

Abstract
As populations age, the prevalence of geriatric neurodegenerative diseases will increase. These diseases generally are multifactorial, arising from complex interactions among genes, environment, concurrent morbidities, treatments, and time. This essay provides a concept for the pathogenesis of Lewy body diseases such as Parkinson disease, by considering them in the context of allostasis and allostatic load. Allostasis reflects active, adaptive processes that maintain apparent steady states, via multiple, interacting effectors regulated by homeostatic comparators—“homeostats.” Stress can be defined as a condition or state in which a sensed discrepancy between afferent information and a setpoint for response leads to activation of effectors, reducing the discrepancy. “Allostatic load” refers to the consequences of sustained or repeated activation of mediators of allostasis. From the analogy of an idling car, the revolutions per minute of the engine can be maintained at any of a variety of levels (allostatic states). Just as allostatic load (cumulative wear and tear) reflects design and manufacturing variations, byproducts of combustion, and time, eventually leading to engine breakdown, allostatic load in catecholaminergic neurons might eventually lead to Lewy body diseases. Central to the argument is that catecholaminergic neurons leak vesicular contents into the cytoplasm continuously during life and that catecholamines in the neuronal cytoplasm are autotoxic. These neurons therefore depend on vesicular sequestration to limit autotoxicity of cytosolic transmitter. Parkinson disease might be a disease of the elderly because of allostatic load, which depends on genetic predispositions, environmental exposures, repeated stress-related catecholamine release, and time.
Keywords: Stress, Allostasis, Catecholamines, Homeostasis, Parkinson disease
Willie Sutton’s Getaway Car
Willie Sutton (1901–1980) was a bank robber. Asked why he robbed banks, he reportedly replied, “because that’s where the money is.” Sutton’s rule, usually applied to medical diagnosis, states, “Consider the obvious first.”
Many observations and experiments over a century fit with the view that loss of catecholaminergic neurons is fundamental to Parkinson disease. Pathological hallmarks of the disease are Lewy bodies and depigmentation of the substantia nigra. Lewy bodies, first described by Friedrich Lewy in 1912, are round, eosinophilic, intracytoplasmic inclusion bodies in neurons such as in the midbrain substantia nigra (the main source of dopamine in the brain) and the pontine locus ceruleus (the main source of norepinephrine in the brain). Depigmentation of the substantia nigra probably results from loss of neurons that contain dopamine, since dopamine spontaneously auto-oxidizes and polymerizes to form polydopamine melanin (literally a substantia nigra). About a half century ago Hornykiewicz first described the neurochemical hallmark of Parkinson disease, which is profound depletion of dopamine in the striatum (especially in the putamen) (Ehringer and Hornykiewicz 1960). The characteristic movement disorder in Parkinson disease therefore is well accepted to result from a nigrostriatal dopaminergic lesion. The disease involves decreased populations of substantia nigra and locus ceruleus neurons. Virtually all currently used, effective drug treatments for Parkinson disease work directly or indirectly by way of stimulating dopamine receptors. The discovery of parkinsonism induced by the dopaminergic neurotoxin MPTP spurred development of new treatments based on protection of dopaminergic neurons. More recently, it has become clear that Parkinson disease involves several non-motor manifestations, among which are cardiac and extracardiac noradrenergic denervation (Sharabi et al. 2008). Finally, the post-mortem brain of patients dying with end-stage Parkinson disease contains diffusely decreased tissue norepinephrine concentrations (Goldstein et al. 2011).
Taken together, when it comes to Parkinson disease, Willie Sutton would say the “money” is in catecholamines. If we knew what caused the loss of catecholaminergic neurons, we would know what causes Parkinson disease— and have better ideas about how to slow or prevent the disease process.
In this essay, I propose a concept to help answer several key questions about the pathogenesis of Parkinson disease and other Lewy body diseases, such as pure autonomic failure and Lewy body dementia.
Only a very small percent of neurons are catecholaminergic. Why are they so susceptible?
All genotypic abnormalities identified so far that cause familial PD have no obvious relationship to catecholamines. How do generalized abnormalities lead to specific loss of catecholaminergic neurons?
The protein alpha-synuclein is present in all neurons, and precipitated alpha-synuclein is a prominent component of Lewy bodies (Spillantini et al. 1997). Why does alpha-synuclein tend to precipitate in the cytoplasm of catecholaminergic neurons in Lewy body diseases?
PD involves other monoaminergic systems. Why does alpha-synuclein tend to precipitate in the cytoplasm of other monoaminergic neurons?
Why is PD a disease of the elderly?
To address these questions, let us consider Willie Sutton’s “getaway car.” Picture the getaway car at the curb outside the bank. What appears to be different about a getaway car, compared to all other cars parked in front of it or behind it? The answer is that the getaway car is in “idle.” The engine is on, and the transmission is in “park.” Why is it idling? Willie Sutton would say, “because it’s a getaway car.” At the critical time, you have to get away quick by shifting into gear. There is no time to turn on the ignition, and if the car doesn’t start during the escape, the robbery is a failure.
Idling getaway cars provide obvious advantages, but there is a cost. If there were a design or manufacturing flaw, or the oil had the wrong formulation, or the idle were set wrong—and if you waited long enough—then the engine could fail.
The internal combustion engine of a car uses energy to generate movement. Fuel from a fuel injector is combusted by electrically generated sparks in the piston chamber. Oxygen is required from the reaction, and the fuel line carries the fuel from the gas tank to the injector. A controller—the driver—determines the idle setting by the accelerator and whether the engine is in “drive” or “park.” Oil is pumped into the engine, to lubricate the pistons. Engines also include a waste management system. The combustion is never perfect, and toxic byproducts of the oxidation are led through a catalytic converter, with nontoxic waste exhausted via the tail pipe. The spent oil must also be replaced, because of debris accumulating over time in the oil, changes in the lubricant properties from the intense heat, and loss of oil over time.
If there were a design or manufacturing flaw in the engine, a faulty catalytic converter, a clog in the fuel line, spark plug misfiring, or contamination of the oil or fuel, then with continued use the engine of the getaway car would function with progressively decreased efficiency. Just from the wear and tear of the car being in idle all that time, the engine’s lifespan probably would be shortened. Harmful deposits would build up inside. If you waited long enough, and there were enough built-in and added on harmful factors at work, so much “gunk” might built up that the engine could fail entirely.
If you conducted an “autopsy” on the failed engine, you would find a buildup of gunk inside. Would the buildup of gunk itself kill the engine? Not necessarily. If you knew that throughout the use of the car you had mistakenly used oil of very high viscosity, or if you knew that the fuel was contaminated, or if you knew there were a faulty catalytic converter, you could reasonably predict that the engine would fail prematurely and you would know how to prevent the buildup of gunk; however, from analysis of the gunk alone you would not be able to identify the cause of the buildup or the cause of the engine failure. Moreover, even if none of the these factors alone caused a problem, together they could kill the engine and build up deposits.
Catecholaminergic Neurons and Getaway Cars
Chemical neurotransmission uses energy in the form of ATP, and electrical signals along axons release neurotransmitters from vesicles in the nerve terminals by exocytosis. The transmitters occupy receptors on effector cells, resulting in phenomena such as movement.
Catecholamine systems are like idling engines in getaway cars, enabling rapid starting and stopping, both physically and mentally. They work in emergencies such as “fight or flight” situations but also to subserve automatic, involuntary, unconscious, everyday behaviors such as standing up, initiating locomotion, and adjusting to altered environmental temperature.
Catecholamines stored in vesicles are released episodically by altered nerve traffic to the terminals; however, the turnover of neuronal monoamines under resting conditions is determined mainly not by release but by net leakage from the vesicles into the cytoplasm and imperfect recycling via vesicular monoamine transporters (VMAT) back into the vesicles (Eisenhofer et al. 2004).
What good does it do to have “leaky vesicles?” At first glance the high rate of leakage of dopamine from vesicles into the cytoplasm and the high rate of reuptake back into the vesicles by way of the VMAT would seem like a waste of energy. My colleague for many years at the NIH, Graeme Eisenhofer, came up with an insightful explanation, “gearing down” (Eisenhofer et al. 2004). If there were a stable pool of vesicles, then an emergency evoking sustained transmitter release would rapidly dissipate that pool. It would be impossible for synthesis of transmitter from scratch (the rate of which can only about double) to keep up with the irreversible loss from the tissue (the rate of which can go up many-fold). But if there were continuous leakage of catecholamines from the vesicles, and continuous replacement by ongoing synthesis, then the neurons could maintain a high rate of release of transmitter for a much longer time. For the same reason that you would keep a getaway car in idle, the brain keeps catecholaminergic systems in idle.
In neurons there is not only rapid signal transmission along the axons from the cell bodies to the terminals but also slow transmission of vesicles and vesicle-related proteins via axonal transport. Signals such as from occupation of neurotrophic factor receptors are transmitted retrogradely from the terminals to the cells bodies. Axonal transport therefore is a two-way street.
In summary, there have always been strong survival advantages to rapid initiation of movement and sustained performance before exhaustion. Catecholaminergic neurons are always in “idle.” Catecholamines leak passively continuously from vesicles into the cytoplasm, where they are “combusted.” Catecholamines are taken back up actively into vesicles via the VMAT and proton pump. Having “leaky vesicles” enables sudden activity by “shifting gears” and stamina by “gearing down.”
Catecholamine Autotoxicity
It is no coincidence that in Parkinson disease, a key manifestation is the inability to initiate movement quickly. Patients often complain also of decreased stamina, both mental and physical, and about 90% of patients have symptoms attributable to autonomic failure (Kaufmann and Biaggioni 2003). To a large extent these symptoms reflect loss of catecholaminergic neurons in the brain and periphery. This section presents a schema for the pathogenetic processes that lead to catecholamine depletion in Lewy body diseases. The central idea is that catecholamines are “suicide chemicals” that are autotoxic when in the cytoplasm.
Cytosolic catecholamines are potentially toxic by two mechanisms. The first is spontaneous auto-oxidation. The extraordinary susceptibility of catecholamines in solution to undergo oxidation was the basis for both early colorimetric and modern electrochemical methods to assay catecholamine levels in biological fluids such as plasma. This is also why solutions of catecholamines used therapeutically always contain substantial amounts of anti-oxidants such as ascorbic acid or sulfites.
The products of oxidation of the catechols in catecholamines are quinones. Quinones are thought to be toxic because of formation of covalent bonds with biological molecules such as proteins and by reduction of oxygen to superoxide and other reactive oxygen species, resulting in lipid peroxidation and membrane disruption (Creveling 2000). As noted above, oxidized catecholamines also tend to polymerize, forming polydopamine and polynorepinephrine (Lee et al. 2007); however, the role and significance of polycatecholamines in catecholamine autotoxicity remain unknown.
Cytosolic catecholamines (and serotonin) also undergo enzymatic oxidation by monoamine oxidase type A (MAO-A), which is localized to the outer mitochondrial membrane. The actions of MAO-A on dopamine and norepinephrinec result in formation of aldehydes—dihydroxyphenylacetaldehyde (DOPAL) from dopamine and dihydroxyphenylglycolaldehyde (DOPEGAL) from norepinephrine.
Aldehydes are highly toxic compounds. Exposure to DOPAL destroys dopaminergic neurons (Panneton et al. 2010). Aldehyde-induced toxicity occurs by several routes, including interacting with and altering functions of intracellular proteins. The protein reactivity of DOPAL depends on both the aldehyde and the catechol (Rees et al. 2009).
Predictably, there are multiple, redundant genes for detoxifying aldehydes. Humans have 19 different genes encoding aldehyde dehydrogenase (ALDH), which converts the aldehydes to acids—dihydroxyphenylacetic acid (DOPAC) from DOPAL and dihydroxymandelic acid from DOPEGAL. Hydrogen peroxide is formed as a necessary consequence of MAO acting on catecholamines. In the setting of metal ions, hydrogen peroxide generates highly reactive hydroxyl radicals.
Enzymes are literally catalytic converters. A prediction from the getaway car analogy is that genetic or acquired decreases in ALDH activity could lead to buildup toxic monoamine aldehydes. Consistent with this view, human striatum contains DOPAL, and in patients with end-stage Parkinson disease, post-mortem putamen DOPAL:DOPAC ratios are higher than in control subjects, in line with decreased ALDH activity (Goldstein et al. 2011).
The major products of lipid peroxidation, 4-hydroxynonenal and malondialdehyde, inhibit ALDH, and ALDH inhibition builds up tissue contents of DOPAL (Jinsmaa et al. 2009). Because DOPAL is toxic, and products of the toxicity inhibit ALDH, induction of a positive feedback loop might accelerate degeneration and death of dopaminergic neurons.
Synuclein and Gunk
Lewy bodies contain high concentrations of aggregated alpha-synuclein. Based on studies of patients with inherited Parkinson disease from mutation of the alpha-synuclein gene or replication of the normal gene, alpha-synucleinopathy can cause both nigrostriatal dopaminergic and sympathetic noradrenergic neuronal loss (Singleton et al. 2004; Goldstein et al. 2001).
There are multiple means by which alpha-synucleinopathy may produce relatively selective loss of catecholaminergic neurons. Synuclein overabundance increases cytosolic dopamine concentrations (Mosharov et al. 2006), possibly by inhibiting vesicular uptake of cytosolic dopamine (Park et al. 2007). Cytosolic DA interacts with calcium and alpha-synuclein to destroy substantia nigra neurons (Mosharov et al. 2009). Synuclein aggregates in axons (“Lewy neurites”) might also interfere with axonal transport of vesicles or vesicle-related proteins or impede retrograde transport of neurotrophic signals. DOPAL potently oligomerizes alpha-synuclein and can precipitate synuclein in catecholamine-producing cells (Burke et al. 2008), setting the stage for another positive feedback loop.
Unfortunately, the world’s literature on DOPAL consists of only about 50 articles, and only a handful of these have involved actual measurements of endogenous DOPAL and related catechols in brain tissue. The catecholaldehyde hypothesis predicts that in less susceptible dopaminergic neurons, such as in the tuberinfundibular system, there is less production or more efficient detoxification of DOPAL than in striatal dopaminergic terminals.
Since neuronal uptake via cell membrane catecholamine transporters and sequestration of cytosolic catecholamine via vesicular monoamine transporters are energy-requiring processes, any of a variety of intrinsic errors of mitochondrial function or metabolic insults posed by exogenous toxins that decrease activities of these transporters could result in correlated decreases in neuronal uptake and vesicular sequestration.
Why is Parkinson’s a Disease of Old People?
Parkinson’s is well known to be a disease mainly of the elderly. The getaway car analogy and a few concepts of scientific integrative medicine (Goldstein 2006) help understand why this should be.
Levels of multiple monitored variables are kept stable by negative feedback loops. By analogy to the getaway car, a decrease in outside temperature would lead to a decrease in engine pressure, increasing the rate of injection of fuel into the combustion chambers by the fuel pump. If a homeostatic loop has an odd number of (−) signs, then during exposure to a continuous perturbing influence, the monitored variable will attain a stable level. Physiological homeostatic systems entail negative feedback regulation of monitored variables, such as core temperature, blood pressure, serum osmolality, and metabolic rate. Conceptually, each system depends on a comparator—a homeostat—to compare afferent information about the monitored variables with set points or other criteria for responding.
Although levels of monitored variables in a system regulated by negative feedback attain apparent steady-states, the actual level of the monitored variable can be adjusted, by changing the set point or other instructions for responding. Allostasis refers to this “other sameness.” For instance, the idle rate can be set to a higher number of revolutions per minute. Stress can be defined as a condition or state in which a sensed discrepancy between afferent information and a setpoint for response leads to activation of effectors, reducing the discrepancy.
The flexibility afforded by allostasis comes at the cost of wear and tear—allostatic load. Allostatic load refers to the consequences of sustained or repeated activation of mediators of allostasis. From the analogy of the idling getaway car, the revolutions per minute of the engine can be maintained at any of a variety of levels (allostatic states). Just as allostatic load, reflecting effects of manufacturing flaws, byproducts of combustion, usage history, and time lead eventually to engine breakdown, allostatic load in catecholaminergic neurons might lead eventually to loss of the neurons.
As wear and tear progresses, system efficiency declines, requiring the effector to be activated more of the time to maintain the engine pressure, in turn accelerating the wear and tear on the engine. In other words, allostatic load can lead eventually to a positive feedback loop and system failure.
The amount of wear and tear depends on four factors: design, manufacturing, usage history, and time. The usage history depends on the error signal over time. In terms of neurodegenerative diseases, design corresponds to genetics, manufacturing to gene expression, and usage history to the frequencies, durations, and severities of release and reuptake of catecholamines.
Allostatic load links stress with degenerative diseases. Activation of effectors to counter threats to homeostasis produces wear and tear determining the level of the monitored variable and on the effectors themselves. Wear and tear, combined with planned obsolescence, decreases effector efficiency. Eventually, even with the effectors activated continuously, the monitored variable drifts from the allostatic setting. Finally, when the effectors fail, the organism can no longer mount a stress response at all.
Over time, a combination of abnormalities could eventually kill catecholamine cells. There might be a faulty catalytic converter. Metabolic breakdown of dopamine results in formation of toxic byproducts, which in turn normally are detoxified by enzymes. If the detoxifying enzymes malfunctioned, the toxic byproducts would accumulate. There might be “over-revving,” corresponding to excessive turnover of catecholamines, such as by decreased efficiency of recycling via cell membrane or vesicular transporters. There might be a deviation in the oil, such as a tendency to break down at high temperature, corresponding to a hyperfunctional polymorphism of the alpha-synuclein gene. The fuel might contain a contaminant, corresponding to a harmful environmental agent such as a pesticide or a toxin converted to a harmful biochemical in the body. There might even be “planned obsolescence,” corresponding to accelerated aging of nerve cells in general. All these processes, combined over a lifetime, could build up “gunk,” represented by the Lewy bodies seen in the catecholamine cells of patients with Parkinson disease.
Occasionally Parkinson disease develops in relatively young people, such as the actor Michael J. Fox. The allostatic load model would explain pathogenetic mechanisms in young people in terms of relatively large contributions of genotypic and gene expression abnormalities or effects of environmental exposures or episodes of stress biasing toward excessive wear and tear and induction of deleterious positive feedback loops at a relatively young age (Figs. 1, 2, 3).
Fig. 1.
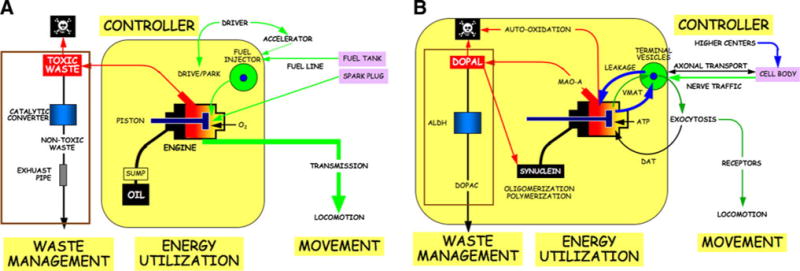
Analogous components of a an internal combustion engine and b a catecholaminergic neuron
Fig. 2.
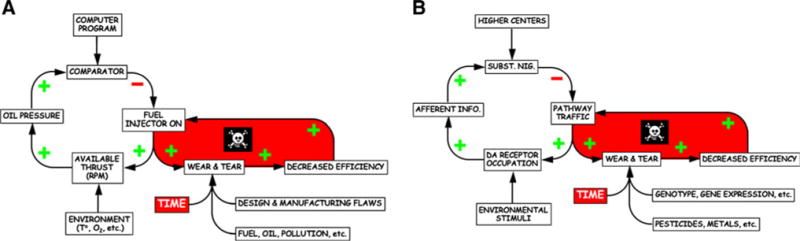
Allostatic load as applied to a an internal combustion engine and b a catecholaminergic neuron. Note system breakdown upon induction of a positive feedback loop
Fig. 3.
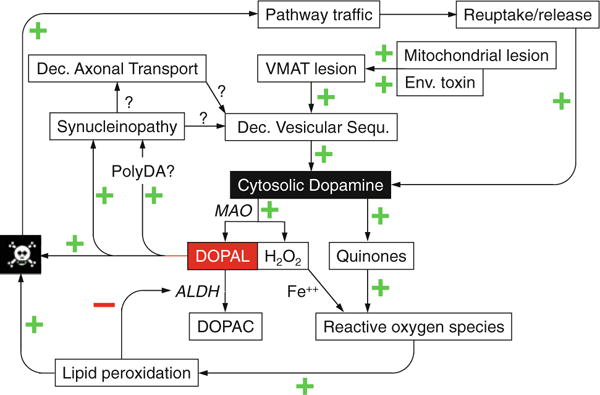
Overview of pathogenetic mechanisms in catecholaminergic neurons. Central to the vulnerability of these neurons are ongoing vesicular leakage and toxicity of cytosolic catecholamines such as dopamine, via spontaneous auto-oxidation and oxidation catalyzed by monoamine oxidase (MAO) to form dihydroxyphenylacetaldehyde (DOPAL). DOPAL is detoxified by aldehyde dehydrogenase (ALDH) to form dihydroxyphenylacetic acid (DOPAC). Other abbreviations: Dec decreased, Env environmental, PolyDA polydopamine, VMAT vesicular monoamine transporter
Epilogue
Dr. Thomas Graboys, a renowned cardiologist and patient with Parkinson disease, orthostatic hypotension, and dementia, has described in an autobiographical book the effects of his disease (Graboys 2008):
As a young intern and resident, and later as an attending cardiologist, I was accustomed to being summoned suddenly in the middle of the night. I could launch myself out of bed, get dressed, and perform at my intellectual peak within moments. I could make life-and-death decisions within seconds of a night-time phone call. Today, I wait for thousands of tiny cellular engines to start themselves so I can rise from the bed and begin another day…
One cannot imagine a more poignant confirmation of the applicability of the getaway car analogy to the pathogenesis of Parkinson disease.
Acknowledgments
Research leading to the concepts in this essay was supported by the intramural research program of the National Institute of Neurological Disorders and Stroke. The author thanks Drs. Yehonatan Sharabi, Basil Eldadah, Graeme Eisenhofer, and Irwin J. Kopin for many valuable discussions.
References
- Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115(2):193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- Creveling CR. The role of catechol quinone species in cellular toxicity. FP Graham Publishing Co.; Johnson City: 2000. [Google Scholar]
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Wien Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004a;56(3):331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Leaky catecholamine stores: undue waste or a stress response coping mechanism? Ann N Y Acad Sci. 2004b;1018:224–230. doi: 10.1196/annals.1296.027. [DOI] [PubMed] [Google Scholar]
- Goldstein DS. Adrenaline and the inner world: an introduction to scientific integrative medicine. The Johns Hopkins University Press; Baltimore: 2006. [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol. 2011;18:703–710. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Li ST, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of alpha-synuclein. Ann Intern Med. 2001;135(11):1010–1011. doi: 10.7326/0003-4819-135-11-200112040-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graboys T. Life in the balance. Sterling; New York: 2008. [Google Scholar]
- Jinsmaa Y, Florang VR, Rees JN, Anderson DG, Strack S, Doorn JA. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem Res Toxicol. 2009;22(5):835–841. doi: 10.1021/tx800405v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol. 2003;23(4):351–363. doi: 10.1055/s-2004-817719. [DOI] [PubMed] [Google Scholar]
- Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318(5849):426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bove J, Prou D, Hananiya A, Markov D, Poulsen N, Larsen KE, Moore CM, Troyer MD, Edwards RH, Przedborski S, Sulzer D. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26(36):9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62(2):218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 2010;5(12):e15251. doi: 10.1371/journal.pone.0015251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, Schulz EM, Lee D. Disruption of dopamine homeostasis underlies selective neurodegeneration mediated by alpha-synuclein. Eur J Neurosci. 2007;26(11):3104–3112. doi: 10.1111/j.1460-9568.2007.05929.x. [DOI] [PubMed] [Google Scholar]
- Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22(7):1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharabi Y, Imrich R, Holmes C, Pechnik S, Goldstein DS. Generalized and neurotransmitter-selective noradrenergic denervation in Parkinson’s disease with orthostatic hypotension. Mov Disord. 2008;23(12):1725–1732. doi: 10.1002/mds.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A, Gwinn-Hardy K, Sharabi Y, Li ST, Holmes C, Dendi R, Hardy J, Crawley A, Goldstein DS. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain. 2004;127(Pt 4):768–772. doi: 10.1093/brain/awh081. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]