

Abstract
Post-ganglionic sympathetic neurons innervate secondary lymphoid organs and secrete norepinephrine (NE) as the primary neurotransmitter. NE binds and signals through 5 distinct members of the adrenergic receptor family. In this study, we show elevated expression of the β2-adrenergic receptor (ADRB2) on primary human CD8+ effector memory T cells. Treatment of both human and murine CD8+ T cells with NE decreased IFN-γ and TNF-α secretion and suppressed their cytolytic capacity in response to T-cell receptor (TCR) activation. The effects of NE were specifically reversed by β2-specific antagonists. Adrb2−/− CD8+ T cells were completely resistant to the effects of NE. Further, the ADRB2-specific pharmacological ligand, albuterol, significantly suppressed CTL effector functions in both human and mouse CD8+ T cells. While both TCR activation and stimulation with IL-12 + IL-18 were able to induce inflammatory cytokine secretion, NE failed to suppress IFN-γ secretion in response to IL-12 + IL18. Finally, the long-acting ADRB2-specific agonist, salmeterol, markedly reduced the cytokine secretion capacity of CD8+ T cells in response to infection with vesicular stomatitis virus. This study reveals a novel intrinsic role for ADRB2 signaling in CD8+ T-cell function and underscores the novel role this pathway plays in adaptive T-cell responses to infection.
Keywords: Norepinephrine, β2-adrenergic receptor, CD8+ T cells, cytokine, cytolysis
Introduction
The sympathetic nervous system regulates several key physiological functions, which are acutely mobilized during the “fight-or-flight” response. These effects are achieved by the release of epinephrine (E) and norepinephrine (NE) from the adrenal gland and NE released by adrenergic nerve endings that terminate in multiple organs such as heart, lungs, and gut. Moreover, sympathetic nerve endings also innervate secondary lymphoid organs [1, 2]. For example, the splenic nerve terminates in adrenergic neurons within the spleen that secrete NE upon stimulation. The stimuli include stress, shock, various cytokines, and importantly infection [3–6].
Adrenergic receptors (ADRs), the family of receptors bound by E and NE, are expressed on cells of the immune system. In human leukocytes, the β-ADRs are expressed with binding sites that vary between 500–2000 sites/cell [7, 8]. NK cells express the most binding sites, followed by monocytes, B cells and CD8+ T cells, with CD4+ T cells expressing the lowest. Functional studies have confirmed that β-ADR signaling regulates various functions in distinct cell subsets. For example, B-cell co-stimulatory molecules as well as IgE secreted from IgE-isotype-switched cells are increased [9, 10], while monocytes and macrophages display decreased pro-inflammatory cytokine production in response to NE [11, 12]. In T cells, Th1 cytokine production by CD4+ T cells is decreased with NE [13], and regulatory T-cell function is also enhanced with ADRB2 ligands [14].
Recent studies have begun to focus on the effects of NE on CD8+ T-cell responses. Grebe et al determined the effect of chemical sympathectomy with 6-hydroxydopamine on anti-influenza CD8+ T-cell responses [5]. In their study, blocking the secretion of NE before influenza infection increased the IFN-γ+CD8+ T-cell response, and blocking ADRB2 signaling with ADRB2-specific antagonists paralleled their observations in sympathectomized animals. Kalinichenko et al, demonstrated a role for NE in inhibiting the generation of anti-tumor CD8+ T cells [15]. The failure to generate CD8+ T cells was mediated in part by suppression of TNF-α from CD4+, CD8+ and F4/80+ cells by NE signaling. This observation was supported by a recent study demonstrating that the long-acting β2-agonist, salmeterol, could inhibit both cytokine secretion and cytotoxic activity in human CD8+ T cells [16]. In contrast, a recent study demonstrated differential sensitivity to NE signaling between naïve and memory CD8+ T cells, and activation of the ADRB2 led to increased inflammatory cytokine secretion, which may reflect differences in various subsets of CD8+ T cells [17]. In some situations, external conditions that stimulate sympathetic signaling can significantly modulate immune responses [18]. For example, thermoregulation through responses to external temperature or internal fever augment cytokine secretion and lytic activity in CD8+ T cells [19]. Further, in mice, housing temperature dramatically influences T cell-mediated GVHD and anti-tumor responses, which are regulated directly through the ADRB2 [20–23].
In this study, we comprehensively addressed the role of adrenergic signaling on multiple aspects of CD8+ T cell effector functions that emanate from both T cell receptor signaling and cytokine-mediated pathways. We found that NE dominantly suppresses inflammatory cytokine secretion and lytic activity by exclusively signaling through the ADRB2. This suppression was specific for TCR-induced functions as stimulation with IL-12 + IL-18 was unaffected by NE to suppress IFN-γ secretion. Taken together, we reveal an important role for ADRB2 signaling to modulate the magnitude of the CD8+ T cell effector function to infection.
Results
NE signaling via ADRB2 inhibits cytokine production and cytolytic activity of human CD8+ T cells
Our previous studies revealed significant gene expression differences between effector memory (CCR7loCXCR3hi; TEM) and central memory/naïve (CCR7hiCXCR3lo; TCM/N) CD8+ T cells in healthy human donors [24]. These data are available from GEO under accession number GSE27337. Within this dataset we found the β2-adrenergic receptor (ADRB2) to be differentially expressed between TEM and TCM/N, with TEM cells having higher expression of ADBR2 (Fig. 1A, left panel). This was confirmed using qPCR (Fig. 1A, right panel). Further, other adrenergic receptor family member mRNAs were below the limit of detection through the microarray analysis. In order to determine the role that the ADRB2 may play in effector function, CD8+ T cells were isolated from PBMCs of healthy human donors and acutely activated in the absence or presence of NE with plate-bound anti-CD3 and anti-CD28 Abs. IFN-γ and TNF-α secretion was assessed 24 hours later and found to be suppressed by NE at both 1 and 5 µM concentrations (Fig. 1B). Of note, we consistently observed that the magnitude of suppression was much greater for TNF-α than for IFN-γ at all concentrations of NE. The kinetics of this suppression was rapid, as NE decreased both IFN-γ and TNF-α cytokine secretion as early as 2 hours post-stimulation (Fig. 1C). As part of the CD8+ T-cell functional assessment, we tested the ability of NE to modulate cytolytic activity using a re-directed lysis assay. We observed significant killing activity at all effector to target ratios that was significantly suppressed by NE (Fig. 1D).
Figure 1. NE suppresses human CD8+ T-cell cytokine secretion and lytic activity.

(A) ADRB2 mRNA expression was assessed by microarray (left, [24]) and qPCR (right) in CD8+CCR7hiCXCR3lo and CD8+CCR7loCXCR3hi cells from peripheral blood of healthy human donors. (B and C) CD8+ T cells were isolated and activated (B) with anti-CD3 and anti-CD28 antibodies for the indicated time points or (C) for 24 hours with increasing concentrations of NE. IFN-γ and TNF-α in the supernatants were measured by ELISA. (D) CD8+ T cells were isolated and incubated with 51Cr-labeled and anti-CD3-coated target cells at increasing effector to target ratios. 51Cr release was measured by scintillation counting 12 hours later. (B, D) Data are shown as mean +/− SEM of 3 replicates and are representative of 3 experiments performed with similar results. (C) The kinetic experiment was performed once. *p<0.05, ** p<0.01, ***p<0.001, ****p<0.0001 compared to CCR7hi or to 0 µM NE, by 2-way ANOVA with Bonferroni post-test.
Although the other ADR family member mRNAs were lowly expressed as assessed by microarray (GSE27337 [24]), it was possible that NE was suppressing CD8+ T-cell effector functions through multiple receptors. Specific adrenergic receptor antagonists were used to determine which receptor was involved in the suppression of CD8+ T-cell function. Phentolamine, a pan α-ADR antagonist, failed to reverse the inhibition of cytokine secretion by NE, indicating that the α-ADR family members were not involved in this pathway (Fig. 2A). In contrast, nadolol, a pan β-ADR antagonist, completely reversed the effects of NE (Fig. 2A). The β-ADR family consists of three members: β1 (ADRB1), β2 (ADRB2), and β3 (ADRB3). As such, we found that atenolol, an ADRB1-specific antagonist, failed to reverse the effects NE, even at the highest concentration tested (Fig. 2B). However, the ADRB2-specific antagonist ICI-118,551 completely reversed the suppressive effects of NE with concentrations as low as 100 nM, confirming an exclusive role for the ADRB2 in suppressing CD8+ T-cell effector function. In order to further confirm this hypothesis, we utilized the ADRB2-specific ligand albuterol. Similar to the effect seen with NE, albuterol significantly suppressed cytokine secretion in human CD8+ T cells at 100-fold lower concentrations than NE due to differences in binding affinities (Fig. 2C).
Figure 2. NE suppresses human cytokine secretion in CD8+ T cells through the ADRB2.

(A) Human CD8+ T cells were isolated and activated with anti-CD3 and anti-CD28 antibodies and NE for 24 hours in the presence of milliQ water (Ctrl), or a pan-α (phentolamine), or a pan-β (nadolol) adrenergic receptor antagonist. IFN-γ and TNF-α in the supernatants were measured by ELISA. (B) CD8+ T cells were isolated and activated as in (A), in the presence of phentolamine (10µM), or increasing concentrations of a β1-specific (atenolol, 0.1µM to 5µM), or a β2-specific (ICI-118,551, 0.1µM to 5µM) adrenergic receptor antagonist. TNF-α production was measured at 24 hours by ELISA. (C) CD8+ T cells were isolated and activated as in (A), in the presence of methanol (V.C.) or increasing concentrations of albuterol or NE, and IFN-γ and TNF-α production measured at 24 hours. Data shown are presented as mean +/− SEM of 3 replicates and are representative of (A and C) 2 experiments and (B) 3 experiments, all from individual donors. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 compared to 0 µM NE or V.C, by 2-way (A and B) or 1-way (C) ANOVA with Bonferroni post-test.
NE signaling inhibits cytokine production of murine CD8+ T cells in response to CD3 stimulation
Although few studies have addressed the effect of NE on CD8+ T-cell responses in mice [5, 15], there remains little understanding of the specificity for ADRB2 signaling and its intrinsic effects on CD8+ T-cell function. Therefore we decided to expand our studies to address whether the effects of ADRB2 signaling were conserved in murine CD8+ T cells, as suggested by studies of chemical sympathectomy during viral infections [5]. Mouse CD8+ T cells from spleen and LNs of naïve C57Bl/6 mice were activated with plate-bound anti-CD3/anti-CD28 Abs for 24 hours with increasing concentrations of NE. As observed in human CD8+ T cells, NE suppressed IFN-γ and TNF-α secretion in a dose dependent manner in mouse CD8+ T cells (Fig. 3A). Likewise, the effects of NE were found to reduce IFN-γ and TNF-α mRNA levels in response to anti-CD3 stimulation (Fig. 3B). In order to determine if ADRB2 signaling could interfere with antigen-specific cytokine secretion, CD8+ T cells from TCR transgenic clone 4 (Cl4) mice were tested, which have a TCR specific for the influenza HA peptide (IYSTVASSL). Spleen cells from Cl4 mice were incubated for 24 hours with IYSTVASSL in the presence or absence of NE. Both IFN-γ and TNF-α as well as IL-2 secretion were suppressed by NE (Fig. 3C). Further, both NE and the β2-specific agonist albuterol suppressed cytokine secretion from mouse Cl4 CD8+ T cells (Fig. 3D), suggesting that the suppression was mediated through ADRB2-dependent signaling. To confirm this, we activated purified CD8+ T cells from Balb/c mice with plate-bound anti-CD3/anti-CD28 Abs in the presence or absence of NE or albuterol, and in the presence of different ADR antagonists. As expected, only nadolol and ICI-118,551 was able to revert the effect of NE and albuterol on mouse CD8+ T-cell cytokine secretion (Fig. 3E). This result was confirmed with purified CD8+ T cells from ADRB2−/− animals as neither NE nor albuterol were able to suppress cytokine secretion in genetically ADRB2-deficient CD8+ T cells (Fig. 3E). Notably, the effect of NE on IFN-γ secretion was dependent upon the strength of stimulation, while TNF-α was responsive to NE regardless of stimulation strength (Supporting Information Fig. 1).
Figure 3. ADRB2 signaling modulates mouse CD8+ T-cell function.
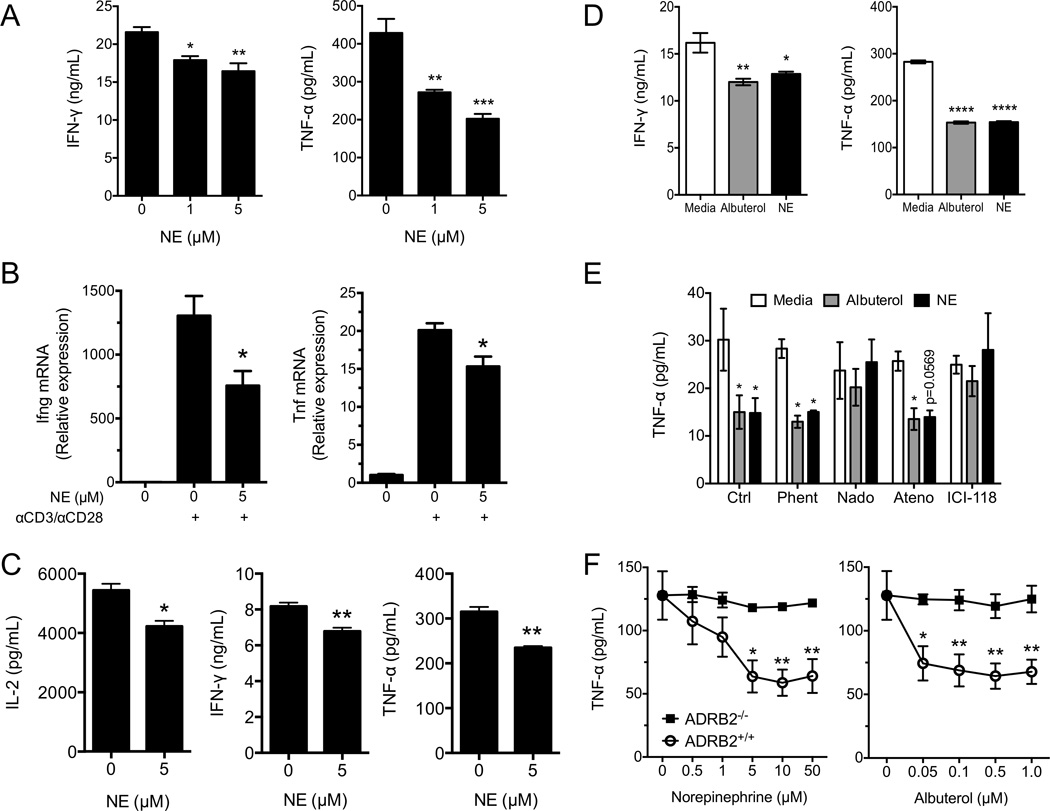
(A) Purified CD8+ T cells from C57Bl/6 mice were activated with anti-CD3 and anti-CD28 antibodies and increasing concentrations of NE for 24 hours. IFN-γ and TNF-α in the supernatants were measured by ELISA. (B) CD8+ T cells from Cl4 mice were activated with anti-CD3 and anti-CD28 antibodies in the presence or absence of NE for 12 hours. RNA was harvested and Ifng and Tnf transcripts were quantified by qPCR. (C) Splenocytes from Cl4 mice were incubated overnight with the HA peptide IYSTVASSL, in the absence of exogenous IL-2 and in the presence or absence of NE. IL-2, IFN-γ and TNF-α were measured from the supernatants at 24 hours. (D) Splenocytes from Cl4 mice were incubated with the HA peptide IYSTVASSL in the presence or absence of albuterol or NE. IFN-γ and TNF-α in the supernatants were measured at 18 hours. (D) CD8+ T cells from Balb/cJ mice were activated as in (A), in the presence of albuterol or NE and in the presence of milliQ water (Ctrl), or a pan-α (phentolamine), a pan-β (nadolol), a β1-specific (atenolol), or a β2-specific (ICI-118,551) adrenergic receptor antagonist (100 nM each). IFN-γ (not shown) and TNF-α in the supernatants were measured. (E) CD8+ T cells from ADRB2+/+ or ADRB2−/− Balb/cJ mice were isolated and activated as in (A), in the presence of increasing concentrations of NE or albuterol. IFN-γ (not shown) and TNF-α in the supernatants were measured. Data are presented as mean +/− SEM of triplicate determinations and are representative of (A and D) 3 experiments, (B, E, and F) 2 experiments, and (C) 1 experiment. * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 compared to 0 µM NE or media, by Student’s t-test (C), or 1-way (A, B, and D) or 2-way (E and F) ANOVA with Bonferroni post-test.
NE suppresses CD8+ T-cell function independently of nicotinic acetylcholine receptor signaling
Although ADRB2 signaling was required for cytokine suppression by NE, it was possible that the effects were indirect through the induction of a negative regulatory pathway. Indeed, Rosas-Ballina et al demonstrated that memory-like CD4+ T cells could secrete acetylcholine in response to high concentrations of NE [25]. Acetylcholine can decrease pro-inflammatory cytokine secretion of macrophages by signaling through the α7 nicotinic acetylcholine receptor [26–28]. To test whether NE was driving acetylcholine-mediated suppression of cytokine secretion, OT-I spleen cell suspensions were activated with the CD8+ T-cell specific ovalbumin peptide (SIINFEKL) in the presence or absence of NE and increasing concentrations of methyllycaconitine (an α7 nicotinic acetylcholine receptor antagonist). As shown, methyllycaconitine failed to revert the effect of NE, even at concentrations as high as 10 µM (Fig. 4A). The ADRB2 signals through the adenylyl cyclase pathway in many cell types, and elevated cAMP levels have been shown to inhibit cytokine expression in CD4+ T cells [29, 30]. As expected, forskolin markedly suppressed TNF-α secretion in CD8+ T cells in response to TCR stimulation (Fig. 4B). However, attempts to block this suppression with two distinct adenylyl cyclase inhibitors, 2’5’-dideoxyadenosine and KH7, failed to reverse the inhibitory effects of NE on TNF-α secretion. These results indicate that while cAMP mobilization profoundly blocks TCR-mediated cytokine expression, additional pathways may interface with adenylyl cyclase activation by NE to suppress CD8+ T-cell effector function. Nonetheless, that data support a direct role for ADRB2 signaling that does not require autocrine responses to acetylcholine.
Figure 4. ADRB2 signaling suppresses mouse CD8+ T-cell cytokine secretion independent of acetylcholine or cAMP.
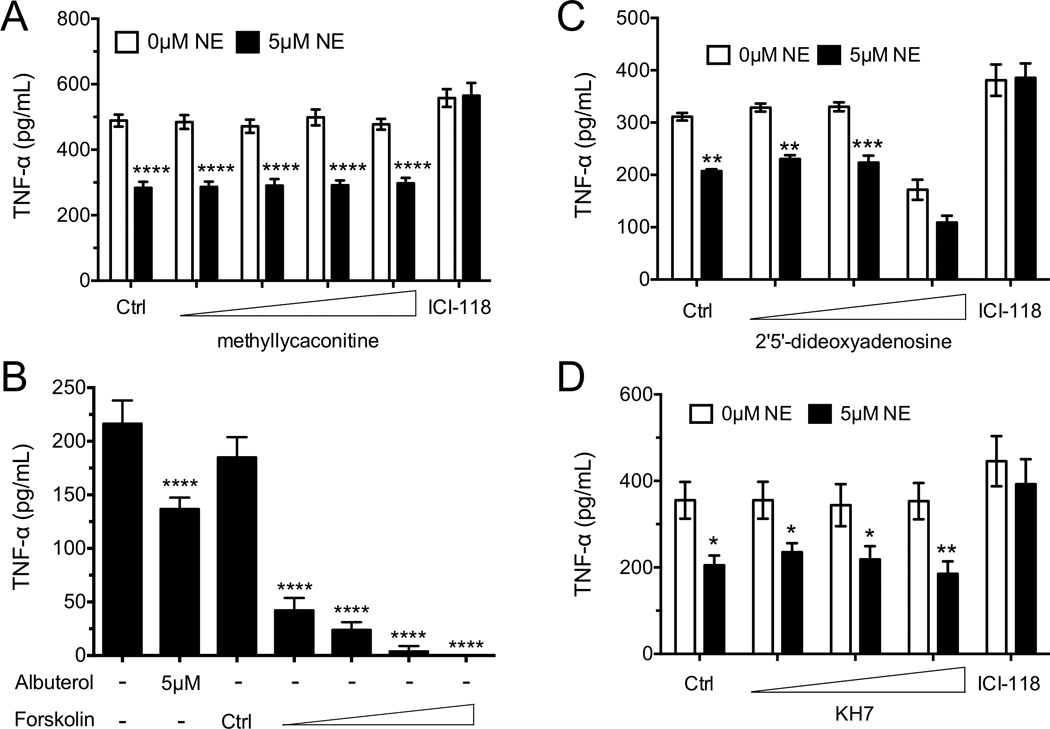
(A) Splenocytes from OT-I mice were isolated and incubated 23 hours with the OVA peptide SIINFEKL and NE with increasing concentrations of an α7 nicotinic receptor antagonist (methyllycaconitine) or a β2 adrenergic receptor antagonist (ICI-118,551). TNF-α in the supernatants was measured. (B) Splenocytes from Cl4 mice were incubated overnight with the HA peptide IYSTVASSL and in the presence or absence of albuterol or increasing concentrations of the cAMP stimulator forskolin. IFN-γ and TNF-α were measured. (C) Splenocytes from Cl4 mice were incubated with HA peptide in the presence or absence of NE, and with increasing concentrations of an adenylyl cyclase inhibitor (2’5’-dideoxyadenosine;) or a β2 adrenergic receptor antagonist (ICI-118,551). (D) CD8+ T cells from Balb/cJ mice were isolated and stimulated with anti-CD3 and anti-CD28 Abs (3 µg/mL each) for 24 hours in the presence or absence of NE and increasing concentrations of a soluble adenylyl cyclase inhibitor (KH7) or a β2 adrenergic receptor antagonist (ICI-118,551). Data are presented as mean +/− SEM of triplicate determinations and are representative of (A and D) a single experiment with pooled data from 3 mice measured separately, (B and C) 2 experiments with pooled data from 3 mice measured separately in each experiment. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 compared to 0µM NE, No Albuterol, or Ctrl, 2-way (A, C and D) or 1-way (B) ANOVA with Bonferroni post-test.
Long-acting β2 agonists suppress cytokine secretion capacity of CD8+ CTLs in vivo
Upon responding to infection, naïve CD8+ T cells divide in response to both antigen and cytokine signals delivered by antigen presenting cells. IL-12 potently drives their differentiation into primary effector cells, which later contract into a small pool of memory cells as the infection resolves. While NE acutely inhibited TCR-mediated effector activity (described above), it was possible that ADRB2 signaling could also influence their development into effector cells in response to IL-12, either by altering their proliferative capacity or by regulating their capacity to secrete inflammatory cytokines upon restimulation. To address the role of NE in altering proliferation, VPD450-labeled Cl4 cells were activated with HA peptide in the absence or presence of NE over a period of 3 days. Dividing cells were tracked based upon VPD450 dilution. NE did not significantly alter the proportion of cells that expanded, nor was there a delay in their progression from one cell division to the next (Fig. 5A). Further, no differences were observed in the rate of cell division between WT and ADRB2-deficient cells (Fig. 5A). To assess the effects of NE during effector cell differentiation, human CD8+ T cells were activated with anti-CD3/anti-CD28 in the absence or presence of IL-12 for 7 days, and NE was added to these cultures at the initiation of priming. IL-12 significantly enhanced the ability of these cells to secrete IFN-γ upon restimulation, while TNF-α secretion remained unaffected. Further, addition of NE to these cultures during priming did not impact their ability to secrete either IFN-γ or TNF-α upon recall stimulation.
Figure 5. ADRB2 signaling does not affect CD8+ T-cell proliferation or programming.
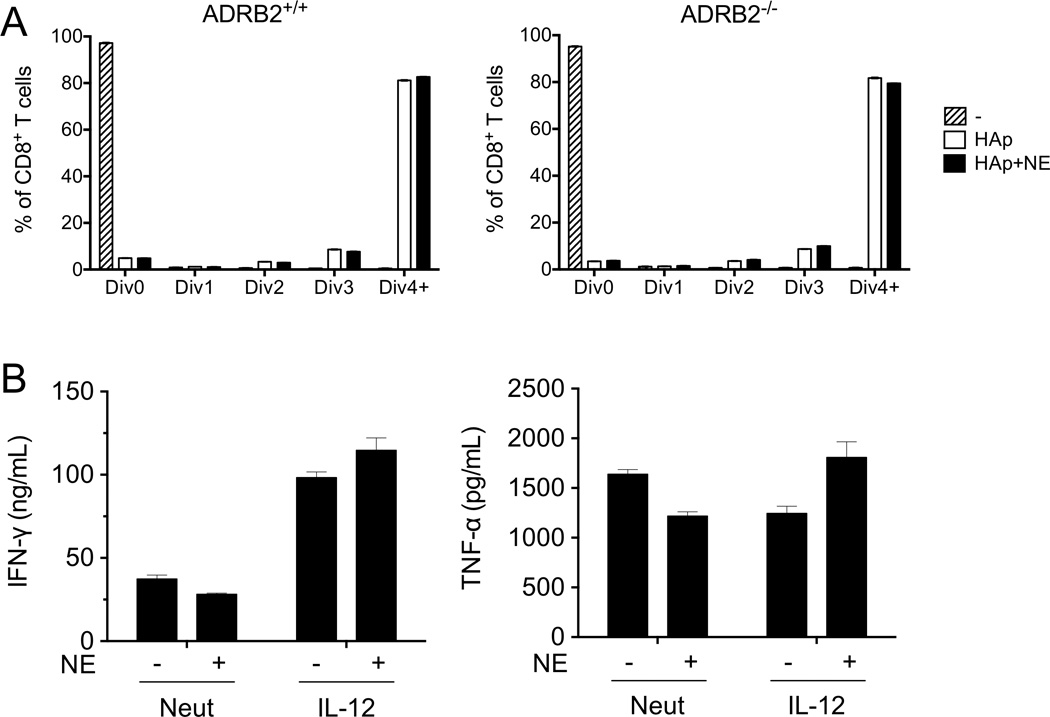
(A) Splenocytes from ADRB2-sufficient or -deficient Cl4 mice were isolated and incubated for 3 days in the presence or absence of the HA peptide IYSTVASSL and NE. Proliferation was measured by dilution of a proliferation dye by flow cytometry. (B) Human CD8+CD45RA+ cells were purified from peripheral blood and activated with plate-bound anti-CD3 and anti-CD28 antibodies, either neutralizing cytokines (Neut) or in the presence of rhIL-12 (IL-12) and in the presence or absence of NE for 7 days. On day 8, cells were restimulated with plate-bound anti-CD3 for 24 hours. IFN-γ and TNF-α in the supernatant were measured by ELISA. Data are presented as mean +/− SEM of triplicate determinations and are representative of (A) 3 experiments with at least 1 mouse in each experiment, and (B) 2 independent experiments from separate donors. Comparisons of NE treatments to controls were not significant as assessed by 2-way ANOVA with Bonferroni post-test.
Two main pathways regulate the acute expression of cytokine genes in effector T cells. In the context of antigen recognition, TCR signaling drives rapid transcription of IFN-γ and TNF-α. However, CD8+ T cells can respond innately by secreting inflammatory cytokines in response to combined stimulation with IL-12 and IL-18 in the absence of cognate antigen [31, 32]. To test the effects of ADRB2 signaling on IL-12/IL-18-induced cytokine secretion, Cl4 T cells were stimulated with IL-12, IL-18, or both in the absence or presence of NE. Surprisingly, in contrast to TCR activation, IL-12/IL-18 stimulation selectively induced IFN-γ, but not TNF-α secretion. However, NE did not significantly inhibit IFN-γ secretion from these cultures. Collectively, these data suggest that NE signaling selectively suppresses acute TCR-driven IFN-γ and TNF-α secretion from CD8+ T cells, but does not influence either their proliferation or their development into effectors in response to IL-12.
Although the in vitro priming experiments described above suggested that NE did not directly block effector cell development, it was possible that ADRB2 signaling could influence their development in vivo during pathogen responses. Utilizing an adoptive cell transfer model followed by viral infection, we tested this indirect mechanism by treating recipient mice with the long-acting β2-specific agonist salmeterol (Fig. 6A). OT-I cells were transferred i.v. into naïve C57Bl/6 hosts, and the mice were infected i.v. with VSV-OVA. Salmeterol was administered i.p. for three consecutive days, beginning on the day of infection. Spleens were then harvested on day 7 post-infection to assess OT-I cell expansion and cytokine secretion upon restimulation. As expected, OT-I cells expanded upon VSV-OVA infection (Fig. 6B). Salmeterol did not affect either the percentage of antigen-specific cells, or the total number of expanded cells present at day 7 (Fig. 6B), consistent with results from in vitro proliferation assays (Fig. 5A). Although the expansion of antigen-specific cells was unaffected by treatment with salmeterol, their programmed cytokine secretion capacity was significantly reduced by approximately 25% (Fig. 6C). Considering that ADRB2 signaling by stimulation with NE in vitro did not alter effector cell development directly (Fig. 5B), it was likely that the effect of salmeterol on cytokine secretion capacity was mediated by an indirect cell-extrinsic mechanism. Thus, the primary role of ADRB2 signaling during CD8+ T-cell priming in vivo may reside within innate cells perhaps through decreasing IL-12 secretion [33], while the intrinsic role of this pathway directly suppresses cytokine secretion and lytic activity once cells have developed into primary effectors.
Figure 6. ADRB2 signaling modulates cytokine secretion programming in mouse CD8+ T cells in vivo.
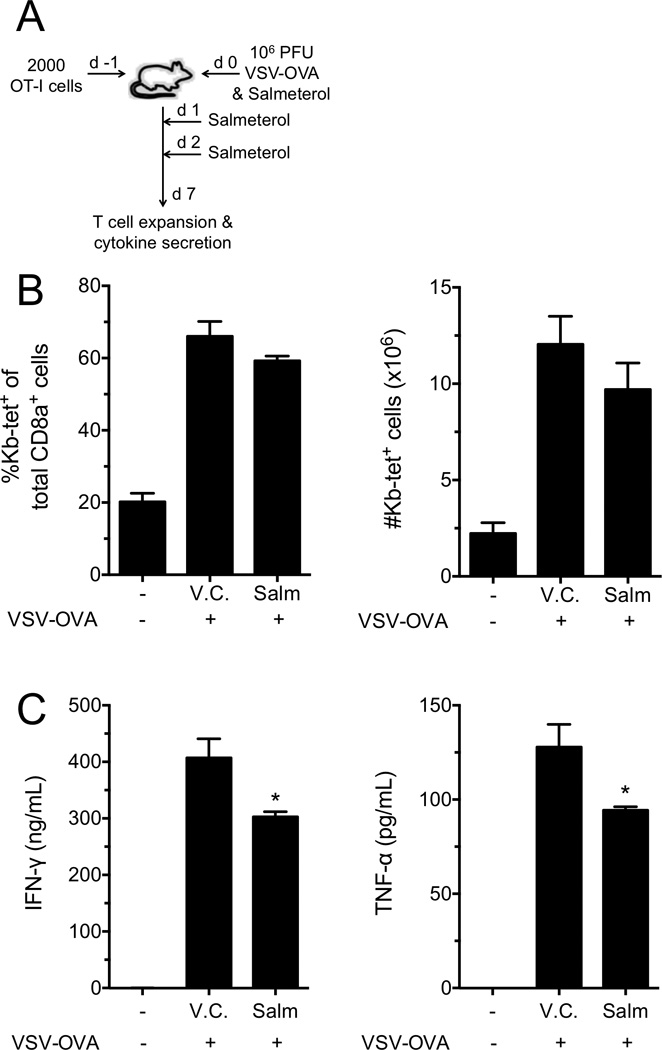
(A) OT-I T cells were transferred (2000/recipient) into naïve C57Bl/6 mice followed by infection with 106 PFU VSV-OVA the next day (day 0). DMSO (V.C.) or Salmeterol was given i.p. on days 0, 1 and 2. (B) Spleens were harvested on d7, and OT-I-specific cellular expansion was determined by flow cytometry. Data display percent of Kb-tet+ cells of the total CD8+ T-cell pool (left) and the total numbers of Kb-tet+ cells within the spleen (right). (C) IFN-γ and TNF-α secretion were measured from splenocytes by flow cytometry after 24 hours restimulation with SIINFEKL peptide. Data are presented as mean +/− SEM of triplicate determinations and are representative of 2 independent experiments with 4 mice per treatment group in each experiment.. *p<0.05 compared to V.C, 1-way ANOVA with Bonferroni post-test.
Discussion
The interface between the nervous and immune systems has recently gained significant interest, and the connection between these systems is quite intimate given that secondary lymphoid organs are innervated by post-ganglionic neurons [1, 2]. These innervations end in adrenergic neurons, which secrete NE upon stimulation [3, 4]. The majority of studies have focused on the effects of nervous system cues on innate cells, and ADRB2 signaling has been shown to decrease pro-inflammatory cytokine secretion in macrophages [12]. Dendritic cells are also affected by ADRB2 signaling, which inhibits antigen cross-presentation [33]. And mast cells fail to degranulate upon stimulus in the presence of ADRB2 ligands [34–37]. Overall, the ADRB2 acts to dampen the innate immune response by decreasing both cytokine secretion and other components of cell-mediated functions.
However, not all reports show a negative regulation of immune system function by adrenergic signals. Additional studies have demonstrated that B cells increase CD86 expression and secrete more IgE, without affecting IgE isotype switching, when ADRB2 ligands are present [9, 10]. As far as CD4+ T cells responding to ADRB2 signaling, the effects depend on the population in question. High concentrations of NE have been shown to induce memory-like CD4+ T cells to secrete acetylcholine [25]. Furthermore, regulatory CD4+ T cells respond positively to ADRB2 ligands by increasing their suppressive activity, as well as their induction of iTregs from naïve CD4+ T cells [14]. In contrast, NE acutely decreases cytokine production from Th1 cells, while leaving cytokine secretion from Th2 cells unaffected [13, 38]. This is due to chromatin modifications within the ADRB2 promoter that allow gene expression in Th1 cells while silencing it in Th2 cells [39]. In contrast, polarization of naïve CD4+ T cells under Th1 conditions in the presence of NE increased their capacity to secrete IFN-γ [40].
Several studies have examined the effects of ADRB2 signaling on CD8+ T cells. One study showed adrenergic signaling, specifically through the ADRB2, inhibited the production of CD8+IFN-γ+ cells upon influenza A infection [5]. However, the intrinsic effect of ADRB2 signaling on CD8+ T cells was not addressed in this study. A second study examined the effect of NE on cytokine and gene expression in different subsets of memory CD8+ T cells [17]. NE was shown to decrease IFN-γ and IL-2 expression, consistent with results from our data presented here, as well as other previous studies [15, 16]. Importantly, Zalli et al recently demonstrated that relatively high concentrations 50 µM) of the long-acting β2-agonist salmeterol could inhibit human CD8+ T-cell effector functions [16], which was consistent with our in vivo findings. In contrast, Slota et al recently reported an increase in other cytokines, namely, TNF-α, IL-1α, IL-6 and CCL-2 [17]. The observation that TNF-α was increased by ADRB2 signaling was inconsistent with the results shown in our current study, as well as in others [15]. The biological relevance of IL-1α and IL-6 gene and cytokine expression on CD8+ T cells remains to be elucidated. Nonetheless, it is important to point out that the memory populations studied by Slota et al do not behave the way we currently understand these populations. Mainly, effector memory and central memory CD8+ T cells were shown to secrete similar levels of cytokines in their study. This is opposite to the currently understood notion that effector memory cells retain acute and increased effector functions, while central memory cells have much lower capacity for immediate effector function but retain the ability to rapidly proliferate to give rise to new effector cells. The way that memory cell populations were identified and isolated in their study could account for some of the discrepancies seen between their studies and others that are in agreement with our current findings.
Despite the possibility that high concentrations of NE can elicit acetylcholine production from T cells, we found no role for this pathway in mediating cytokine suppression in primary CD8+ T cells. Rather, we found that mimicking the canonical signaling pathway of ADRB2 via the cAMP stimulator forskolin was able to downregulate cytokine secretion from CD8+ T cells, which has been shown before for CD4+ T cells [29, 30]. However, despite testing different adenylyl cyclase inhibitors, we were unable to reverse the inhibition of cytokine secretion by NE. This could be due to ineffective activity by the inhibitors and the toxic effects of the inhibitors on cytokine secretion when used at higher concentrations. Finally, salmeterol was able to reduce cytokine secretion programming of CD8+ T cells in vivo, in the context of a viral infection. This is in agreement with the results from a chemical sympathectomy model with viral infection [5, 6]. However, this effect is more likely due to indirect effects on antigen presenting cells since we showed no effect of NE on CD8+ T-cell programming, nor proliferation, in vitro.
In summary, we have described an intrinsic effect of ADRB2 signaling on human and mouse CD8+ T cell effector functions. NE, the natural ligand for ADRB2 in immune organs, as well as pharmacological ADRB2 ligands, were able to suppress IFN-γ and TNF-α cytokine secretion acutely when cells were stimulated through their T-cell receptor. This is the first report to demonstrate a differential suppression for TNF-α modulation, which was considerably more responsive to NE, while the effect of NE on IFN-γ depends more on T cell receptor signal strength, as evidenced by titration of T-cell receptor stimulation and IL-12/IL-18 induced IFN-γ secretion. Additionally, NE was able to suppress cytolytic capacity of human CD8+ T cells ex vivo.
Materials and Methods
Mice
C57Bl/6, OT-I, Balb/cJ, Clone4-Tg (Cl4) and Adrb2−/− mice were housed in specific pathogen free conditions at the University of Texas Southwestern Medical Center Animal Research Center facilities. Adrb2−/− mice [41] were a kind gift from Dr. Virginia Sanders (Ohio State University), and Clone4-Tg (Cl4) mice [42] were purchased from Jax mice (Jackson laboratory). All experiments involving mice in this study were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
Human subjects
Informed consent was obtained from adult healthy donors according to the guidelines established by the Internal Review Board of the University of Texas Southwestern Medical Center and the Declaration of Helsinki. Peripheral blood was then collected by venipuncture.
CD8+ T cell isolation and culture
For the isolation of human CD8+ T cells, peripheral blood mono-nuclear cells (PBMCs) were obtained by ficoll density centrifugation of whole blood from healthy human donors. CD8+ T cells were purified using a negative isolation kit (BD Biosciences). Alternatively, cells were isolated using a FACSAria sorter (BD Biosciences). For mouse, CD8+ T cells were purified from spleen and lymph nodes using a negative isolation kit (Invitrogen) or by sorting. For cytokine secretion, human purified CD8+ T cells were cultured with plate-bound anti-CD3/anti-CD28 (clones OKT3/CD28.2, 3µg/mL each) in complete IMDM supplemented with 10% FBS. Cells were cultured at 1×106/mL and supplemented with rhIL2 (200 U/mL). For differentiation, human CD8+CD45RA+ cells were isolated by sorting and stimulated as above for 7 days, with a 1:10 split on d3, under neutralized or IL-12 polarization conditions as previously described [24, 43]. Cells were then rested overnight without rhIL2 and restimulated with plate-bound anti-CD3 (OKT3, 3µg/mL). Mouse purified CD8+ T cells were cultured at 1×106/mL with plate bound anti-CD3/anti-CD28 (clones 2C11/37.51, 3µg/mL each for ELISA, 2µg/mL each for RNA gene expression). Cl4 and OT-I spleen cell suspensions were cultured at 2×106/mL, or as indicated, with the CD8+ T cell specific Influenza A hemagglutinin (HA) peptide (IYSTVASSL, 0.5µM, or as indicated), or the OVA peptide (SIINFEKL, 10nM), in supplemented media as indicated above. For proliferation assays, Cl4 splenocytes were labeled with VPD450 according to manufacturer’s instructions and incubated with IYSTVASSL (0.05µM) for 3 days. For restimulation of spleen single cell suspensions from infection experiments, cells were incubated at 2×106/mL, and supplemented with rhIL2 (200 U/mL), in the presence or absence of the CD8+ T-cell specific ovalbumin peptide (SIINFEKL, 10nM).
Antagonists and ADRB2 ligands
Phentolamine hydrochloride (Sigma, P7547), nadolol (Sigma, N1892), atenolol (Sigma, A7655), and ICI-118,551 hydrochloride (SelleckChem, 0821), were used in this study as antagonists for different adrenergic receptors and were dissolved in milliQ water and filter-sterilized. Three different ADRB2 agonists were used. Norepinephrine bitartrate (Sigma, A0937) was dissolved in complete IMDM supplemented with 10% FBS. Albuterol (Salbutamol sulfate, SelleckChem, S2507) was dissolved in either DMSO (Fig. 2C) or complete IMDM supplemented with 10% FBS (all others). Salmeterol (Tocris, 1660) was dissolved in DMSO. The α7 nicotinic acetylcholine receptor antagonist, methyllycaconitine, was dissolved in milliQ water. Forskolin was dissolved in DMSO and used to stimulate cAMP production. The corresponding vehicle control was used for each experiment.
qPCR
Total RNA was isolated 12 hours after stimulation using the RNeasy® Mini Kit with DNaseI treatment (Qiagen), according to manufacturer’s instructions. Reverse transcription was then performed with the ABI High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Primers directed against Ppia were used for reference of mouse gene expression. Relative gene expression was calculated using the 2−ΔΔCt method [44].
Mouse primer sequences: Ppia: For 5’-TTATTCCAGGATTCATGTGCCAGGG-3’, Rev 5’-TCATGCCTTCTTTCACCTTCCCAA-3’; Ifng: For 5’-ACAATCAGGCCATCAGCAACAAC-3’, Rev 5’-CAGCGACTCCTTTTCCGCTTC-3’; Tnf: For 5’-CTGTAGCCCACGTCGTAGCA-3’, Rev 5’-AGCAAATCGGCTGACGGTGT-3’.
ELISA
Human IFN-γ and TNF-α cytokine secretion was determined 24 hours after cell activation (with anti-CD3/anti-CD28 Abs), unless otherwise stated. ELISA MAX kits (BioLegend) were used according to manufacturer’s instructions. For mouse cytokine secretion assays, supernatants were harvested 24 hours after stimulation (with anti-CD3/anti-CD28 Abs, or IYSTVASSL) or 21–24 hours after restimulation with SIINFEKL. ELISA MAX kit (BioLegend) was used for mouse TNF-α analysis, according to manufacturer’s instructions. The mouse IFN-γ ELISA used coating antibody (BD Biosciences Rat anti-Mouse IFN-γ, Clone R4-6A2, 551216; used at 3 µg/mL), standards (eBioscience rmIFN-γ, 14-8311-63; top standard at 8 ng/mL), detection antibody (BioLegend biotin-Rat anti-Mouse IFN-γ, Clone XMG1.2, 505804; used at 0.5 µg/mL), and streptavidin (BioLegend, 405210; used 1:1000) following BioLegend’s ELISA MAX mouse TNF-α protocol.
Flow cytometry
PMBCs from healthy human donors were labeled with anti-CD8α-PE (BD Biosciences, 555635), and anti-CD45RA-FITC (BD Biosciences, 555488), or anti-CCR7-PE (BD Biosciences, 552176), anti-CXCR3-AlexaFluor488 (BD Biosciences, 558047), and anti-CD8α-AlexaFluor647 (BD Biosciences, 557708) antibodies for cell sorting. Mouse spleen and lymph node single cell suspensions were stained with anti-CD8α-PE (BioLegend, 100708) for cell sorting. For proliferation experiments, cell suspensions were stained with anti-CD8α-APC (BioLegend, 100712), AnnexinV-FITC (BioLegend, 640906), and Ghost Dye-Red780 (Tonbo, 13-0865), and Violet Proliferation Dye 450 (VPD450, BD Biosciences, 562158).
Antibody redirected lysis assay
A re-directed killing assay [45] was performed by labeling THP-1 target cells with 100–500µCi of 51Cr (Na2[51Cr]O4) for 90min. Cells were then washed and left unlabeled or labeled with anti-CD3 Ab (OKT3; 1.5µg/mL) for 20min, to provide antigen-receptor dependent stimulation. Cells were then washed twice and resuspended at 10,000 cells/mL. 1,000 labeled THP-1 target cells were incubated with human CD8+ T cells at increasing effector:target (E:T) ratios for 12–16hrs at 37 °C, in complete IMDM with 10% FBS and supplemented with rhIL2 (200 U/mL), in the presence or absence of NE. Cytolytic capacity was determined by scintillation counting of 51Cr release into the supernatant. Percent specific lysis was calculated with the formula: (Sample − Spontaneous) ÷ (Maximum − Spontaneous) × 100. Where “Spontaneous” is determined by incubating labeled THP-1 cells alone, and “Maximum” is determined by adding detergent to labeled THP-1 cells incubated alone.
Vesicular stomatitis virus infections
For infections, C57Bl/6 mice received 2,000 OT-I cells intra-venously (i.v.) 1 day before i.v. infection with 106 plaque forming units (PFU) of Vesicular Stomatitis Virus expressing chicken ovalbumin (VSV-OVA). Salmeterol, a long-acting ADRB2 agonist, was given at a dose of 40µg/mouse intra-peritoneally (i.p.) on the day of infection, as well as days 1 and 2 post-infection. Control mice received sterile DMSO i.p. Spleens were harvested on day 7 post-infection and single cell suspensions made using frosted glass slides for analysis.
Statistical analysis
Two different statistical tests were performed using GraphPad Prism 6 software. For simple pair-wise comparisons, a Student’s two-tailed t-test was used. For experiments with two samples in different groups, a two-way ANOVA was used followed by a Bonferoni post-test for pair-wise comparisons within the groups. Differences were considered significant when p≤0.05.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Michelle Gill for excellent advice and for providing useful reagents. We thank Dr. Virginia Sanders (Ohio State University) for the kind gift of the ADRB2−/− (Balb/cJ) and for her excellent advice on their use. We thank Angela Mobley and the Flow Cytometry facility for excellent cell sorting expertise. This work was supported by the NIH: T32AI005284 (L.D.E.), T32GM008203 (L.D.E.) and R01AI56222 (J.D.F.), and by the Careers in Immunology Fellowship from the American Association of Immunologists (D.A.).
Footnotes
CONFLICT OF INTEREST
The authors declare no financial or commercial conflict of interest.
REFERENCES
- 1.Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol. 1985;135:755s–765s. [PubMed] [Google Scholar]
- 2.Felten DL, Ackerman KD, Wiegand SJ, Felten SY. Noradrenergic sympathetic innervation of the spleen: I. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J Neurosci Res. 1987;18:28–36. 118–121. doi: 10.1002/jnr.490180107. [DOI] [PubMed] [Google Scholar]
- 3.Shimizu N, Hori T, Nakane H. An interleukin-1 beta-induced noradrenaline release in the spleen is mediated by brain corticotropin-releasing factor: an in vivo microdialysis study in conscious rats. Brain Behav Immun. 1994;8:14–23. doi: 10.1006/brbi.1994.1002. [DOI] [PubMed] [Google Scholar]
- 4.Francis J, MohanKumar SM, MohanKumar PS. Correlations of norepinephrine release in the paraventricular nucleus with plasma corticosterone and leptin after systemic lipopolysaccharide: blockade by soluble IL-1 receptor. Brain Res. 2000;867:180–187. doi: 10.1016/s0006-8993(00)02311-8. [DOI] [PubMed] [Google Scholar]
- 5.Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5300–5305. doi: 10.1073/pnas.0808851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grebe KM, Takeda K, Hickman HD, Bailey AL, Embry AC, Bennink JR, Yewdell JW. Cutting edge: Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. Journal of immunology. 2010;184:540–544. doi: 10.4049/jimmunol.0903395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maisel AS, Fowler P, Rearden A, Motulsky HJ, Michel MC. A new method for isolation of human lymphocyte subsets reveals differential regulation of beta-adrenergic receptors by terbutaline treatment. Clin Pharmacol Ther. 1989;46:429–439. doi: 10.1038/clpt.1989.161. [DOI] [PubMed] [Google Scholar]
- 8.Maisel AS, Knowlton KU, Fowler P, Rearden A, Ziegler MG, Motulsky HJ, Insel PA, Michel MC. Adrenergic control of circulating lymphocyte subpopulations. Effects of congestive heart failure, dynamic exercise, and terbutaline treatment. J Clin Invest. 1990;85:462–467. doi: 10.1172/JCI114460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe A, Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. Journal of immunology. 2000;165:680–690. doi: 10.4049/jimmunol.165.2.680. [DOI] [PubMed] [Google Scholar]
- 10.Pongratz G, McAlees JW, Conrad DH, Erbe RS, Haas KM, Sanders VM. The level of IgE produced by a B cell is regulated by norepinephrine in a p38 MAPK- and CD23-dependent manner. J Immunol. 2006;177:2926–2938. doi: 10.4049/jimmunol.177.5.2926. [DOI] [PubMed] [Google Scholar]
- 11.Kizaki T, Shirato K, Sakurai T, Ogasawara JE, Oh-ishi S, Matsuoka T, Izawa T, Imaizumi K, Haga S, Ohno H. Beta2-adrenergic receptor regulate Toll-like receptor 4-induced late-phase NF-kappaB activation. Mol Immunol. 2009;46:1195–1203. doi: 10.1016/j.molimm.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Grailer JJ, Haggadone MD, Sarma JV, Zetoune FS, Ward PA. Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J Innate Immun. 2014;6:607–618. doi: 10.1159/000358524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. Journal of immunology. 1997;158:4200–4210. [PubMed] [Google Scholar]
- 14.Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, Reis VO, Keller AC, Brum PC, Basso AS. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol. 2013;43:1001–1012. doi: 10.1002/eji.201243005. [DOI] [PubMed] [Google Scholar]
- 15.Kalinichenko VV, Mokyr MB, Graf LH, Jr, Cohen RL, Chambers DA. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J Immunol. 1999;163:2492–2499. [PubMed] [Google Scholar]
- 16.Zalli A, Bosch JA, Goodyear O, Riddell N, McGettrick HM, Moss P, Wallace GR. Targeting ss2 adrenergic receptors regulate human T cell function directly and indirectly. Brain Behav Immun. 2015;45:211–218. doi: 10.1016/j.bbi.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Slota C, Shi A, Chen G, Bevans M, Weng NP. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. 2015;46:168–179. doi: 10.1016/j.bbi.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol. 2015;15:335–349. doi: 10.1038/nri3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mace TA, Zhong L, Kilpatrick C, Zynda E, Lee CT, Capitano M, Minderman H, Repasky EA. Differentiation of CD8+ T cells into effector cells is enhanced by physiological range hyperthermia. J Leukoc Biol. 2011;90:951–962. doi: 10.1189/jlb.0511229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kokolus KM, Capitano ML, Lee CT, Eng JW, Waight JD, Hylander BL, Sexton S, Hong CC, Gordon CJ, Abrams SI, Repasky EA. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci U S A. 2013;110:20176–20181. doi: 10.1073/pnas.1304291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eng JW, Reed CB, Kokolus KM, Repasky EA. Housing temperature influences the pattern of heat shock protein induction in mice following mild whole body hyperthermia. Int J Hyperthermia. 2014;30:540–546. doi: 10.3109/02656736.2014.981300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leigh ND, Kokolus KM, O'Neill RE, Du W, Eng JW, Qiu J, Chen GL, McCarthy PL, Farrar JD, Cao X, Repasky EA. Housing Temperature-Induced Stress Is Suppressing Murine Graft-versus-Host Disease through beta2-Adrenergic Receptor Signaling. J Immunol. 2015;195:5045–5054. doi: 10.4049/jimmunol.1500700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eng JW, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, Ma WW, Repasky EA, Hylander BL. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat Commun. 2015;6:6426. doi: 10.1038/ncomms7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood. 2011;118:3890–3900. doi: 10.1182/blood-2011-05-357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdes-Ferrer SI, Patel NB, Chavan S, Al-Abed Y, Yang H, Tracey KJ. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. 2009;15:195–202. doi: 10.2119/molmed.2009.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al-Abed Y, Tracey KJ, Pavlov VA. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–574. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, Joshi S, Wang H, Andersson U, Chavan SS, Tracey KJ. alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014;20:350–358. doi: 10.2119/molmed.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snijdewint FG, Kalinski P, Wierenga EA, Bos JD, Kapsenberg ML. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J Immunol. 1993;150:5321–5329. [PubMed] [Google Scholar]
- 30.Grader-Beck T, van Puijenbroek AA, Nadler LM, Boussiotis VA. cAMP inhibits both Ras and Rap1 activation in primary human T lymphocytes, but only Ras inhibition correlates with blockade of cell cycle progression. Blood. 2003;101:998–1006. doi: 10.1182/blood-2002-06-1665. [DOI] [PubMed] [Google Scholar]
- 31.Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN-gamma secretion induced by IL-12 and IL-18. Eur J Immunol. 2002;32:2807–2816. doi: 10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 32.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198:1583–1593. doi: 10.1084/jem.20031051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herve J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, Blancou P. beta2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol. 2013;190:3163–3171. doi: 10.4049/jimmunol.1201391. [DOI] [PubMed] [Google Scholar]
- 34.Bissonnette EY, Befus AD. Anti-inflammatory effect of beta 2-agonists: inhibition of TNF-alpha release from human mast cells. J Allergy Clin Immunol. 1997;100:825–831. doi: 10.1016/s0091-6749(97)70280-x. [DOI] [PubMed] [Google Scholar]
- 35.Chong LK, Suvarna K, Chess-Williams R, Peachell PT. Desensitization of beta2-adrenoceptor-mediated responses by short-acting beta2-adrenoceptor agonists in human lung mast cells. Br J Pharmacol. 2003;138:512–520. doi: 10.1038/sj.bjp.0705050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsuji T, Kato T, Kimata M, Miura T, Serizawa I, Inagaki N, Nagai H. Differential effects of beta2-adrenoceptor desensitization on the IgE-dependent release of chemical mediators from cultured human mast cells. Biol Pharm Bull. 2004;27:1549–1554. doi: 10.1248/bpb.27.1549. [DOI] [PubMed] [Google Scholar]
- 37.Gebhardt T, Gerhard R, Bedoui S, Erpenbeck VJ, Hoffmann MW, Manns MP, Bischoff SC. beta2-Adrenoceptor-mediated suppression of human intestinal mast cell functions is caused by disruption of filamentous actin dynamics. Eur J Immunol. 2005;35:1124–1132. doi: 10.1002/eji.200425869. [DOI] [PubMed] [Google Scholar]
- 38.Ramer-Quinn DS, Baker RA, Sanders VM. Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production. Journal of immunology. 1997;159:4857–4867. [PubMed] [Google Scholar]
- 39.McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM. Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain, behavior, and immunity. 2011;25:408–415. doi: 10.1016/j.bbi.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]
- 41.Sanders VM, Kasprowicz DJ, Swanson-Mungerson MA, Podojil JR, Kohm AP. Adaptive immunity in mice lacking the beta(2)-adrenergic receptor. Brain, behavior, and immunity. 2003;17:55–67. doi: 10.1016/s0889-1591(02)00056-9. [DOI] [PubMed] [Google Scholar]
- 42.Lo D, Freedman J, Hesse S, Palmiter RD, Brinster RL, Sherman LA. Peripheral tolerance to an islet cell-specific hemagglutinin transgene affects both CD4+ and CD8+ T cells. European journal of immunology. 1992;22:1013–1022. doi: 10.1002/eji.1830220421. [DOI] [PubMed] [Google Scholar]
- 43.Chowdhury FZ, Estrada LD, Murray S, Forman J, Farrar JD. Pharmacological inhibition of TPL2/MAP3K8 blocks human cytotoxic T lymphocyte effector functions. PLoS One. 2014;9:e92187. doi: 10.1371/journal.pone.0092187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood. 2009;113:5516–5525. doi: 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.