

Abstract
2-Aminoimidazole (2-AI)-based compounds have been shown to efficiently disrupt biofilm formation, disperse existing biofilms, and resensitize numerous multidrug-resistant bacteria to antibiotics. Using Pseudomonas aeruginosa and Staphylococcus aureus, we provide initial pharmacological studies regarding the application of a 2-AI as a topical adjuvant for persistent dermal infections. In vitro assays indicated that the 2-AI H10 is nonbactericidal, resensitizes bacteria to antibiotics, does not harm the integument, and promotes wound healing. Furthermore, in vivo application of H10 on swine skin caused no gross abnormalities or immune reactions. Taken together, these results indicate that H10 represents a promising lead dermal adjuvant compound.
Keywords: transdermal absorption, antimicrobial activity, skin irritation, synergism, oroidin derivative, drip-flow reactor, ESKAPE pathogens
Introduction
Bacteria have the ability to switch between a free-living, planktonic state to a surface-attached, multicellular sessile state known as a biofilm. To complicate the growing trend of antibiotic resistance and a seemingly inevitable postantibiotic era,1 bacteria in a biofilm are remarkably more resilient than their planktonic counterparts.2 In the biofilm state, bacteria display differential gene expression and are up to 1,000-fold more tolerant to antibiotics and host immune responses.3–5 Moreover, biofilms are directly involved an estimated 65% of all nosocomial infections.6 Because bacteria spend approximately 80% of their time in the biofilm state and exist ubiquitously in nature,3,7 it is imperative that the involvement of biofilms in antibiotic tolerance and pathogenesis is more fully understood.
Approximately 80 % of all bacterial infections involve biofilms; they account for the persistent colonization of indwelling medical devices and are ultimately liable for the mortality and morbidity of nearly all cystic fibrosis (CF) patients.6,8–10 CF patients are frequently plagued with Pseudomonas aeruginosa biofilms that colonize the pleural membrane, and 80% of adults with CF are burdened with chronic P. aeruginosa infections.7,10–12 While it accounts for a smaller surface area than the lungs, the skin is considered the largest organ in the body and is the first physical barrier encountered by pathogens – thus it is acutely susceptible to biofilm colonization. When wounds become colonized with biofilm-forming bacteria, the ability to heal is compromised and they can become very difficult to treat.13 Leg ulcers, a common chronic wound often found in immunocompromised patients,14 are predominantly colonized by P. aeruginosa (88%) and Staphylococcus aureus (33%),3 two of the most prevalent species found in chronic wound biofilms.15,16 Furthermore, resistance to the antibiotics routinely used to treat biofilms only augments the crisis. In 2011, for example, nearly 80,500 people acquired methicillin-resistant S. aureus infections in the USA, and approximately 11,300 people died as a result of methicillin-resistant S. aureus infections, which is more than the number of people who die annually from human immunodeficiency virus/acquired immune deficiency syndrome, Salmonella poisoning, kidney infection, influenza, acute bronchitis, and Hodgkin’s lymphoma combined.17,18
The current standard of care for chronic wounds are large doses of antibiotics, mechanical debridement, manual irrigation, and the application of specialized dressings on the affected area.19 However, following initial treatment, these wounds often relapse.3 While they persist on biotic surfaces, biofilms can also inhabit nutrient-poor abiotic surfaces such as catheters and prosthetic devices like joints and heart valves.20,21 One reason for relapse is the existence of persister cells, which lay dormant under the protective exopolymeric substance coating the biofilm.22 Another reason for delayed wound healing could be secretion of bioactive compounds by the biofilm bacteria, which can be isolated as biofilm-conditioned media (BCM). Several reports have established that BCM from both P. aeruginosa and S. aureus inhibited cellular proliferation.23,24 These two species are members of the Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa, and Enterobacter (ESKAPE) family of pathogens that have highly documented resistance and thus have threatening potential for transmission and pathogenicity.25
Following an essentially 40-year halt in new antibiotic deployment, resistance was rapidly developed to the two new narrow-spectrum antibiotics released in the early 2000s.26 Within the many classes of antibiotics, vancomycin and the polymyxins are often drugs of last resort but remain at the forefront of antibiotic therapy; however, resistant strains of even these drugs are appearing.27–29 As an alternative to traditional bacteriostatic/bactericidal antibiotics, a class of small molecules has been developed that resensitizes bacteria to current antibiotics30 as well as inhibits and disperses preformed biofilms.31–33 These small-molecule inhibitors contain a 2-aminoimidazole (2-AI) moiety that is the minimum core pharmacophore in the bromoageliferins, which the Agelas conifera marine sponge utilizes to minimize biofilm colonization by marine bacteria such as Rhodospirillum salexigens.34,35
These small-molecule inhibitors are cell permeable and bind a class of bacterial proteins called the response regulators (RRs).36 More specifically, pull-down assays identified the master controller of biofilm formation, BfmR, as an RR-binding partner of a 2-AI in A. baumannii, another ESKAPE pathogen.36 RRs play an integral role in a signal transduction pathway called the two-component system (TCS), and the majority of RRs are DNA-binding proteins.37–40 Within a TCS, a membrane-bound histidine kinase detects and integrates extracellular signals to phosphorylate the appropriate RR, and for DNA-binding RRs, this either activates or represses gene transcription.41 Beyond providing environmental surveillance and controlling survival responses, TCSs are essential elements of the virulence, biofilm formation, and antibiotic resistance responses of bacterial pathogens.42–45
Controlling the activities of TCSs for therapeutic advantage has garnered considerable interest,46,47 but nearly all medicinal efforts thus far have focused on affecting the TCS histidine kinases.48–51 A representative RR-associated compound from the 2-AI family, 2-amino-N-undecyl-H-imidazole-5-butanamide (H10; Figure 1, inset), a “reverse amide” class of antibiofilm agents, was chosen for topical application.31,32 These compounds have previously functioned synergistically with conventional antibiotics to eradicate bacteria within a biofilm state and have resensitized multidrug-resistant planktonic bacteria to numerous antibiotics.30 The compound H10 was specifically selected to represent the best balance between biofilm inhibition and dispersion across multiple species. The goal of this study was to further implicate H10 as a favorable representative lead within the 2-AI family. Here, we demonstrate the nonbactericidal nature of H10, assay its biofilm inhibition ability, establish any synergism, and determine its suitability as a topical biofilm treatment via histopathological characterization following dermal application to swine skin.
Figure 1.
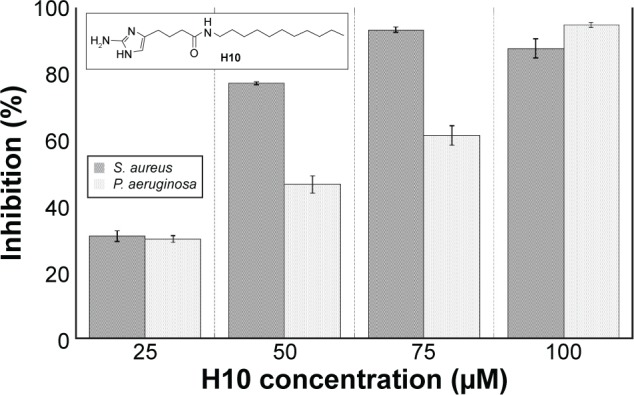
Biofilm inhibition assay for H10 against S. aureus and P. aeruginosa.
Notes: Inhibition assay performed to determine the working concentration for H10 in S. aureus and P. aeruginosa. The data are given as the mean ± SD. (Inset) Chemical structure of 2-amino-N-undecyl-H-imidazole-5-butanamide (H10).
Abbreviations: SD, standard deviation; S. aureus, Staphylococcus aureus; P. aeruginosa, Pseudomonas aeruginosa.
Materials and methods
Antibiofilm activity assays
Overnight cultures of P. aeruginosa PAO1 and S. aureus ATCC BAA-44 were diluted to an optical density (OD600) of 0.01 in the appropriate biofilm growth media (10% brain–heart infusion broth with 5% fetal bovine serum for P. aeruginosa and tryptic soy broth with 0.5% glucose for S. aureus). H10 was added to a series of bacterial suspensions to achieve a wide range of concentrations (for half-maximal effective concentration [IC50] calculation) from a 100 mM stock in 100% dimethyl sulfoxide (DMSO). Later, final concentrations of 25, 50, 75, and 100 μM were used for the inhibition assays. The resulting suspensions were transferred into 96-well polyvinyl chloride plates (100 μL per well) that were covered with self-sealing plastic wrap (Glad Press’n Seal; The Clorox Company, Oakland, CA, USA) prior to incubation at 37°C. P. aeruginosa and S. aureus were incubated for 6 and 24 hours, respectively. Following incubation, the media was discarded and the plates were rinsed with water to remove any planktonic or loosely attached bacteria. Crystal violet (110 μL of 0.1%) was added to each well and the plates were incubated at room temperature to stain the remaining biomass. The plates were then rinsed and air-dried. Ethanol (EtOH; 150 μL) was added to each well to solubilize the remaining crystal violet, and 150 μL of the solubilized crystal violet was transferred to a new plate to measure the OD540. Each H10 concentration was assessed using two rows of eight wells. An average OD540 was determined for each individual row. Then, the average of the two rows for each H10 concentration was determined. IC50 values were calculated, and the percent inhibition was determined by comparing this average to the OD540 of the untreated wells. Dispersion assays for half-maximal effective concentration (EC50) determination were performed similarly to the inhibition assays, except the biofilms were allowed to form and were gently rinsed prior to 24-hour treatment with various concentrations of H10 (diluted in biofilm growth medium). The plates were then rinsed and processed for crystal violet staining, and the EC50 values were determined.
Growth curves
Overnight cultures of P. aeruginosa PAO1 and S. aureus ATCC BAA-44 were diluted to an OD600 of 0.02 in fresh Luria-Bertani (LB; Fisher, Pittsburgh, PA, USA) medium or tryptic soy broth, respectively. The bacterial suspensions were treated with 12.5 and 50 μM H10 for S. aureus and P. aeruginosa, respectively, using a 10 mM stock solution (prepared in 100% DMSO). For the growth control, the additional bacterial suspension contained an equal volume of DMSO. The test cultures were transferred into a 96-well plate (200 μL per well, each column containing a different test sample). OD600 measurements were collected and the plates were then shaken (180 rpm) at 37°C. The plates were briefly removed from the shaker at 1, 2, 4, 6, 8, 24, and 32 hours for subsequent OD600 measurements.
Single-species biofilm drip-flow reactor assays
Previously published drip-flow reactor (DFR) protocols were used to grow biofilms for the evaluation of H10.52,53 The DFR (Biosurface Technologies, Belgrade, MT, USA) was equipped with hydroxyapatite (HA)-coated glass slides (Clarkson Chromatography, South Williamsport, PA, USA) and autoclaved to ensure sterility. The DFR was initially placed in a 37°C incubator under aerobic conditions and attached to a medium reservoir. Approximately 20 minutes before inoculation, the reactor was placed in a horizontal position for sterile medium (1% brain–heart infusion broth; Thermo Fisher Scientific, Waltham, MA, USA) to be dripped in and collect over the coupons to form a conditioning layer on the surface of the HA-coated slide. Next, each of the four channels of the reactor was inoculated with 1 mL of an overnight culture (approximately 108 CFU/mL) of either S. aureus 29213 (American Type Culture Collection, Manassas, VA, USA) or P. aeruginosa mPAO1 (www.genome.washington.edu/UWGC, University of Washington, Seattle, WA, USA). The reactor was then raised to a 10° angle and sterile medium was dripped through the reactor at a total rate of 40 mL/h (10 mL/h per coupon) for 72 hours.
After 3 days of growth, the biofilms were treated with H10, which was provided as a 100 mM stock in DMSO (100%; Sigma-Aldrich Co., St Louis, MO, USA) that was stored at −20°C until use. Prior to treatment, the stock solution was diluted to 100 μM with sterile phosphate-buffered saline (PBS), and fresh solutions were prepared for every experiment. For treatment, flow to the reactor was halted and the reactor placed in a horizontal position. The cover over each channel was removed and 25 mL of each treatment solution (PBS or H10) was applied and then incubated at 37°C for 24 hours. After treatment, the solutions were drained and each channel of the reactor was rinsed with PBS (20 mL) to remove any planktonic bacteria.
Viable plate counts were obtained by placing the treated HA-coated slides in 50 mL conical vials and then removing the biofilms from the slide surface by first repeatedly rinsing and scraping the biofilm from the slide using a sterile Teflon policeman into 10 mL of PBS and disaggregating the bacteria using a sequence of vortex (30 seconds), sonicate (2 minutes), and vortex (30 seconds) to produce a bacterial suspension. The suspension was then serially diluted with PBS and plated on tryptic soy agar (TSA; Thermo Fisher Scientific). The plates were incubated at 37°C for 24 hours and the number of colony-forming units (CFUs) counted. The number of CFU per unit area (cm2) was calculated based on the dilution and surface area of the slide.
DFR experiment with penicillin/streptomycin and H10
A single-species DFR experiment was constructed as described in previously, using S. aureus 29213. As before, H10 (100 μM) was added after a 3-day growth period, but here it was administrated in combination with penicillin/streptomycin (P/S; 100 U/mL and 100 μg/mL, respectively; Thermo Fisher Scientific). Control solutions included PBS alone and P/S alone. Viable plate counts were obtained and analyzed as before.
Human fibroblast cell culture
Human foreskin fibroblasts (hFFs) were isolated from newborn foreskin using previously described methods54 and in accordance with the University of Washington Institutional Review Board. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% newborn calf serum and P/S. All cultures were maintained in a humidified 5% CO2 incubator at 37°C, and the control experiments were conducted identically but without P/S.
Biofilm-conditioned medium
BCM was produced by using the S. aureus biofilms previously grown using DFRs for the analysis of a single species. After the 3-day growth period and the addition of either PBS or H10 for an additional 24 hours growth, each medium was centrifuged at 4,700 rpm to remove bacterial cells. These BCMs were filtered to remove any remaining cells, adjusted to pH 7.4, sterile filtered (0.22 μm), labeled “BCM” and “BCM + H10”, respectively, and stored at −20°C until further use. Prior to experimental use, each BCM was thawed and supplemented with 10% newborn calf serum (Sigma).
Human fibroblast viability evaluation
Viability of the hFFs following BCM exposure was assessed using 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT; Sigma). After the final scratch closure assay image (72 hours) was obtained, the culture medium was removed and replaced with 300 μL of fresh DMEM (without phenol red; Life Technologies, Carlsbad, CA, USA) and 60 μL of XTT. After 4 hours, the A450 was measured. Control hFF cultures (+ control) were exposed to standard culture medium, and blank, cell-free samples containing only cell culture medium and XTT were used as the negative control. All cultures were evaluated in quadruplicate. Data are presented as the mean ± the standard deviation (SD).
Scratch migration/closure
hFFs were grown in 24-well plates (30,000 cells/well) for 2 days, after which 80%–90% confluence was reached. The cultures were then scratched with a 200 μL plastic pipette tip, washed twice with PBS, and treated with 300 μL of either BCM or BCM + H10. The scratched cultures were then imaged to obtain the initial scratch area. Then, the hFF cultures were incubated in a humidified 5% CO2 incubator at 37°C. Every 24 hours, the cultures were imaged and returned to the incubator, and the assay was terminated after 72 hours (four total images). Control hFF cultures were exposed to standard culture medium instead of BCM, and all conditions were tested in quadruplicate.
All scratch images were captured using a 4× objective on a Fisher Scientific Micromaster™ (Thermo Fisher Scientific) light microscope. The images were analyzed and percent scratch area closed was calculated for each time point using the Metamorph® image analysis software (Molecular Devices, Sunnyvale, CA, USA). The scratch assays were performed in duplicate, and each set of experiments used BCM generated from separate DFR runs. Data are presented as the mean ± SD.
Statistical analysis
Statistical significance was determined using analysis of variance with a Tukey’s honest significant difference post hoc test where α=0.05 and P≤0.05 were considered significant.
Porcine skin irritation assay
According to a North Carolina State University Institutional Animal Care and Use Committee (IACUC)-approved protocol (13-088-B), two 30–50 lb sibling female weanling pigs with maximal dorsal surface area were obtained. After a 4 days minimum environmental acclimatization period to the College of Veterinary Medicine facilities, each pig dorsum was clipped 1 day prior to the initial dose. Each dorsum included three controls (one positive, two negative) in Hill Top chambers (Hill Top Research, St Petersburg, FL, USA): poloxamer 407 (P407) only (–control), EtOH only (–control), and P407 +20% SDS (+ control). The remaining dorsal area contained three Hill Top chambers for 5 mM doses of H10 that were administered in 24-hour increments for the first 72 hours of the experiment (Figure 2). Each dosing schedule began at 0 hour and the chambers remained in continuous contact with the skin until the end of the experimental period. For each collection time (48, 72, and 168 hours), two patches were evaluated, where the patch was lifted, the dosed area was scored on the Draize55 scale, the area was photographed, and an 8-mm punch biopsy was taken from the direct center of the area. The duplicate doses were split across two animals. Any wounds were minimally treated (two interrupted stitches) and covered with an empty Hill Top chamber. At the end of the study (7 days), the animals were euthanized according to IACUC protocols, and remaining samples were taken at will from the surrounding areas. All animal handling-procedures were carried out according to NCSU institutional guidelines as well as at least two federal statutes. NCSU is a registered research facility under the Animal Welfare Act (#55-R-005) and has an approved Animal Welfare Assurance Statement with the Office of Laboratory Animal Welfare (#A3331-01).
Figure 2.
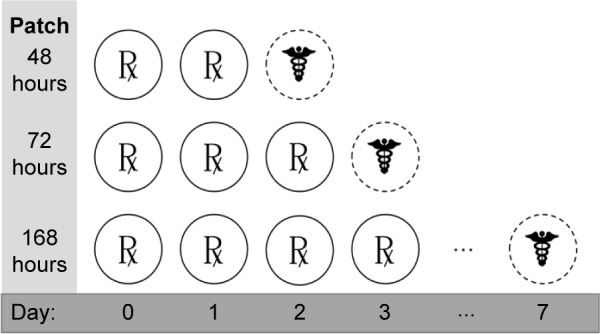
Swine skin dosing and histology schedule.
Notes: Doses (represented by ℞) of H10 (5 mM) were administered to all test sites at 0 hour (day 0). After 24 hours, another dose was administered to all sites. On day 2, another dose was given to all sites except “48 hours”, which was imaged, biopsied, closed, and sealed (represented by ☤). On day 3, a final dose was administered for “168 hours”, while “72 hours” was imaged, biopsied, closed, and sealed. Finally, after 7 days, the final “168 hours” site was examined.
Results
Preliminary cytotoxicity
Using a 25–100 μM range of H10 (Figure 1, inset), S. aureus and P. aeruginosa were grown in compound-enriched media to establish the approximate concentration at which the inhibition of biofilm formation was most effective (Figure 1). To allow for sufficient biofilm growth, P. aeruginosa and S. aureus were incubated for 6 and 24 hours, respectively. For both species, 25 μM H10 inhibited less than 40% of the biofilm-forming ability. However, for S. aureus, this inhibition increased significantly at 50 μM and approached 95% at 75 μM. In contrast, the maximal P. aeruginosa inhibition did not occur until 100 μM H10. To further characterize its pharmacodynamics, the IC50 and EC50 values for H10 against each species were determined. In the context of these experiments, the IC50 is the H10 concentration that inhibits 50% of biofilm formation, while the EC50 is the H10 concentration that disperses 50% of a preformed biofilm. For S. aureus, the H10 IC50 and EC50 values were 12 and 100 μM, respectively, and 31 and 46 μM for P. aeruginosa.
Because imposing selective pressure on bacteria can result in adaptation to avoid cell death, it is important that the 2-AI compounds are not bactericidal. Therefore, concentrations of H10 near the IC50, 12.5 μM (S. aureus) and 50 μM (P. aeruginosa), were used to evaluate the ability of planktonic bacteria to grow in the presence of this compound. In both cases, there was no significant decrease in growth compared to the controls (Figure 3).
Figure 3.
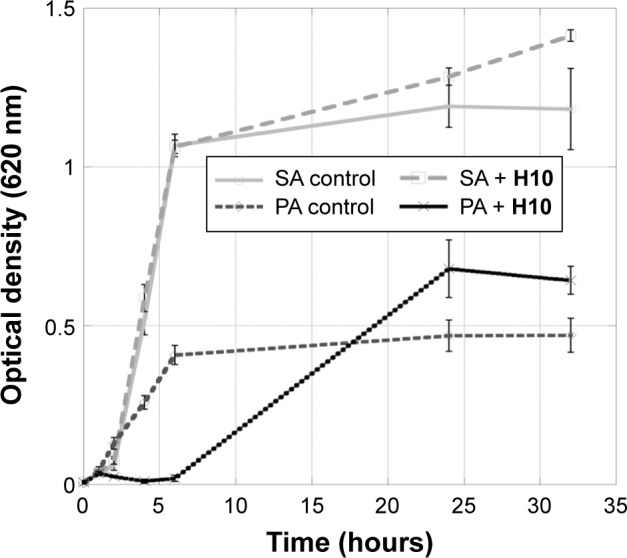
Growth curves for S. aureus and P. aeruginosa in the presence of H10.
Notes: Representative growth curves for S. aureus (light gray lines) and P. aeruginosa (dark gray lines) in the presence of H10 at a concentration greater than the IC50. Data are presented as the mean ± SD of each group. The boxes and circles represent the control values and treatment with H10 at the indicated concentration, respectively.
Abbreviations: IC50, half maximal inhibitory concentration; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; SD, standard deviation.
Biofilm dispersion
To replicate the previously observed biofilm dispersion of 2-AIs,30,32,33,36 3-day biofilms grown in a DFR were treated for 24 hours with H10, and instead of indirectly monitoring viability, this experiment directly calculated the number of CFUs. As an ASTM E2647-08 Standard Test Method,56 the DFR model afforded a low shear, high gas transfer environment for growing biofilms of both species.57,58 For CFU determination, the bacteria were grown with in concentration of H10 well beyond the IC50 (100 μM). While there was no statistically significant difference between the control and H10 experimental groups for P. aeruginosa, there was a marked CFU decrease for S. aureus (P<0.019; Figure 4), indicating that H10 was able to disperse the preformed biofilm.
Figure 4.
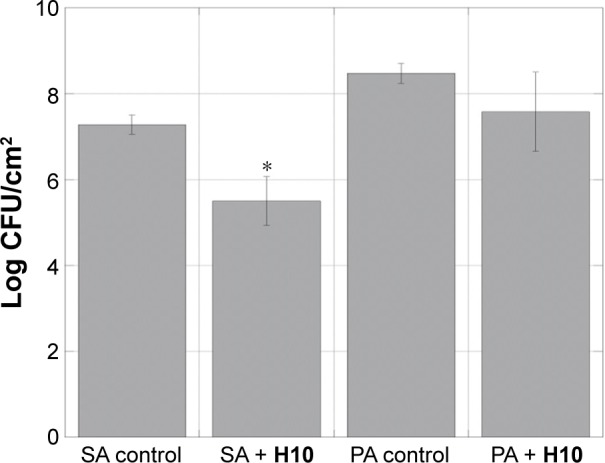
Preformed biofilms are dispersed in the presence of H10.
Notes: Viable cell counts of S. aureus and P. aeruginosa 3-day biofilms following treatment with H10 (24 hours), showing dispersion of a preformed biofilm. The data are given as the mean ± SD, n=3. The asterisk (*) indicates values with significant difference from the other group within the species (P≤0.020).
Abbreviations: CFU, colony-forming unit; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; SD, standard deviation.
Synergism
Further dispersion studies were conducted to compare the observed decrease in the S. aureus log CFU count upon H10 addition with that of a clinically relevant P/S combination, as well as investigate how the compounds function in concert. This particular strain is an antibiotic-resistant isolate, where the typical drug cocktails employed in the clinic are not effective. While both H10 and P/S decreased the cell viability from 7.84±0.56 log CFU/cm2 to 5.98±0.19 and 6.46±0.07, respectively, the synergistic action of both compounds resulted in a marked decrease of almost 4 log CFU/cm2 (P<0.007), effectively resensitizing antibiotic-resistant S. aureus to antibiotics (Figure 5).
Figure 5.
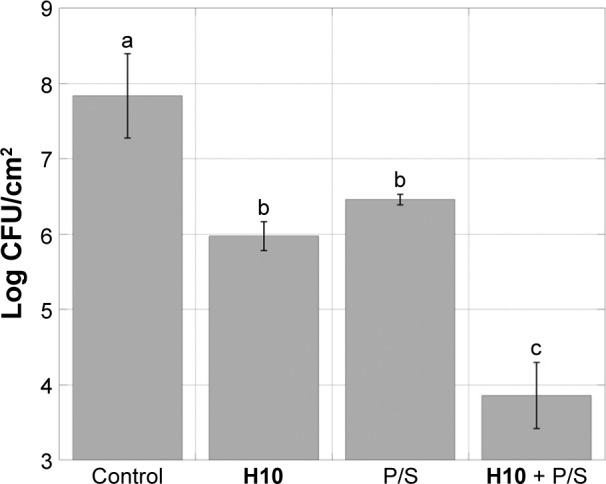
Synergism of H10 with antibiotic cocktail in S. aureus.
Notes: When S. aureus is grown in a combination of H10 and a P/S cocktail, the antibiotic-resistant S. aureus 29213 strain becomes resensitized to antibiotic treatment. The data are given as the mean ± SD, n=3. Statistically significant differences between columns (Tukey’s HSD intervals at P<0.05) are marked with different letters (a–c).
Abbreviations: CFU, colony-forming unit; P/S, penicillin/streptomycin; S. aureus, Staphylococcus aureus; SD, standard deviation; HSD, honest significant difference.
Fibroblast viability
A suitable topical adjuvant for biofilm-based infections must be able to treat the infection while leaving the surrounding tissues unharmed during treatment. Therefore, a fibroblast viability assay was conducted to determine if H10 had any negative consequences on eukaryotic cells. The hFF cultures exposed to BCM were significantly less viable than both the controls (P<0.001) and the BCM + H10-exposed cultures (P<0.0014; Figure 6). Moreover, there was no statistically significant difference between the control and BCM + H10 cultures (P<0.2363), suggesting that H10 altered the S. aureus biofilm metabolism/compound secretion such that the deleterious effects of the BCM on hFFs were eradicated.
Figure 6.
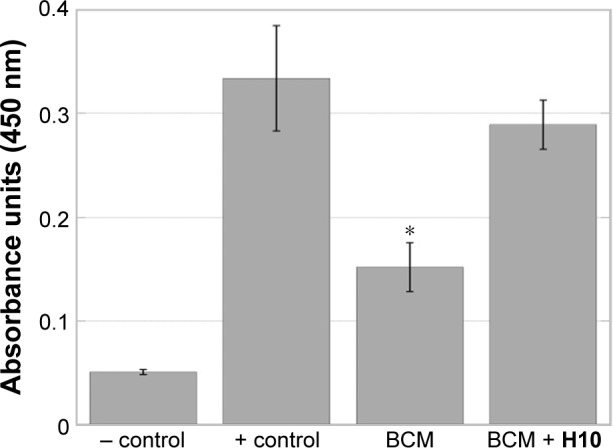
hFF viability assay in BCM.
Notes: In hFF cells, BCM from S. aureus grown in the presence of H10 (BCM + H10) does not exhibit the deleterious effects of S. aureus BCM. The data are given as the mean ± SD, n=3. The asterisk (*) indicates statistically significant difference from both the control and BCM + H10 (P≤0.002).
Abbreviations: BCM, biofilm-conditioned medium; hFF, human foreskin fibroblast; S. aureus, Staphylococcus aureus.
Scratch closure
After ensuring that H10 did not affect fibroblast viability in vitro, a scratch closure assay was used to evaluate performance in a wound model that approximated an in vivo system. After 48 hours, the hFFs exposed to BCM from the untreated biofilm had begun to die and the scratch widened, and by 72 hours, most of the BCM-exposed cells were dead (Figure 7). In contrast, the cultures exposed to BCM from an H10-treated biofilm (BCM + H10) were healthy enough to proliferate and migrate to instigate scratch closure. Across all time points, there were no statistically significant differences in scratch closure between the control and the BCM + H10 cultures, whereas the last two “BCM” time points were markedly different (P≤0.001).
Figure 7.
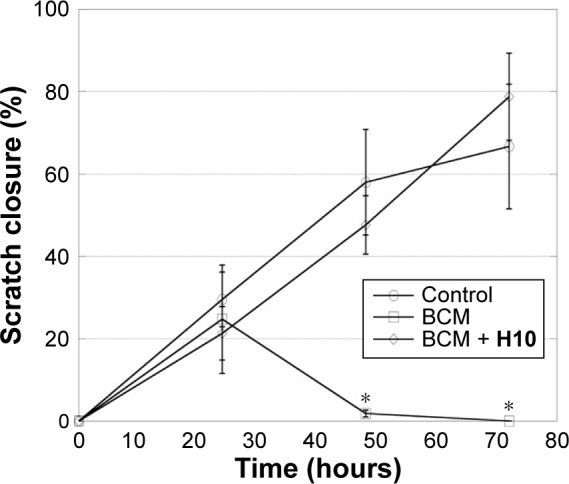
Percent scratch area closed for the in vitro scratch assay.
Notes: Results are shown for two experimental groups, where hFFs were exposed to either antibiotic-resistant Staphylococcus aureus BCM or BCM from these cells grown in the presence of H10 (BCM + H10). The results for one control group are also shown. The data are given as the mean ± SD, n=4. The asterisk (*) indicates values with significant difference from the other groups at the same time point (P≤0.001).
Abbreviations: hFFs, human foreskin fibroblasts; BCM, biofilm-conditioned medium; SD, standard deviation.
Porcine skin irritation
To expand upon the in vitro assays, swine skin was utilized as the in vivo model for human skin. In these experiments, 5 mM H10 was applied directly to the skin. Despite application at orders of magnitude greater than the therapeutic dose, H10 failed to produce any significant observable response by histopathological examination, even after 7 days of constant contact (Figure 2). Overall, H10 did not cause any gross abnormalities in the porcine skin model nor did it stimulate any significant immune reaction (Table 1).
Table 1.
Histopathological evaluation of H10 epidermal/dermal toxicity
| Condition | Endpoint | Replicate | Epidermal histopathology
|
Dermal histopathology
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inflammation
|
Hydropic
|
Necrosis
|
Inflammation
|
Edema
|
Hemorrhage
|
|||||||||
| E | L | E | L | E | L | E | L | E | L | E | L | |||
| 25% P407 control |
|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 20% SDS in EtOH |
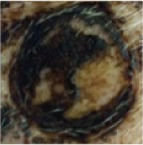
|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | ||
| 5% DMSO in 25% P407 |
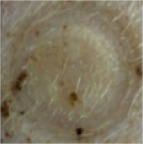
|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 5 mM H10 in 25% P407 48 hours |
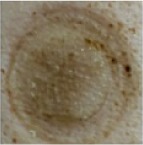
|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 5 mM H10 in 25% P407 72 hours |

|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0–1 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | ||
| 5 mM H10 in 25% P407 168 hours |
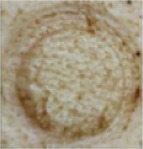
|
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0–1 | 0 | 0 | 0 | 0 | ||
Notes: Effects of acute dermal exposure to H10 in a swine model. Histopathology slides prepared from punch biopsies were scored for the presence of inflammatory cells on a Draize scale of 0–4, where 0= absent, 1= minimal, 2= mild, 3= moderate, and 4= marked. Duplicate doses were split across two animals, and the image included for each condition represents the photograph taken at the end of treatment, prior to biopsy.
Abbreviations: E, eosinophils; L, lymphocytes; P407, poloxamer 407; SDS, sodium dodecyl sulfate; EtOH, ethanol; DMSO, dimethyl sulfoxide.
Discussion
To first establish that H10 inhibits biofilm formation, S. aureus and P. aeruginosa were exposed to varying concentrations of the compound. For both species, >90% biofilm inhibition was achieved. However, compound-induced eradication of biofilms could be due to compound toxicity. For application as an adjuvant, it is also important that H10 is not bactericidal, as this could induce resistance mechanisms. Growth curves for both species did not indicate H10 toxicity. After establishing that H10 is not bactericidal, the biofilm dispersion capability was determined using viable cell plate counts (log CFU/cm2) from a DFR assay, which was the most suitable method to grow biofilms for CFU determination.58 In both CF and chronic wounds, biofilms form at a liquid–air interface, which the DFR models well. It also approximates “plug flow”, where nutrient concentrations and cell densities change along the coupon, which can be directly applicable to catheters and the other aforementioned indwelling medical devices.59 Following the growth curves and inhibition assays, 3-day S. aureus and P. aeruginosa biofilms were treated with H10 for 24 hours and monitored for dispersion (decrease in log CFU/cm2). For S. aureus, the H10 treatment group was significantly lower than the control, and while the changes in P. aeruginosa in the presence of H10 suggested a treatment-derived decrease, it was not statistically significant. Therefore, H10 may treat infections containing S. aureus more effectively, but suitability for treating P. aeruginosa infections cannot be discredited. Nonetheless, H10 inhibits and disperses biofilm formation in a nonbactericidal manner.
An exciting prospect of adjuvants is that they may reinfuse the chemotherapeutic arsenal with highly effective antibiotics that are widely available and cost-effective. It was the sulfa drugs and penicillin in the 1930s and 1940s that brought infections from the cause of 52.7% of all deaths in 1900 to only 2.7% in 2010.1 The drug cocktail tested herein against S. aureus 29213, a multidrug-resistant strain, was P/S. A similar DFR model was used as with the single-species experiments, but a third experimental group received both H10 and P/S. This combination therapy exhibited a synergistic effect, where considerably greater log reductions were observed for the combination than for either treatment alone. Thus, H10 resensitizes antibiotic-resistant bacteria to previously obsolete antibiotics.
After establishing that H10 is not toxic to its bacterial target(s), toxicity toward human cells must also be investigated; an adjuvant is useless if it cannot treat the infection without harming the surrounding tissues. Fibroblasts are the principal dermal cells and are integral to the wound healing process.60 Therefore, hFFs were used in a scratch closure assay, where the hFFs supplemented with BCM failed to close properly, as Kirker et al61 observed previously. However, when fed BCM from bacteria grown in the presence of H10, the scratch closure was indiscernible from the control. Therefore, the effect of H10 on a growing S. aureus culture is enough to alter the BCM such that it does not inhibit in vitro scratch closure. Moreover, Jeffery Marano et al24 characterized the contents of BCM from S. aureus and found multiple protein components in the active fraction of the BCM. Preliminary sequencing data were obtained to identify the potential proteins and several of these components have been correlated with delayed wound healing.24 While none of them were TCS proteins, their transcription may be controlled by upstream RRs.
The final step to establish this 2-AI variant as suitable adjuvant for dermal bacterial infections was to ensure that there were no major histopathological changes in an animal dermal model. Weanling pigs are an accepted animal model for approximating human skin, because they have a similar multilayered epidermis and repeatedly exhibit comparable histological and biochemical properties.62,63 Using the appropriate controls, an epidermal/dermal response to topical compound application was elicited, but it was for SDS, not H10. H10 did not cause any gross abnormalities nor did it provoke significant immune reaction; therefore, H10 is not harmful in an in vivo topical application model.
Conclusion
This study demonstrated that H10 is nonbactericidal, inhibits and disperses biofilms, is synergistic, and resensitizes bacteria to previously obsolete antibiotics. Furthermore, the lack of in vitro scratch closure inhibition suggests that the compound may promote wound healing. Moreover, it exhibited suitability as a topical biofilm treatment. Further investigation of H10 would include studies in the efficacy of in vivo biofilm eradication.
Acknowledgments
This work was supported by NIH grants RO1 GM55769 and R41 AI092952 01A1, as well as the V Foundation for Cancer Research.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Lipsky BA. Diabetic foot infections: current treatment and delaying the ‘post-antibiotic era’. Diabetes Metab Res Rev. 2016;32(Suppl 1):246–253. doi: 10.1002/dmrr.2739. [DOI] [PubMed] [Google Scholar]
- 2.Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet. 2001;358(9276):135–138. doi: 10.1016/s0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 3.Percival SL, Hill KE, Malic S, Thomas DW, Williams DW. Antimicrobial tolerance and the significance of persister cells in recalcitrant chronic wound biofilms. Wound Repair Regen. 2011;19(1):1–9. doi: 10.1111/j.1524-475X.2010.00651.x. [DOI] [PubMed] [Google Scholar]
- 4.Foreman A, Holtappels G, Psaltis AJ, et al. Adaptive immune responses in Staphylococcus aureus biofilm-associated chronic rhinosinusitis. Allergy. 2011;66(11):1449–1456. doi: 10.1111/j.1398-9995.2011.02678.x. [DOI] [PubMed] [Google Scholar]
- 5.Rasmussen TB, Givskov M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int J Med Microbiol. 2006;296(2–3):149–161. doi: 10.1016/j.ijmm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284(5418):1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 7.Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15(2):167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mah TF, O’Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9(1):34–39. doi: 10.1016/s0966-842x(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 9.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23(1):146–158. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 10.Yoon SS, Hennigan RF, Hilliard GM, et al. Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev Cell. 2002;3(4):593–603. doi: 10.1016/s1534-5807(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 11.Costerton JW. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001;9(2):50–52. doi: 10.1016/s0966-842x(00)01918-1. [DOI] [PubMed] [Google Scholar]
- 12.Ciofu O, Tolker-Nielsen T, Jensen PØ, Wang H, Høiby N. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev. 2015;85:7–23. doi: 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 13.Bjarnsholt T, Kirketerp-Møller K, Jensen PØ, et al. Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen. 2008;16(1):2–10. doi: 10.1111/j.1524-475X.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 14.Bionda N, Fleeman RM, de la Fuente-Núñez C, et al. Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur J Med Chem. 2016;108:354–363. doi: 10.1016/j.ejmech.2015.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowd SE, Sun Y, Secor PR, et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008;8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James GA, Swogger E, Wolcott R, et al. Biofilms in chronic wounds. Wound Repair Regen. 2008;16(1):37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 17.Dantes R, Mu Y, Belflower R, et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013;173(21):1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochanek KD, Murphy SL, Xu J. Deaths: final data for 2011. Natl Vital Stat Rep. 2015;63(3):1–120. [PubMed] [Google Scholar]
- 19.Cowan LJ, Stechmiller JK, Phillips P, Yang Q, Schultz G. Chronic Wounds, Biofilms and Use of Medicinal Larvae. Ulcers. 2013;2013:7. doi: 10.1155/2013/487024. Article ID 487024. [DOI] [Google Scholar]
- 20.Peters G, Locci R, Pulverer G. Microbial colonization of prosthetic devices. II. Scanning electron microscopy of naturally infected intravenous catheters. Zentralbl Bakteriol Mikrobiol Hyg B. 1981;173(5):293–299. [PubMed] [Google Scholar]
- 21.Villers D, Espaze E, Coste-Burel M, et al. Nosocomial Acinetobacter baumannii infections: microbiological and clinical epidemiology. Ann Intern Med. 1998;129(3):182–189. doi: 10.7326/0003-4819-129-3-199808010-00003. [DOI] [PubMed] [Google Scholar]
- 22.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 23.Kirker KR, Secor PR, James GA, Fleckman P, Olerud JE, Stewart PS. Loss of viability and induction of apoptosis in human keratinocytes exposed to Staphylococcus aureus biofilms in vitro. Wound Repair Regen. 2009;17(5):690–699. doi: 10.1111/j.1524-475X.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeffery Marano R, Jane Wallace H, Wijeratne D, William Fear M, San Wong H, O’Handley R. Secreted biofilm factors adversely affect cellular wound healing responses in vitro. Sci Rep. 2015;5:13296. doi: 10.1038/srep13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infectious Diseases Society of America. Spellberg B, Blaser M, et al. Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis. 2011;52(Suppl 5):S397–S428. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3(9):541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 27.Neu HC. The crisis in antibiotic resistance. Science. 1992;257(5073):1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 28.Liu YY, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–168. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 29.Lee JY, Howden BP. Vancomycin in the treatment of methicillin-resistant Staphylococcus aureus – a clinician’s guide to the science informing current practice. Expert Rev Anti Infect Ther. 2015;13(7):855–869. doi: 10.1586/14787210.2015.1041924. [DOI] [PubMed] [Google Scholar]
- 30.Rogers SA, Huigens RW, Cavanagh J, Melander C. Synergistic effects between conventional antibiotics and 2-aminoimidazole-derived antibiofilm agents. Antimicrob Agents Chemother. 2010;54(5):2112–2118. doi: 10.1128/AAC.01418-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballard TE, Richards JJ, Wolfe AL, Melander C. Synthesis and antibiofilm activity of a second-generation reverse-amide oroidin library: a structure-activity relationship study. Chemistry. 2008;14(34):10745–10761. doi: 10.1002/chem.200801419. [DOI] [PubMed] [Google Scholar]
- 32.Richards JJ, Ballard TE, Melander C. Inhibition and dispersion of Pseudomonas aeruginosa biofilms with reverse amide 2-aminoimidazole oroidin analogues. Org Biomol Chem. 2008;6(8):1356–1363. doi: 10.1039/b719082d. [DOI] [PubMed] [Google Scholar]
- 33.Richards JJ, Huigens RW, III, Ballard TE, Basso A, Cavanagh J, Melander C. Inhibition and dispersion of proteobacterial biofilms. Chem Commun. 2008;(14):1698–1700. doi: 10.1039/b719802g. [DOI] [PubMed] [Google Scholar]
- 34.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Rittschof D, Rinehart KL. Bioactive bromopyrrole metabolites from the Caribbean sponge Agelas conifera. J Org Chem. 1991;56(9):2965–2975. [Google Scholar]
- 35.Yamada A, Kitamura H, Yamaguchi K, et al. Development of chemical substances regulating biofilm formation. B Chem Soc Japan. 1997;70(12):3061–3069. [Google Scholar]
- 36.Thompson RJ, Bobay BG, Stowe SD, et al. Identification of BfmR, a response regulator involved in biofilm development, as a target for a 2-aminoimidazole-based antibiofilm agent. Biochemistry. 2012;51(49):9776–9778. doi: 10.1021/bi3015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci. 2001;26(6):369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 38.Narayanan A, Kumar S, Evrard AN, Paul LN, Yernool DA. An asymmetric heterodomain interface stabilizes a response regulator-DNA complex. Nat Commun. 2014;5:3282. doi: 10.1038/ncomms4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lou YC, Weng TH, Li YC, et al. Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat Commun. 2015;6:8838. doi: 10.1038/ncomms9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maris AE, Kaczor-Grzeskowiak M, Ma Z, Kopka ML, Gunsalus RP, Dickerson RE. Primary and secondary modes of DNA recognition by the NarL two-component response regulator. Biochemistry. 2005;44(44):14538–14552. doi: 10.1021/bi050734u. [DOI] [PubMed] [Google Scholar]
- 41.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 42.Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr Opin Microbiol. 2006;9(2):143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Stephenson K, Hoch JA. Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacol Ther. 2002;93(2–3):293–305. doi: 10.1016/s0163-7258(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 44.Kuroda M, Kuwahara-Arai K, Hiramatsu K. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem Biophys Res Commun. 2000;269(2):485–490. doi: 10.1006/bbrc.2000.2277. [DOI] [PubMed] [Google Scholar]
- 45.Belcheva A, Golemi-Kotra D. A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem. 2008;283(18):12354–12364. doi: 10.1074/jbc.M710010200. [DOI] [PubMed] [Google Scholar]
- 46.Stephenson K, Hoch JA. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr Med Chem. 2004;11(6):765–773. doi: 10.2174/0929867043455765. [DOI] [PubMed] [Google Scholar]
- 47.Stephenson K, Hoch JA. Two-component and phosphorelay signal-transduction systems as therapeutic targets. Curr Opin Pharmacol. 2002;2(5):507–512. doi: 10.1016/s1471-4892(02)00194-7. [DOI] [PubMed] [Google Scholar]
- 48.Barrett JF, Goldschmidt RM, Lawrence LE, et al. Antibacterial agents that inhibit two-component signal transduction systems. Proc Natl Acad Sci U S A. 1998;95(9):5317–5322. doi: 10.1073/pnas.95.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephenson K, Hoch JA. Histidine kinase-mediated signal transduction systems of pathogenic microorganisms as targets for therapeutic intervention. Curr Drug Targets Infect Disord. 2002;2(3):235–246. doi: 10.2174/1568005023342443. [DOI] [PubMed] [Google Scholar]
- 50.Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol. 2010;13(2):232–239. doi: 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 51.Matsushita M, Janda KD. Histidine kinases as targets for new antimicrobial agents. Bioorg Med Chem. 2002;10(4):855–867. doi: 10.1016/s0968-0896(01)00355-8. [DOI] [PubMed] [Google Scholar]
- 52.Desrosiers M, Myntti M, James G. Methods for removing bacterial biofilms: in vitro study using clinical chronic rhinosinusitis specimens. Am J Rhinol. 2007;21(5):527–532. doi: 10.2500/ajr.2007.21.3069. [DOI] [PubMed] [Google Scholar]
- 53.Xu KD, Stewart PS, Xia F, Huang CT, McFeters GA. Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl Environ Microbiol. 1998;64(10):4035–4039. doi: 10.1128/aem.64.10.4035-4039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukano Y, Knowles NG, Usui ML, et al. Characterization of an in vitro model for evaluating the interface between skin and percutaneous biomaterials. Wound Repair Regen. 2006;14(4):484–491. doi: 10.1111/j.1743-6109.2006.00138.x. [DOI] [PubMed] [Google Scholar]
- 55.Draize J. Appraisal of the Safety of Chemicals in Foods, Drugs and Cosmetics. Austin, TX: Association of Food and Drug Officials, US, Texas State Department of health; 1959. Dermal toxicity; pp. 46–59. [Google Scholar]
- 56.ASTM E2647-08 Standard Test Method for Quantification of a Pseudomonas aeruginosa Biofilm Grown Using a Drip Flow Biofilm Reactor with Low Shear and Continuous flow. ASTM International; West Conshohocken, PA: 2008. [Google Scholar]
- 57.Buckingham-Meyer K, Goeres DM, Hamilton MA. Comparative evaluation of biofilm disinfectant efficacy tests. J Microbiol Methods. 2007;70(2):236–244. doi: 10.1016/j.mimet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 58.Goeres DM, Hamilton MA, Beck NA, et al. A method for growing a biofilm under low shear at the air–liquid interface using the drip flow biofilm reactor. Nat Protoc. 2009;4(5):783–788. doi: 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 59.Curtin JJ, Donlan RM. Using bacteriophages to reduce formation of catheter-associated biofilms by Staphylococcus epidermidis. Antimicrob Agents Chemother. 2006;50(4):1268–1275. doi: 10.1128/AAC.50.4.1268-1275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pérez-Díaz M, Alvarado-Gomez E, Magaña-Aquino M, et al. Anti-biofilm activity of chitosan gels formulated with silver nanoparticles and their cytotoxic effect on human fibroblasts. Mater Sci Eng C Mater Biol Appl. 2016;60:317–323. doi: 10.1016/j.msec.2015.11.036. [DOI] [PubMed] [Google Scholar]
- 61.Kirker KR, James GA, Fleckman P, Olerud JE, Stewart PS. Differential effects of planktonic and biofilm MRSA on human fibroblasts. Wound Repair Regen. 2012;20(2):253–261. doi: 10.1111/j.1524-475X.2012.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Godin B, Touitou E. Transdermal skin delivery: predictions for humans from in vivo, ex vivo and animal models. Adv Drug Deliv Rev. 2007;59(11):1152–1161. doi: 10.1016/j.addr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 63.Lebeaux D, Chauhan A, Rendueles O, Beloin C. From in vitro to in vivo models of bacterial biofilm-related infections. Pathogens. 2013;2(2):288–356. doi: 10.3390/pathogens2020288. [DOI] [PMC free article] [PubMed] [Google Scholar]