In addition, the cells were processed for immunofluorescence staining to evaluate the expression of KDR/VEGF receptor 2, PECAM1/CD31, CD34, and PROM1/CD133 in fluorescence-activated cell sorting (Fig. S3B).
Cell proliferation assays
Cell proliferation was checked by the xCelligence Real-Time Cell Analyzer instrument (RTCA, ACEA Biosciences, San Diego, CA, USA). BMNCs were seeded on the E-plate, a specialized plate used with RTCA. Each well contained a gold microelectrode array to measure the electrical impedance in real time. Forty-eight h after seeding, nonadherent cells were removed, and the adherent cells were kept cultured on the E-plate. The medium in each well was changed every second day. The cells were underwent different treatments and the cell growth was monitored for more than 7 d.
Cell counting kit-8 (CCK-8; Beyotime Biotechnology, C0038) was employed to measure the cell proliferation as well. The cells were plated onto 96-well culture plates and underwent different treatments, then WST-8 dye (10 μl) was added to each well. After 4 h incubation at 37°C, the absorbance at 450 nm of each well was measured in a microplate reader (Emax, Molecular Devices, E13456, CA, USA). All experiments were performed in triplicates and repeated 3 times.
Fluorescence Ca2+ measurements in intact cells
Kd is the dissociation constant of fluo3 for Ca2+ at room temperature (400 nM).67,68
Gene silencing
Lentiviral vector (LV) carrying Atg7 shRNA was constructed by Hanbio Technology (Shanghai, China) and LV carrying Stim1 shRNA was constructed by Gene Pharma (Shanghai, China). LV was added to the cells at a multiplicity of infection of 100 at 2 d of seeding BMNCs. The transfection medium was changed 48 h later, and cells were cultured in fresh medium continuously. Real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) and western blots were used to detect the efficiency of knockdown of Atg7 or Stim1 in EPCs.
GFP-mRFP-LC3 adenoviral vector monitor the autophagy flux
EPCs were plated on glass-bottomed cell culture dishes and infected by adenoviral vectors containing GFP-mRFP-LC3 (HanBio Technology) according to the manufacturer's instruction. EPCs were replaced with fresh medium and then incubated for 24 h. EPCs were observed under an LCSM to confirm the infection efficiency and autophagy flux was determined by evaluating the number of YFP and RFP puncta.
RT-qPCR
Total RNA was isolated from EPCs using RNAiso reagent (Takara, D312). Isolated RNA was reverse transcribed using PrimeScript RT reagent Kit (Takara, RR047A) to cDNA following manufacturer's protocol. Quantitative PCR was performed using SYBR Premix Ex Taq II kit (Takara, RR820A), with the following primers (Takara): Stim1, forward: 5′CTTGGCCTGGGATCTCAGAG3′, reverse: 5′TCAGCCATTGCCTTCTTGCC3′; Atg7, forward: 5′GGCACCCAAAGACATCAAGG3′, reverse: 5′GTGTTGTGCAGGGTTCCCAT3′; Gapdh, forward: 5′ATGCCATCACTGCCACTC3′, reverse: 5′GGGTAGGAACACGGAAGG3′.
Immunoblots
After being rinsed 3 times by ice-cold phosphate-buffered saline, EPCs were lysed with cell lysis buffer (Pierce, 89900) supplemented with 0.5 mM PMSF and 2 mM sodium orthovanadate. Following the centrifugation (14000 g, 15 min), protein concentrations were detected by the BCA assay (Beyotime Biotechnology, P0012). The same mass of total protein were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk in TBS (Boster Biological Technology, AR0031) containing 0.5% Tween-20 (Solarbio, T8220) and then membrane-bound proteins were probed with primary antibodies against MAP1LC3B, SQSTM1, ATG7, ATG12–ATG5 conjugate, STIM1, TRPC1, ORAI1, CAMKK2, p-CAMKK2, RPS6KB, p-RPS6KB, MTOR, p-MTOR or ACTB, followed by HRP-conjugated secondary antibodies. The bands of protein were visualized by chemiluminescence detection and quantified by Image Quant TL software (GE Healthcare, Sweden).
Apoptosis analysis
EPC apoptosis rate was evaluated by ANXA5/annexin V-PE apoptosis detection kit I (BD Biosciences, 559763). After exposure to 60 μg/ml ox-LDL 0, 12 and 24 h, EPCs were washed 3 times with 3 ml cold PBS and then resuspended in 1× binding buffer (BD Biosciences, 51-66121E) at a concentration of 1 × 106 cells/ml. The solution was transfered to a 2 ml culture tube plus 5 μl ANXA5-PE (BD Biosciences, 51-65875X) and 5 μl 7-AAD (BD Biosciences, 51-68981E), then incubated for 20 min at 25°C in the dark. Flow cytometry (Beckman Coulter, MoFlo XDP, CA, USA) was performed to detect apoptosis of EPCs.
Mitochondrial membrane potential analysis
Disruption of mitochondrial membrane potential (MMP) was assessed by mitochondrial membrane potential assay kit with JC-1 (Beyotime Biotechnology, C2006). Briefly, EPCs of 5 to 7 d were seeded on the glass-bottomed cell culture dishes, then exposed to 60 μg/ml ox-LDL 0, 12 or 24 h. Washing the cells twice with PBS and incubating with JC-1 working solution for 20 min at 37°C in the dark. Then EPCs were rinsed with cold JC-1 staining buffer twice. We monitored the red fluorescence with excitation at 525 nm and emission at 590 nm as well as the green fluorescence with excitation at 490 nm and emission at 530 nm in an LCSM. Then we compared the ratio of red and green fluorescence in different groups.
Statistical analysis
Statistical analysis was performed using ANOVA followed by a t test corrected for multiple comparisons (Least-Significant-Difference). Nonparametric ANOVA (Kruskall–Wallis) followed by the Dunn multiple comparison post-hoc test was used when one or more data sets did not show Gaussian distribution. Values of P < 0.05 were considered statistically significant.
Supplementary Material
Abbreviations
- 2-APB
2-aminoethyl diphenylborinate
- 3-MA
3-methyladenine
- 7-AAD
7-Aminoactinomycin D
- ACTB
actin, β
- AMPK
AMP-activated protein kinase
- ANXA5
annexin V
- ATG5
autophagy-related 5
- ATG7
autophagy-related 7
- ATG12
autophagy-related 12
- Baf
bafilomycin A1
- BECN1
Beclin 1, autophagy related
- BMNCs
bone marrow-derived mononuclear cells
- [Ca2+]i
intracellular calcium concentration
- CAMKK2
calcium/calmodulin-dependent protein kinase kinase 2, β
- CCK-8
cell counting kit-8
- CQ
chloroquine
- DiI-acLDL
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labeled acetylated LDL
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- ERK
extracellular-regulated kinase
- EPC
endothelial progenitor cell
- FGF2
fibroblast growth factor 2
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- I/R
ischemia and reperfusion
- IGF1
insulin-like growth factor 1
- IgG
immunoglobulin G
- ITGAV-ITGB5
integrin αvβ5 heterodimeric protein
- ITPR3
inositol 1,4,5-trisphosphate receptor 3
- KDR
kinase inset domain protein receptor
- LCSM
laser confocal scanning microscope
- LV
lentiviral vector
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MAPK
mitogen-activated protein kinase
- MDA
malondialdehyde
- MMP
mitochondrial membrane potential
- Mt
mitochondrial
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- OLR1/LOX-1
oxidized low density lipoprotein (lectin-like) receptor 1
- ORAI1
ORAI1 calcium release-activated calcium modulator 1
- ox-LDL
oxidized low-density lipoprotein
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- PECAM1
platelet/endothelial cell adhesion molecule 1
- PMSF
phenylmethanesulfonyl fluoride
- PPARG
peroxisome proliferator activated receptor gamma
- PROM1
prominin 1
- RFP
red fluorescent protein
- rHs
recombinant Homo sapiens
- ROS
reactive oxygen species
- RPS6KB/p70S6K
ribosomal protein S6 kinase
- RTCA
real-time cell analyzer
- shRNA
short hairpin RNA
- SDS
sodium dodecyl sulfate
- SELE
selectin
- SOCCs
store-operated calcium channels
- SOCE
store-operated calcium entry
- SQSTM1/p62
sequestosome 1
- STIM1
stromal interaction molecule 1
- TBARS
thiobarbituric acid reactive substances
- TRPC1
transient receptor potential cation channel 1, subfamily C, member 1
- UEA
ulex europaeus agglutinin
- VEGF
vascular endothelial growth factor
- VSMCs
vascular smooth muscle cells
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof. Hui Dong, Prof. Jiqin Lian and Dr. Zhenhong Ni for their valuable advice on the manuscript revision.
Funding
This work was supported by the National Natural Science Foundation of China (81370211).
References
- [1].Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997; 275:964-7; PMID:9020076; http://dx.doi.org/ 10.1126/science.275.5302.964 [DOI] [PubMed] [Google Scholar]
- [2].Cuadrado-Godia E, Regueiro A, Nunez J, Diaz-Ricard M, Novella S, Oliveras A, Valverde MA, Marrugat J, Ois A, Giralt-Steinhauer E, et al.. Endothelial progenitor cells predict cardiovascular events after atherothrombotic stroke and acute myocardial infarction. A PROCELL substudy. PLoS One 2015; 10:e132415; http://dx.doi.org/ 10.1371/journal.pone.0132415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 2003; 348:593-600; PMID:12584367; http://dx.doi.org/ 10.1056/NEJMoa022287 [DOI] [PubMed] [Google Scholar]
- [4].Umemura T, Soga J, Hidaka T, Takemoto H, Nakamura S, Jitsuiki D, Nishioka K, Goto C, Teragawa H, Yoshizumi M, et al.. Aging and hypertension are independent risk factors for reduced number of circulating endothelial progenitor cells. Am J Hypertens 2008; 21:1203-9; PMID:18787520; http://dx.doi.org/ 10.1038/ajh.2008.278 [DOI] [PubMed] [Google Scholar]
- [5].Xu JY, Lee YK, Wang Y, Tse HF. Therapeutic application of endothelial progenitor cells for treatment of cardiovascular diseases. Curr Stem Cell Res Ther 2014; 9:401-14; PMID:24947903; http://dx.doi.org/ 10.2174/1574888X09666140619121318 [DOI] [PubMed] [Google Scholar]
- [6].Wang HJ, Zhang D, Tan YZ, Li T. Autophagy in endothelial progenitor cells is cytoprotective in hypoxic conditions. Am J Physiol Cell Physiol 2013; 304:C617-26; PMID:23269239; http://dx.doi.org/ 10.1152/ajpcell.00296.2012 [DOI] [PubMed] [Google Scholar]
- [7].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:1845-6; PMID:23656658; http://dx.doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- [8].Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest 2015; 125:55-64; PMID:25654551; http://dx.doi.org/ 10.1172/JCI73943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ceylan-Isik AF, Dong M, Zhang Y, Dong F, Turdi S, Nair S, Yanagisawa M, Ren J. Cardiomyocyte-specific deletion of endothelin receptor a rescues aging-associated cardiac hypertrophy and contractile dysfunction: Role of autophagy. Basic Res Cardiol 2013; 108:335; PMID:23381122; http://dx.doi.org/ 10.1007/s00395-013-0335-3 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [10].Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281:29776-87; PMID:16882669; http://dx.doi.org/ 10.1074/jbc.M603783200 [DOI] [PubMed] [Google Scholar]
- [11].Li BH, Yin YW, Liu Y, Pi Y, Guo L, Cao XJ, Gao CY, Zhang LL, Li JC. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Dis 2014; 5:e1182; PMID:24743737; http://dx.doi.org/ 10.1038/cddis.2014.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012; 15:545-53; PMID:22445600; http://dx.doi.org/ 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grootaert MO, Da CMP, Bitsch N, Pintelon I, De Meyer GR, Martinet W, Schrijvers DM. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015; 11:2014-32; PMID:26391655; http://dx.doi.org/ 10.1080/15548627.2015.1096485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, Bultynck G, Parys JB. MTOR-controlled autophagy requires intracellular Ca(2+) signaling. PLoS One 2013; 8:e61020; PMID:23565295; http://dx.doi.org/ 10.1371/journal.pone.0061020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].East DA, Campanella M. Ca2+ in quality control. An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013; 9:1710-9; PMID:24121708; http://dx.doi.org/ 10.4161/auto.25367 [DOI] [PubMed] [Google Scholar]
- [16].Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J Biol Chen 2012; 287:38625-36; ; http://dx.doi.org/ 10.1074/jbc.M112.365767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al.. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/ 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- [18].Law BY, Wang M, Ma DL, Al-Mousa F, Michelangeli F, Cheng SH, Ng MH, To KF, Mok AY, Ko RY, et al.. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol Cancer Ther 2010; 9:718-30; PMID:20197400; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0700 [DOI] [PubMed] [Google Scholar]
- [19].Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 2008; 65:3640-52; PMID:18850067; http://dx.doi.org/ 10.1007/s00018-008-8383-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al.. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270-83; PMID:20655468; http://dx.doi.org/ 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al.. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006-17; PMID:19325567; http://dx.doi.org/ 10.1038/cdd.2009.34 [DOI] [PubMed] [Google Scholar]
- [22].Wong A, Grubb DR, Cooley N, Luo J, Woodcock EA. Regulation of autophagy in cardiomyocytes by Ins(1,4,5)P(3) and IP(3)-receptors. J Mol Cell Cardiol 2013; 54:19-24; PMID:23137780; http://dx.doi.org/ 10.1016/j.yjmcc.2012.10.014 [DOI] [PubMed] [Google Scholar]
- [23].Guo RW, Wang H, Gao P, Li MQ, Zeng CY, Yu Y, Chen JF, Song MB, Shi YK, Huang L. An essential role for stromal interaction molecule 1 in neointima formation following arterial injury. Cardiovasc Res 2009; 81:660-8; PMID:19052075; http://dx.doi.org/ 10.1093/cvr/cvn338 [DOI] [PubMed] [Google Scholar]
- [24].Kuang CY, Yu Y, Guo RW, Qian DH, Wang K, Den MY, Shi YK, Huang L. Silencing stromal interaction molecule 1 by RNA interference inhibits the proliferation and migration of endothelial progenitor cells. Biochem Biophys Res Commun 2010; 398:315-20; PMID:20599714; http://dx.doi.org/ 10.1016/j.bbrc.2010.06.088 [DOI] [PubMed] [Google Scholar]
- [25].Wang LY, Zhang JH, Yu J, Yang J, Deng MY, Kang HL, Huang L. Reduction of store-operated Ca(2+) entry correlates with endothelial progenitor cell dysfunction in atherosclerotic mice. Stem Cells Dev 2015; 24:1582-90; PMID:25753987; http://dx.doi.org/ 10.1089/scd.2014.0538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lin FY, Tsao NW, Shih CM, Lin YW, Yeh JS, Chen JW, Nakagami H, Morishita R, Sawamura T, Huang CY. The biphasic effects of oxidized-low density lipoprotein on the vasculogenic function of endothelial progenitor cells. PLoS One 2015; 10:e123971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lu C, Yu X, Zuo K, Zhang X, Cao C, Xu J, Wang S, Tang T, Ye M, Pei E, et al.. Tripterine treatment improves endothelial progenitor cell function via integrin-linked kinase. Cell Physiol Biochem 2015; 37:1089-103; PMID:26402060; http://dx.doi.org/ 10.1159/000430234 [DOI] [PubMed] [Google Scholar]
- [28].Tie G, Yan J, Yang Y, Park BD, Messina JA, Raffai RL, Nowicki PT, Messina LM. Oxidized low-density lipoprotein induces apoptosis in endothelial progenitor cells by inactivating the phosphoinositide 3-kinase/Akt pathway. J Vasc Res 2010; 47:519-30; PMID:20431300; http://dx.doi.org/ 10.1159/000313879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kim MY, Liang GH, Kim JA, Choi SS, Choi S, Suh SH. Oxidized low-density lipoprotein- and lysophosphatidylcholine-induced ca mobilization in human endothelial cells. Korean J Physiol Pharmacol 2009; 13:27-32; PMID:19885023; http://dx.doi.org/ 10.4196/kjpp.2009.13.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van Tits LJ, Hak-Lemmers HL, Demacker PN, Stalenhoef AF, Willems PH. Oxidized low-density lipoprotein induces calcium influx in polymorphonuclear leukocytes. Free Radic Biol Med 2000; 29:747-55; PMID:11053776; http://dx.doi.org/ 10.1016/S0891-5849(00)00372-5 [DOI] [PubMed] [Google Scholar]
- [31].Yuan Z, Miyoshi T, Bao Y, Sheehan JP, Matsumoto AH, Shi W. Microarray analysis of gene expression in mouse aorta reveals role of the calcium signaling pathway in control of atherosclerosis susceptibility. Am J Physiol Heart Circ Physiol 2009; 296:H1336-43; PMID:19304945; http://dx.doi.org/ 10.1152/ajpheart.01095.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo AA, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 2012; 21:511-25; PMID:22012985; http://dx.doi.org/ 10.1093/hmg/ddr481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem 2002; 277:15813-8; PMID:11867640; http://dx.doi.org/ 10.1074/jbc.M201075200 [DOI] [PubMed] [Google Scholar]
- [35].Cui Y, Narasimhulu CA, Liu L, Li X, Xiao Y, Zhang J, Xie X, Hao H, Liu JZ, He G, et al.. Oxidized low-density lipoprotein alters endothelial progenitor cell populations. Front Biosci (Landmark Ed) 2015; 20:975-88; PMID:25961537; http://dx.doi.org/ 10.2741/4351 [DOI] [PubMed] [Google Scholar]
- [36].Wu Y, Wang Q, Cheng L, Wang J, Lu G. Effect of oxidized low-density lipoprotein on survival and function of endothelial progenitor cell mediated by p38 signal pathway. J Cardiovasc Pharmacol 2009; 53:151-6; PMID:19188833; http://dx.doi.org/ 10.1097/FJC.0b013e318197c637 [DOI] [PubMed] [Google Scholar]
- [37].Di Santo S, Diehm N, Ortmann J, Volzmann J, Yang Z, Keo HH, Baumgartner I, Kalka C. Oxidized low density lipoprotein impairs endothelial progenitor cell function by downregulation of E-selectin and integrin α(v)beta5. Biochem Biophys Res Commun 2008; 373:528-32; PMID:18590706; http://dx.doi.org/ 10.1016/j.bbrc.2008.06.066 [DOI] [PubMed] [Google Scholar]
- [38].Ingueneau C, Huynh UD, Marcheix B, Athias A, Gambert P, Negre-Salvayre A, Salvayre R, Vindis C. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J Cell Mol Med 2009; 13:1620-31; PMID:20187291; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00593.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hwang JS, Eun SY, Ham SA, Yoo T, Lee WJ, Paek KS, Do JT, Lim DS, Seo HG. PPARdelta modulates oxLDL-induced apoptosis of vascular smooth muscle cells through a TGF-β/FAK signaling axis. Int J Biochem Cell Biol 2015; 62:54-61; PMID:25732738; http://dx.doi.org/ 10.1016/j.biocel.2015.02.014 [DOI] [PubMed] [Google Scholar]
- [40].Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN, Yu BX, Zhou P, Zhang FR, Liu YY, et al.. Inhibition of orai1 Store-Operated calcium channel prevents foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol 2016; 36:618-28; PMID:26916730; http://dx.doi.org/ 10.1161/ATVBAHA.116.307344 [DOI] [PubMed] [Google Scholar]
- [41].Hong D, Bai YP, Gao HC, Wang X, Li LF, Zhang GG, Hu CP. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014; 235:310-7; PMID:24911634; http://dx.doi.org/ 10.1016/j.atherosclerosis.2014.04.028 [DOI] [PubMed] [Google Scholar]
- [42].Vindis C, Elbaz M, Escargueil-Blanc I, Auge N, Heniquez A, Thiers JC, Negre-Salvayre A, Salvayre R. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. Arterioscler Thromb Vasc Biol 2005; 25:639-45; PMID:15618541; http://dx.doi.org/ 10.1161/01.ATV.0000154359.60886.33 [DOI] [PubMed] [Google Scholar]
- [43].Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm 2013; 2013:152786; PMID:23935243; http://dx.doi.org/ 10.1155/2013/152786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Muller C, Salvayre R, Negre-Salvayre A, Vindis C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: Prevention by HDLs. Autophagy 2011; 7:541-3; PMID:21412049; http://dx.doi.org/ 10.4161/auto.7.5.15003 [DOI] [PubMed] [Google Scholar]
- [45].Tie G, Yan J, Messina JA, Raffai RL, Messina LM. Inhibition of p38 mitogen-activated protein kinase enhances the apoptosis induced by oxidized low-density lipoprotein in endothelial progenitor cells. J Vasc Res 2016; 52:361-71; http://dx.doi.org/ 10.1159/000443889 [DOI] [PubMed] [Google Scholar]
- [46].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- [47].Choi SH, Gonen A, Diehl CJ, Kim J, Almazan F, Witztum JL, Miller YI. SYK regulates macrophage MHC-II expression via activation of autophagy in response to oxidized LDL. Autophagy 2015; 11:785-95; PMID:25946330; http://dx.doi.org/ 10.1080/15548627.2015.1037061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Deng X, Fan Y, Xiang D, Mehta JL. Lectin-like ox-LDL receptor-1 (LOX-1)-Toll-like receptor 4 (TLR4) interaction and autophagy in CATH a differentiated cells exposed to angiotensin II. Mol Neurobiol 2015; 51:623-32; PMID:24902807; http://dx.doi.org/ 10.1007/s12035-014-8756-z [DOI] [PubMed] [Google Scholar]
- [49].Zabirnyk O, Liu W, Khalil S, Sharma A, Phang JM. Oxidized low-density lipoproteins upregulate proline oxidase to initiate ROS-dependent autophagy. Carcinogenesis 2010; 31:446-54; PMID:19942609; http://dx.doi.org/ 10.1093/carcin/bgp299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al.. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012; 485:251-5; PMID:22535248; http://dx.doi.org/ 10.1038/nature10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mollace V, Gliozzi M, Musolino V, Carresi C, Muscoli S, Mollace R, Tavernese A, Gratteri S, Palma E, Morabito C, et al.. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int J Cardiol 2015; 184:152-8; PMID:25703423; http://dx.doi.org/ 10.1016/j.ijcard.2015.02.007 [DOI] [PubMed] [Google Scholar]
- [52].Brito PM, Devillard R, Negre-Salvayre A, Almeida LM, Dinis TC, Salvayre R, Auge N. Resveratrol inhibits the mTOR mitogenic signaling evoked by oxidized LDL in smooth muscle cells. Atherosclerosis 2009; 205:126-34; PMID:19108833; http://dx.doi.org/ 10.1016/j.atherosclerosis.2008.11.011 [DOI] [PubMed] [Google Scholar]
- [53].Brinkley TE, Nicklas BJ, Kanaya AM, Satterfield S, Lakatta EG, Simonsick EM, Sutton-Tyrrell K, Kritchevsky SB. Plasma oxidized low-density lipoprotein levels and arterial stiffness in older adults: The health, aging, and body composition study. Hypertension 2009; 53:846-52; PMID:19332658; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.108.127043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Serke H, Vilser C, Nowicki M, Hmeidan FA, Blumenauer V, Hummitzsch K, Losche A, Spanel-Borowski K. Granulosa cell subtypes respond by autophagy or cell death to oxLDL-dependent activation of the oxidized lipoprotein receptor 1 and toll-like 4 receptor. Autophagy 2009; 5:991-1003; PMID:19730000; http://dx.doi.org/ 10.4161/auto.5.7.9507 [DOI] [PubMed] [Google Scholar]
- [55].Yu KY, Wang YP, Wang LH, Jian Y, Zhao XD, Chen JW, Murao K, Zhu W, Dong L, Wang GQ, et al.. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res Cardiol 2014; 109:416; PMID:24847907; http://dx.doi.org/ 10.1007/s00395-014-0416-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Decuypere JP, Paudel RC, Parys J, Bultynck G. Intracellular Ca(2+) signaling: A novel player in the canonical mTOR-controlled autophagy pathway. Commun Integr Biol 2013; 6:e25429; PMID:24265855; http://dx.doi.org/ 10.4161/cib.25429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914-25; PMID:20114074; http://dx.doi.org/ 10.1016/j.cellsig.2010.01.015 [DOI] [PubMed] [Google Scholar]
- [58].Lam D, Kosta A, Luciani MF, Golstein P. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol Biol Cell 2008; 19:691-700; PMID:18077554; http://dx.doi.org/ 10.1091/mbc.E07-08-0823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Harr MW, McColl KS, Zhong F, Molitoris JK, Distelhorst CW. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy 2010; 6:912-21; PMID:20814235; http://dx.doi.org/ 10.4161/auto.6.7.13290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou Y, Pan H, Han W. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIgamma/AKT-mediated pathway. Cancer Lett 2016; 372:226-38; PMID:26803057; http://dx.doi.org/ 10.1016/j.canlet.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sukumaran P, Sun Y, Vyas M, Singh BB. TRPC1-mediated Ca(2)(+) entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis 2015; 6:e1674; PMID:25741599; http://dx.doi.org/ 10.1038/cddis.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ding Z, Liu S, Sun C, Chen Z, Fan Y, Deng X, Wang X, Mehta JL. Concentration polarization of ox-LDL activates autophagy and apoptosis via regulating LOX-1 expression. Sci Rep 2013; 3:2091; PMID:23807658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ding Z, Liu S, Deng X, Fan Y, Wang X, Mehta JL. Hemodynamic shear stress modulates endothelial cell autophagy: Role of LOX-1. Int J Cardiol 2015; 184:86-95; PMID:25697875; http://dx.doi.org/ 10.1016/j.ijcard.2015.01.065 [DOI] [PubMed] [Google Scholar]
- [64].Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E(−)/(−) mice. Sci Rep 2014; 4:5519; PMID:24980430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ding Z, Wang X, Schnackenberg L, Khaidakov M, Liu S, Singla S, Dai Y, Mehta JL. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7 g. Int J Cardiol 2013; 168:1378-85; PMID:23305858; http://dx.doi.org/ 10.1016/j.ijcard.2012.12.045 [DOI] [PubMed] [Google Scholar]
- [66].Decuypere JP, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgo J, Agostinis P, Missiaen L, De Smedt H, Parys JB, et al.. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011; 7:1472-89; PMID:22082873; http://dx.doi.org/ 10.4161/auto.7.12.17909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Walczysko P, Wagner E, Albrechtova JT. Use of co-loaded Fluo-3 and Fura Red fluorescent indicators for studying the cytosolic Ca(2+)concentrations distribution in living plant tissue. Cell Calcium 2000; 28:23-32; PMID:10942701; http://dx.doi.org/ 10.1054/ceca.2000.0132 [DOI] [PubMed] [Google Scholar]
- [68].Zhang L, Yu H, Sun Y, Lin X, Chen B, Tan C, Cao G, Wang Z. Protective effects of salidroside on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Eur J Pharmacol 2007; 564:18-25; PMID:17349619; http://dx.doi.org/ 10.1016/j.ejphar.2007.01.089 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.