

Abstract
Introduction
This commentary outlines a conceptual model for subjective cognitive decline (SCD) in relation to Alzheimer’s disease (AD) biomarkers in the preclinical stages of disease, and a framework for effectively utilizing SCD in secondary prevention clinical trials.
Objective
Mounting evidence supports the notion that SCD is sensitive to encroaching Aβ-amyloid and neurodegeneration. SCD has also been shown to provide additive information of AD-dementia risk beyond what is known about the biomarker status of the individual. Thus, we provide recommendations for the implementing SCD measurement in clinical trials.
Results
We argue that SCD can be measured at three catch points within the course of the clinical trial; firstly, at the initial recruitment and screening phase, secondly, to create more robust estimates of rates of AD-dementia progression, and finally, to measure subjective experiences of cognitive change and quality of life over the course of the trial as a proxy of clinically meaningful functional improvement. We provide recommendations of how SCD can be approached at each of these points
Conclusion
SCD is an important component of the preclinical AD-dementia trajectory. Future studies need to elucidate the interactive influence of Aβ-amyloid and tau on SCD from a spatiotemporal perspective. Even as this evidence accrues, it is clear that SCD can provide unique and additive information about rates of progression and subjectively experienced cognitive change within clinical trials.
Keywords: Cognition, subjective cognitive decline, preclinical, Alzheimer’s disease, amyloid, tau
Introduction
With the advent of preclinical Alzheimer’s disease (AD) secondary prevention trials, identification of clinically and cognitively normal individuals who are risk of AD dementia is paramount, particularly in the earliest stages of disease. Secondary prevention clinical trials primarily target cognitively asymptomatic adults over the age of 65 years who exhibit evidence of biomarkers of interest (Reiman et al. 2016), for instance, neocortical Aβ-amyloid burden in the A4 trial (Sperling et al. 2014). Solely exhibiting an elevated amount of Aβ-amyloid burden, however, does not inexorably indicate progression to AD dementia (Aizenstein et al. 2008; Jagust 2015; Knopman et al. 2003; Rowe et al. 2010). As such, enriching cohorts for cognitive and behavioral AD-related markers are important for effective outcomes in clinical trials (Feldman et al. 2014; Reiman et al. 2016; Sperling et al. 2014). One promising marker is the presence of a subjective concern of cognitive decline (SCD) (Jessen 2014; Jessen et al. 2014). Mounting evidence suggests that increasing SCD is related to greater levels of neocortical Aβ-amyloid burden (Amariglio et al. 2012; Mielke et al. 2012; Perrotin et al. 2012; Zwan et al. 2015), downstream of markers of neurodegeneration (Amariglio et al. 2015b; Chételat et al. 2010; Jessen et al. 2006; Mosconi et al. 2008; Stewart et al. 2011; van der Flier et al. 2004), and cerebrospinal fluid (CSF) markers of Aβ-amyloid, t-tau and p-tau (Stomrud et al. 2007; van Harten et al. 2012; Visser et al. 2009). In those with high Aβ-amyloid burden, the presence of SCD predicts rapid cognitive decline (Pietrzak et al. 2015) and also increases rates of clinical progression to MCI or AD by five-fold in comparison with those with low or no presence of SCD (Buckley et al. 2016).
Neuroimaging markers of neocortical tau burden have been recently introduced to longitudinal ageing studies (Brier et al. 2016; Johnson et al. 2016; Schöll et al. 2016), and will provide researchers with the ability to assess in vivo spatiotemporal relationships between SCD, Aβ-amyloid and tau (Colijn and Grossberg 2015). There are no studies currently reporting these findings, however, some cerebrospinal fluid (CSF) studies suggest that increasing CSF t-tau and/or p-tau is associated with greater SCD (Leoni et al. 2013; van Harten et al. 2012), and particularly in relation with lower CSF Aβ-amyloid (Mosconi et al. 2008; Stomrud et al. 2007; Visser et al. 2009). Downstream markers of tauopathy in AD regions of interest, such as glucose hypometabolism and brain atrophy, have consistently found evidence of a strong relationship with the presence of SCD (Amariglio et al. 2015b; Jessen et al. 2006; Mosconi et al. 2008; Saykin et al. 2006; Scheef et al. 2012). What has not been addressed by previous reviews is how SCD might fit into the preclinical trajectory alongside biomarkers, and how this knowledge can be utilized effectively in clinical trials. One aim of this commentary is to present a conceptual model of SCD and AD biomarkers in the preclinical stages, with the naturally accompanying aim to provide a framework by which SCD can be utilized in secondary prevention clinical trials.
Many recent reviews and meta-analyses have covered the state of SCD research as it currently stands (Jessen et al. 2014; Mitchell et al. 2014; Rabin et al. 2015), and so this will be considered outside the scope of this commentary. We will present the argument that assessing SCD in clinical trials provides unique and nuanced information that is additive to what can be ascertained from AD biomarkers; firstly, assessing SCD is ideal as a front-line screening tool. Secondly, SCD measurement can provide additional information for estimating risk of dementia progression. Thirdly, SCD can be used for tracking the individual’s phenomenological experiences during clinical trials, which is particularly important from the perspective of determining clinically meaningful outcomes from a trial involving clinically asymptomatic participants. The choice of SCD measure also plays an important role in clinical trials, and will be broadly discussed in this commentary.
SCD in relation to Aβ-amyloid and tau biomarkers
A recent review of SCD in relation to AD biomarkers has widely covered the literature (Colijn and Grossberg 2015), however, we will briefly discuss findings that are directly pertinent this commentary. For the most part, greater SCD has been found to relate to high neocortical Aβ-amyloid burden (Amariglio et al. 2012; Amariglio et al. 2015a; Buckley et al. 2016; Mielke et al. 2012; Perrotin et al. 2012; Zwan et al. 2015), CSF fluid markers of Aβ-amyloid (Rolstad et al. 2011; Wolfsgruber et al. 2015), and post-mortem examinations of neuritic plaques (Barnes et al. 2006; Kryscio et al. 2014). Null findings have been reported (Buckley et al. 2013; Hollands et al. 2015), however, it is possible that this incongruency arises from differing SCD methodologies and SUVR thresholds (Colijn and Grossberg 2015; Rabin et al. 2015). Some studies also suggest that the SCD/Aβ-amyloid relationship is amplified by apolipoprotein ε4 (APOEε4) carriership (Rowe et al. 2010; Zwan et al. 2015). SCD has also been shown to be more prevalent in the presence of both Aβ-amyloid and neurodegenerative markers, such as grey matter atrophy, hippocampal atrophy and glucose hypometabolism (Amariglio et al. 2015b; Chételat et al. 2010), supporting the notion of an additive effect of underlying disease on SCD severity. Mielke and colleagues (2012) reported that the presence of SCD reduced the number needed to screen for 100 individuals with high neocortical Aβ-amyloid burden by 37%. An age effect has been reported on this relationship, with Mielke et al. (2012) reporting this effect in 70–79 year olds, with the sensitivity of dropping by approximately 20% in those over 80 years. Other studies have presented similar findings (Zwan et al. 2015), suggesting that the specificity of SCD to the presence of Aβ-amyloid may well decrease with age.
Supporting evidence for the hypothetical model of dynamic biomarkers (Jack et al. 2013) has consistently been found for a relationship between SCD and downstream markers of neurodegeneration, such as brain atrophy and glucose hypometabolism (Amariglio et al. 2015b; Chételat et al. 2010; Jessen et al. 2006; Mosconi et al. 2008; Stewart et al. 2011; van der Flier et al. 2004). In particular, Amariglio and colleagues (2015) demonstrated that the greatest severity of SCD in clinically normal older adults was observed when evidence of both Aβ-amyloid and neurodegeneration (stage 2 of preclinical AD (Sperling et al. 2010)) were present. In CSF studies there is some suggestion that a positive relationship between t-tau and p-tau exists with SCD (Leoni et al. 2013; Mosconi et al. 2008), however, others have reported no convincing relationship (Grambaite et al. 2013). Neuroimaging studies using tau-selective radiotracers have only recently begun, and so findings are yet to be published in relation to SCD.
SCD provides valuable predictive information about risk of cognitive decline (Hohman et al. 2011; Snitz et al. 2015) and future progression to AD dementia (Koppara et al. 2015; Reisberg et al. 2010; Wang et al. 2004). A meta-analysis has suggested an increased annual rate of progression to MCI by 7% or AD dementia by 2% in comparison with those without SCD (Mitchell et al. 2014). In older adults with SCD, significantly reduced CSF Aβ-amyloid and increased t-tau and p-tau has been found in those who progress to AD dementia (van Harten et al. 2012). Some studies, however, have only found an association between CSF t-tau and SCD in those who later exhibit cognitive decline (Rolstad et al. 2013). In older adults with high Aβ-amyloid burden, rates of progression to AD dementia were approximately five times greater in those with high versus low SCD (Buckley et al. 2016). As such, SCD can provide useful predictive information even after biomarker status as been ascertained, which is important from a clinical trial perspective, where robust predictive models of dementia progression are critical for reliably predicting treatment effect (Reiman et al. 2016; Sperling et al. 2015).
What remains to be elucidated is the complex interplay between SCD, Aβ-amyloid and tau in preclinical AD in order to develop temporal sequencing of events. A complex symbiotic, yet regionally-dissociated relationship between neocortical Aβ-amyloid and medial tau deposition has been found in post-mortem studies (Braak and Braak 1991) and is now being replicated in vivo with the advent of tau PET imaging. It is clear that tau deposition in medial temporal regions is related to older age (Schöll et al. 2016), and evidence of entorhinal tau deposition can appear as early as in the second decade of life (Braak et al. 2011). Other isocortical regions of tau, however, are more closely related to Aβ-amyloid burden (Brier et al. 2016; Johnson et al. 2016; Schöll et al. 2016), and are more highly associated with cognitive decline compared with Aβ-amyloid (Johnson et al. 2016; Schöll et al. 2016), suggesting a temporal dependence of tau propagation on Aβ-amyloid (Jack 2014). It is still unclear, however, where SCD sits within the temporal sequencing of pathological changes in the preclinical phase, specifically in relation the first appearance of SCD.
What is currently theorized is that SCD appears downstream of threshold evidence of markers of both Aβ-amyloid and tau (Sperling et al. 2015; Sperling et al. 2010), however, longitudinal studies are yet to be conducted that directly interrogate this question. What is clear, is that SCD manifests at multiple stages, and can also be present in individuals with low Aβ-amyloid (Buckley et al. 2013), which is arguably not considered as a component of the preclinical AD continuum. One could argue that SCD at this stage is not indicative of phenomenological experiences of cognitive change related to AD dementia. An alternative perspective is the following set of possibilities:
That SCD is a much earlier indicator of risk than currently posited, and remains to be investigated (see Figure 1)
That SCD is a much earlier risk indicator, but due to its lack of specificity, is a much more reliable indicator of future dementia progression in the presence of one or more AD biomarker
That AD dementia-related SCD requires Aβ-amyloid to be present in order to be effective in indicating increased risk for future progression. Thus, SCD that appears in individuals without evidence of abnormal AD biomarkers is symptomatic of other psychogenic or organic aetiologies.
Fig 1.
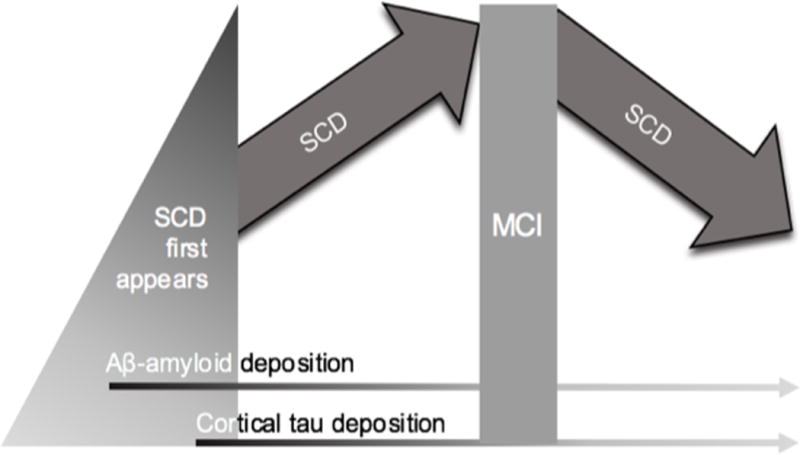
Diagrammatic representation of the initial appearance of SCD in relation to AD biomarkers, and the quadratic relationship between SCD and clinical stages
Taken together, these studies support the notion that SCD is associated with AD biomarkers to some degree, and can provide multiplicative information relating to risk of dementia progression. As such, not only can SCD aid in a more efficient screening process for those with AD biomarkers (particularly Aβ-amyloid as in Mielke et al. 2012), but can also provide more sensitive predictive models of dementia progression (Buckley et al. 2015). The argument provided in the current commentary surrounds the importance of SCD at the front-line and within-trial stages of secondary prevention trials. While there is mounting evidence for an association between Aβ-amyloid and SCD, it is not entirely clear how this relationship interacts with tau deposition. That is, does the early appearance of Aβ-amyloid drive SCD, or is the subsequent interactive relationship between Aβ-amyloid and tau deposition the main player in the manifestation of SCD? Although investigations have been conducted to some extent with CSF studies, the lack of knowledge relating to the influence of interactive effects of region-specific Aβ-amyloid and tau on SCD has still left this question unanswered. This is an important to address in the future as understanding the precise temporal nature of preclinical disease progression is critical for isolating optimal timeframes for clinical intervention.
Preclinical staging criteria for AD propose that SCD occurs around the time of Stage 2, when evidence of both Aβ-amyloid and neurodegeneration are apparent (Sperling et al. 2010). This is supported by findings that show increased reports of SCD in Stage 2 in comparison with Stage 1, where only Aβ-amyloid is present (Amariglio et al. 2015). Studies, however, also support elevated SCD in clinically normal older adults with high compared to low Aβ-amyloid burden (Buckley et al. 2016), supporting the notion of a potential synergistic relationship of Aβ-amyloid and downstream neurodegenerative markers on SCD (see Figure 1: Amariglio et al. 2015; Buckley et al. 2016). We propose that SCD will subtly manifest early in the disease process as neocortical Aβ-amyloid deposition becomes abnormal (Braak and Braak 1999), but will become much more pronounced and reach a peak when tau deposition starts to propagate into cortical associative regions (during Stage 2: see Figure 1). Once cognitive impairment takes hold and the individual moves towards early dementia, SCD will begin to recede in the face of looming cognitive deficits and increasing insight failure (Buckley et al. 2015b; Clare et al. 2010; Gifford et al. 2015a; Rabin et al. 2012; Reisberg and Gauthier 2008). We also posit that SCD in oldest-old individuals (that is, over 80 years) will be less specific to AD-related decline, and as such, this trajectory may not follow the same course, thus reducing SCD reliability in clinical trials.
The utility of SCD within clinical trials
Using SCD as a ‘front-line’ recruitment initiative
In this commentary, we put forward the notion that SCD can reduce the cost of screening individuals with PET imaging by increasing the sensitivity of recruiting ‘true positives’ (Mielke et al. 2012). Clinical trials currently measure SCD during the course of treatment (Sperling et al. 2014), however, no trials are yet specifically recruiting individuals on the basis of SCD. Where SCD comes to the fore is as a self-selecting recruitment mechanism. The presence of SCD forms the initial self-selection recruitment mechanism that can connect an individual to treatment alternatives (Ngandu et al. 2015; Sperling et al. 2014). Utilizing SCD as a ‘first response’ mechanism allows for cost-effective, fast and efficient screening at a population-level prior to conducting expensive neuroimaging screens. In order to make this process as efficient as possible, this initial screening procedure should categorize individuals according to whether or not they satisfy criteria for SCD (see Figure 2).
Fig 2.
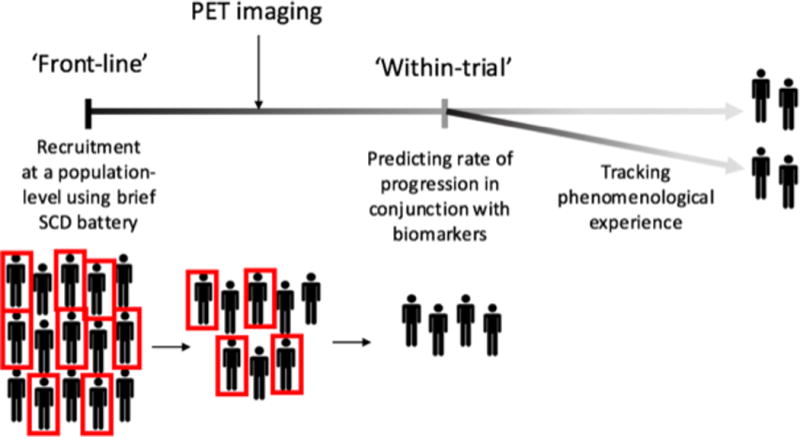
Use of SCD for clinical trials at three stages of the research trajectory
A caveat to this approach is that it may result a relatively heterogeneous SCD population, with a proportion of individuals manifesting AD-related SCD, and others representing the ‘false positive’ (Mitchell et al., 2014). Another important issue relates to the potential biased recruitment of those with heightened awareness of family history of dementia. The advantage to this issue is that individuals with family history are more likely to hold the APOEε4 allele (REF), and evidence also supports an association between SCD and APOEε4 carriership (Dik et al. 2001; Rowe et al. 2010; Zwan et al. 2015), supporting the notion that SCD driven by an awareness of family history may highlight genetic AD risk. The concern, however, is that previous experiences of dementia from a spouse, family member, or friend could motivate anxiety-driven SCD. Studies have shown that negative affect can mask salient associations between SCD and objective cognitive performance (Jonker et al. 2000; Mitchell 2008), and also Aβ-amyloid (Buckley et al. 2013), suggesting that SCD related to anxious hyper-vigilance may introduce complex confounds in the recruitment process. There is some suggestion that increasing levels of anxiety with SCD may be a behavioural marker of rapid cognitive decline in preclinical AD (Pietrzak et al. 2015), however, these findings are relatively novel and a mechanism for disease is forthcoming. Aside from these caveats, we believe that recruiting on the basis of SCD is advantageous. There is much evidence to support the argument that dichotomizing individuals according to SCD increases risk for AD-dementia (Dik et al. 2001; Jessen et al. 2010; Kryscio et al. 2014; Luck et al. 2014; Mitchell et al. 2014; van Oijen et al. 2007; Zwan et al. 2015) and rates of mortality (Luck et al. 2015) in clinically asymptomatic older adults, thus there is precedence for introducing SCD into the recruitment process for clinical trials.
Using SCD as an additional ‘within trial’ predictor of rates of progression
Mounting evidence suggests that gathering information on levels of SCD severity can provide additive information about the risks of dementia progression in those with evidence of AD biomarkers. Within this context, it is likely that level of concern (or level of severity of SCD) will provide unique predictive validity beyond dichotomized groupings, and thus lend more robust estimates to models of rates of AD-dementia progression. SCD severity can be measured in a variety of ways, for instance, according to how many SCD items an individual endorses, or alternatively, according to the magnitude of a total SCD score. An intriguing question is whether endorsing an increasing set of SCD items in a simple ‘dose-response’ type fashion may monotonically predictive of AD-dementia, and whether this could replace the less informative ‘total score’ method (Gifford et al. 2015b; Rabin et al. 2015). The rationale is that each item could be weighted according to AD-risk (Rabin et al. 2015). This question is currently being pursued by the SCD-I, who are currently conducting item-analyses on large sets of data from a range of longitudinal aging studies (Rabin et al. 2015). Regardless, SCD severity has been shown to provide additive information on the risk of clinical progression in those with evidence of abnormal AD biomarkers, and so, we argue that SCD can be utilized within clinical trials for the purposes of ascertaining a more robust predictive algorithm for risk of progression to dementia (see Figure 2).
Using SCD to track phenomenological experiences of cognitive change ‘within trial’
Guidelines from the Food and Drug Administration (FDA) state that evidence of functional benefit from preclinical treatment is an important consideration (Guidance for Industry. Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease. Draft Guidance 2013; Kozauer and Katz 2013). At such early phases of the disease, using traditional tests of functional improvement in activities of daily living (ADLs) to reveal clinically meaningful change over the course of the trial is problematic (Sperling et al. 2015). An alternative approach is to track changes to both ADLs and quality of life, which in this case could be tracked effectively by capturing phenomenological experiences of cognitive change (Amariglio et al. 2015a). It is more beneficial to measure the multidimensional form of SCD throughout the trial, which can provide this unique and nuanced information (Buckley et al. 2015). Subjective monitoring of benefits gained (or otherwise) from treatment is of primary salience to the individual, and can provide accurate information about changes to quality of life and ADLs (Clare et al. 2010) that changes to AD biomarkers alone cannot deliver (see Figure 2 for representation). As such, SCD instruments that reach beyond a simple dichotomous answer will likely lend an important element to outcome measures for clinical trials. Specifically, instruments that can monitor function along a spectrum (i.e. Likert scale response), can allow for the capturing of severity at the cross-sectional level, the level of concern related to SCD at each time point, and also the derivative of change over time. There is an active body of literature that seeks to capture these more nuanced indices of SCD change and severity of concern; one pertinent example is the Cognitive Function Instrument (CFI; Amariglio et al., 2015s) available in the A4 trial, which seeks to capture level of cross-sectional SCD concern along with rate of change of SCD concern over time.
The ultimate question: what measures should be used for SCD in clinical trials?
The argument presented in this commentary is that SCD is an important component of preclinical AD as it is reflective of underlying neurobiological AD change and can be effectively implemented in clinical trials at multiple stages. The question then becomes, how should SCD be measured? We recommend that SCD be approached differently according to the context with which it will be used, i.e. for recruitment, tracking risk or eliciting subjective experiences.
With regards to front-line recruitment, the intention is to effectively screen large portions of the community (that is, anyone over 60 years, and perhaps younger in the future). As such, it is critical for subjective and cognitive screens to be brief and reliable. There is a tradeoff in this requirement, in that greater SCD reliability will likely arise from more comprehensive assessment, which will most likely involve a qualitative component (Buckley et al. 2015a; Buckley et al. 2015c). At a population-level, however, an SCD measure will necessarily need to be short, standardized, and require a categorical response. At the very least, it is likely that front-line assessment of SCD will be involve self-identification. We believe that adhering to SCD-I criteria is the best recommendation for this front-line approach; that is, probing (a) for recent memory change (within a year), (b) whether these changes are concerning, and perhaps (c) ascertaining whether the individual has considered seeing a doctor about these concerns (Jessen et al. 2014). These simple questions assay a range of pertinent information that is most clinically-relevant for future progression to dementia, and is most likely to filter out SCD that is ‘non-organic’ in nature (see Figure 1).
When considering SCD measurement within clinical trials, the focus becomes more on reliably ascertaining the level of SCD severity and capturing nuanced phenomenological experiences of cognitive change. Determining SCD severity for the purposes of predicting rates of progression has been demonstrated in validated SCD questionnaires (Amariglio et al. 2015a; Mitchell et al. 2014; Rabin et al. 2015). Other validated measures exist, however, do not have a consistent track record in relation to preclinical AD-related SCD (Mitchell et al. 2008; Rabin et al. 2015). As mentioned above, efforts are underway within the SCD-I to produce a standardized and sensitive questionnaire for AD dementia-related SCD. As such, it is too soon to make a forgone conclusion about the best measures of SCD severity. What is clear, however, is that tracking SCD severity can provide important additive information about the rate of progression in clinically asymptomatic older adults with evidence of Aβ-amyloid burden (Buckley et al. 2016; Pietrzak et al. 2015). As such, monitoring of this facet of SCD within the trial is important for providing a more refined predictive estimate of progression.
Finally, tracking the phenomenological experience of SCD will be useful for ascertaining treatment effect within the trial, and the subjective changes in quality of life throughout the trial. The measurement of phenomenological experience is not well served by dichotomous outcomes or other purely quantitative questionnaire measures. We have found that rich and nuanced information can be gleaned about the experience of cognitive change from qualitative measurement (Buckley et al. 2015a; Buckley et al. 2015c). Qualitative measures, however, can result in idiosyncratic measurement and interpretation, and thus requires an element of quantification. In order to ameliorate these issues, efforts are also underway to develop a semi-structured interview of SCD which can capture the qualitative elements or ‘themes’ of phenomenological experience, which are indicative of AD-dementia risk (Buckley et al. 2015c). In addition, the A4 clinical trial currently includes an outcome measure, the CFI (Amariglio et al. 2015a), that captures both ADLs and metacognitive elements of quality of life (Sperling et al. 2014). It is a previously validated measure (Amariglio et al. 2015a), which is currently the first SCD instrument adapted as an outcome measure on a clinical trial. With these types of measures, we foresee the ability to build an SCD profile for each individual, which can inform changes in the subjective experience of cognitive change, ADLs and quality of life.
Conclusion
In summary, this commentary presents a conceptual model of the relationship between SCD and AD biomarkers in the preclinical stages of disease, highlighting the advantages of including SCD measures to secondary prevention clinical trials. We argue that SCD likely makes a subtle appearance during Stage 1 of preclinical AD, when Aβ-amyloid deposition has reached a threshold of burden, but will likely become more prevalent and severe during Stage 2, when downstream markers of neurodegeneration take hold. SCD will be strongly, and potentially predominantly, influenced by tau burden and other measures of neurodegeneration, and increase in severity alongside increasing tau deposition until the mid to late stages of mild cognitive impairment where level of insight gradually begins to decline.
With regard to clinical trials, SCD can be harnessed for recruitment efficiency, for predicting rates of AD-dementia progression and tracking phenomenological experiences of cognitive change. We present evidence to support each of these claims and provide some recommendations for measurement approaches. Future work will need to determine how SCD relates to tau deposition and Aβ-amyloid interactively. SCD holds great promise in the early detection and prevention of AD dementia, and recent work in this field has made leaps and bounds towards standardized SCD operationalization and measurement. The field will now need to focus on improving measurement and implementation of SCD approaches to secondary prevention clinical trials.
Acknowledgments
Dr Buckley is funded by the National Medical and Health Research Council (NHMRC) Dementia Research Fellowship (APP1105576).
References
- Aizenstein H, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amariglio RE, et al. Subjective cognitive complaints and amyloid burden in cognitively normal older individuals. Neuropsychologia. 2012;50:2880–2886. doi: 10.1016/j.neuropsychologia.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amariglio RE, Donohue MC, Marshall GA, et al. Tracking early decline in cognitive function in older individuals at risk for alzheimer disease dementia: The alzheimer’s disease cooperative study cognitive function instrument. JAMA Neurology. 2015a;72:446–454. doi: 10.1001/jamaneurol.2014.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amariglio RE, et al. Subjective cognitive concerns, amyloid-β, and neurodegeneration in clinically normal elderly. Neurology. 2015b;85:56–62. doi: 10.1212/wnl.0000000000001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes L, Schneider J, Boyle P, Bienias J, Bennett D. Memory complaints are related to Alzheimer disease pathology in older persons. Neurology. 2006;67:1581–1585. doi: 10.1212/01.wnl.0000242734.16663.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DRMD, Ghebremedhin EMD, Del Tredici KMDP. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. Journal of Neuropathology and Experimental Neurology. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Brier MR, et al. Local and Distributed PiB Accumulation Associated with Development of Preclinical Alzheimer’s Disease. Neurobiology of Aging. 2016 doi: 10.1016/j.neurobiolaging.2015.10.025. doi: http://dx.doi.org/10.1016/j.neurobiolaging.2015.10.025. [DOI] [PMC free article] [PubMed]
- Buckley R, et al. Phenomenological characterisation of memory complaints in preclinical and prodromal Alzheimer’s disease. Neuropsychology. 2015a;29:571–581. doi: 10.1037/neu0000156. [DOI] [PubMed] [Google Scholar]
- Buckley R, et al. Factors affecting memory complaints in the AIBL aging study: biomarkers, memory, affect, and age. Int Psychogeriatr. 2013;25:1307–1315. doi: 10.1017/S1041610213000665. [DOI] [PubMed] [Google Scholar]
- Buckley R, et al. Self and informant memory concerns align in healthy memory complainers and in early stages of mild cognitive impairment but separate with increasing cognitive impairment. Age and ageing. 2015b;44:1012–1019. doi: 10.1093/ageing/afv136. [DOI] [PubMed] [Google Scholar]
- Buckley RF, et al. Subjective memory decline predicts greater rates of clinical progression in preclinical Alzheimer’s disease. Alzheimer’s & Dementia Preprint. 2016 doi: 10.1016/j.jalz.2015.12.013. [DOI] [PubMed] [Google Scholar]
- Buckley RF, Saling MM, Frommann I, Wolfsgruber S, Wagner M. Subjective Cognitive Decline from a Phenomenological Perspective: A Review of the Qualitative Literature. Journal of Alzheimer’s Disease. 2015c;48:S125–S140. doi: 10.3233/JAD-150095. [DOI] [PubMed] [Google Scholar]
- Chételat G, et al. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain. 2010;133:3349–3358. doi: 10.1093/brain/awq187. [DOI] [PubMed] [Google Scholar]
- Clare L, Whitaker CJ, Nelis SM. Appraisal of Memory Functioning and Memory Performance in Healthy Ageing and Early-Stage Alzheimer’s. Disease Aging, Neuropsychology, and Cognition. 2010;17:462–491. doi: 10.1080/13825580903581558. [DOI] [PubMed] [Google Scholar]
- Colijn MA, Grossberg GT. Amyloid and Tau Biomarkers in Subjective Cognitive Impairment. Journal of Alzheimer’s Disease. 2015:1–8. doi: 10.3233/JAD-150180. [DOI] [PubMed] [Google Scholar]
- Dik MG, Jonker C, Comijs HC, Bouter LM, Twisk JW, van Kamp GJ, Deeg DJ. Memory complaints and APOE-epsilon4 accelerate cognitive decline in cognitively normal elderly. Neurology. 2001;57:2217–2222. doi: 10.1212/wnl.57.12.2217. [DOI] [PubMed] [Google Scholar]
- Feldman HH, et al. Alzheimer’s disease research and development: a call for a new research roadmap. Annals of the New York Academy of Sciences. 2014;1313:1–16. doi: 10.1111/nyas.12424. [DOI] [PubMed] [Google Scholar]
- Gifford KA, et al. Inclusion of an Informant Yields Strong Associations between Cognitive Complaint and Longitudinal Cognitive Outcomes in Non-Demented Elders. Journal of Alzheimer’s Disease. 2015a;43:121–132. doi: 10.3233/JAD-131925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford KA, Liu D, Romano RR, Jones RN, Jefferson AL. Development of a subjective cognitive decline questionnaire using item response theory: A pilot study Alzheimer’s & Dementia: Diagnosis. Assessment & Disease Monitoring. 2015b;1:429–439. doi: 10.1016/j.dadm.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grambaite R, Hessen E, Auning E, Aarsland D, Selnes P, Fladby T. Correlates of Subjective and Mild Cognitive Impairment: Depressive Symptoms and CSF Biomarkers Dementia and Geriatric Cognitive. Disorders Extra. 2013;3:291–300. doi: 10.1159/000354188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidance for Industry. Alzheimer’s Disease: Developing Drugs for the Treatment of Early Stage Disease. Draft Guidance. 2013 [Google Scholar]
- Hohman T, Beason-Held L, Lamar M, Resnick S. Subjective cognitive complaints and longitudinal changes in memory and brain function. Neuropsychology. 2011;25:125–130. doi: 10.1037/a0020859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollands S, et al. Amyloid-β Related Memory Decline is not Associated with Subjective or Informant Rated Cognitive Impairment in Healthy Adults. Journal of Alzheimer’s Disease. 2015;43:677–686. doi: 10.3233/JAD-140678. [DOI] [PubMed] [Google Scholar]
- Jack CR. PART and SNAP Acta neuropathologica. 2014;128:773–776. doi: 10.1007/s00401-014-1362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Is amyloid-β harmful to the brain? Insights from human imaging studies. Brain. 2015 doi: 10.1093/brain/awv326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen F. Subjective and objective cognitive decline at the pre-dementia stage of Alzheimer’s disease Eur Arch. Psychiatry Clin Neurosci. 2014;264:3–7. doi: 10.1007/s00406-014-0539-z. [DOI] [PubMed] [Google Scholar]
- Jessen F, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 2014;10:844–852. doi: 10.1016/j.jalz.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen F, et al. Volume reduction of the entorhinal cortex in subjective memory impairment. Neurobiol Aging. 2006;27:1751–1756. doi: 10.1016/j.neurobiolaging.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Jessen F, et al. Prediction of Dementia by Subjective Memory Impairment Effects of Severity and Temporal Association With Cognitive Impairment. Archives of General Psychiatry. 2010;67:414–422. doi: 10.1001/archgenpsychiatry.2010.30. [DOI] [PubMed] [Google Scholar]
- Johnson KA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Annals of neurology. 2016;79:110–119. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker C, Geerlings MI, Schmand B. Are memory complaints predictive for dementia? A review of clinical and population-based studies. Int J Geriatr Psychiatry. 2000;15:983–991. doi: 10.1002/1099-1166(200011)15:11<983::aid-gps238>3.0.co;2-5. 10.1002/1099-1166(200011)15:11<983::AID-GPS238>3.0.CO;2-5 [pii] [DOI] [PubMed] [Google Scholar]
- Knopman D, et al. Neuropathology of cognitively normal elderly. Journal of Neuropathology & Experimental Neurology. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- Koppara A, et al. Cognitive performance before and after the onset of subjective cognitive decline in old age Alzheimer’s & Dementia: Diagnosis. Assessment & Disease Monitoring. 2015;1:194–205. doi: 10.1016/j.dadm.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozauer N, Katz R. Regulatory innovation and drug development for early-stage Alzheimer’s disease New England. Journal of Medicine. 2013;368:1169–1171. doi: 10.1056/NEJMp1302513. [DOI] [PubMed] [Google Scholar]
- Kryscio RJ, et al. Self-reported memory complaints: Implications from a longitudinal cohort with autopsies. Neurology. 2014;83:1359–1365. doi: 10.1212/wnl.0000000000000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V, et al. Diagnostic power of 24S-hydroxycholesterol in cerebrospinal fluid: candidate marker of brain health. Journal of Alzheimer’s Disease. 2013;36:739–747. doi: 10.3233/JAD-130035. [DOI] [PubMed] [Google Scholar]
- Luck T, Luppa M, Matschinger H, Jessen F, Angermeyer MC, Riedel-Heller SG. Incident subjective memory complaints and the risk of subsequent dementia. Acta Psychiatr Scand. 2014;131:290–296. doi: 10.1111/acps.12328. [DOI] [PubMed] [Google Scholar]
- Luck T, Röhr S, Jessen F, Villringer A, Angermeyer MC, Riedel-Heller SG. Mortality in Individuals with Subjective Cognitive Decline: Results of the Leipzig Longitudinal Study of the Aged (LEILA75+) Journal of Alzheimer’s Disease. 2015:1–10. doi: 10.3233/JAD-150090. [DOI] [PubMed] [Google Scholar]
- Mielke MM, et al. Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology. 2012;79:1570–1577. doi: 10.1212/WNL.0b013e31826e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AJ. The clinical significance of subjective memory complaints in the diagnosis of mild cognitive impairment and dementia: a meta-analysis. Int J Geriatr Psychiatry. 2008;23:1191–1202. doi: 10.1002/gps.2053. [DOI] [PubMed] [Google Scholar]
- Mitchell AJ, Beaumont H, Ferguson D, Yadegarfar M, Stubbs B. Risk of dementia and mild cognitive impairment in older people with subjective memory complaints: meta-analysis. Acta Psychiatr Scand. 2014;130:439–451. doi: 10.1111/acps.12336. [DOI] [PubMed] [Google Scholar]
- Mosconi L, et al. Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol Psychiatry. 2008;63:609–618. doi: 10.1016/j.biopsych.2007.05.030. doi:S0006-3223(07)00564-1 [pii] 10.1016/j.biopsych.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngandu T, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. The Lancet. 2015;385:2255–2263. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- Perrotin A, Mormino EC, Madison CM, Hayenga AO, Jagust WJ. Subjective cognition and amyloid deposition imaging: A pittsburgh compound b positron emission tomography study in normal elderly individuals. Arch Neurol. 2012;69:223–229. doi: 10.1001/archneurol.2011.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzak RH, et al. Trajectories of memory decline in preclinical Alzheimer’s disease: results from the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing. Neurobiology of Aging. 2015;36:1231–1238. doi: 10.1016/j.neurobiolaging.2014.12.015. doi: http://dx.doi.org/10.1016/j.neurobiolaging.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Rabin LA, et al. Subjective Cognitive Decline in Older Adults: An Overview of Self-Report Measures used Across 19 International Research Studies. Journal of Alzheimer’s Disease Preprint. 2015:1–25. doi: 10.3233/JAD-150154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin LA, Wang C, Katz MJ, Derby CA, Buschke H, Lipton RB. Predicting Alzheimer’s Disease: Neuropsychological Tests, Self-Reports, and Informant Reports of Cognitive Difficulties. Journal of the American Geriatrics Society. 2012;60:1128–1134. doi: 10.1111/j.1532-5415.2012.03956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. CAP-advancing the evaluation of preclinical Alzheimer disease treatments. Nat Rev Neurol. 2016;12:56–61. doi: 10.1038/nrneurol.2015.177. http://www.nature.com/nrneurol/journal/v12/n1/abs/nrneurol.2015.177.html-supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. International Psychogeriatrics. 2008;20:1–16. doi: 10.1017/S1041610207006412. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Shulman MB, Torossian C, Leng L, Zhu W. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6:11–24. doi: 10.1016/j.jalz.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolstad S, Berg AI, Bjerke M, Blennow K, Johansson B, Zetterberg H, Wallin A. Amyloid-Beta42 is Associated with Cognitive Impairment in Healthy Elderly and Subjective Cognitive Impairment. Journal of Alzheimer’s Disease. 2011;26:135–142. doi: 10.3233/JAD-2011-110038. [DOI] [PubMed] [Google Scholar]
- Rolstad S, Berg AI, Bjerke M, Johansson B, Zetterberg H, Wallin A. Cerebrospinal fluid biomarkers mirror rate of cognitive decline. Journal of Alzheimer’s Disease. 2013;34:949–956. doi: 10.3233/JAD-121960. [DOI] [PubMed] [Google Scholar]
- Rowe CC, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Saykin AJ, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic. MCI Neurology. 2006;67:834–842. doi: 10.1212/01.wnl.0000234032.77541.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheef L, et al. Glucose metabolism, gray matter structure, and memory decline in subjective memory impairment. Neurology. 2012;79:1332–1339. doi: 10.1212/WNL.0b013e31826c1a8d. [DOI] [PubMed] [Google Scholar]
- Schöll M, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89:971–982. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitz BE, Small BJ, Wang T, Chang C-CH, Hughes TF, Ganguli M. Do Subjective Memory Complaints Lead or Follow Objective Cognitive Change? A Five-Year Population Study of Temporal Influence. Journal of the International Neuropsychological Society. 2015;21:732–742. doi: 10.1017/S1355617715000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Amariglio R, Marshall G, Rentz D. Establishing Clinical Relevance in Preclinical Alzheimer’s Disease. The journal of prevention of Alzheimer’s disease. 2015;2:85. doi: 10.14283/jpad.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, et al. Criteria for preclinical Alzheimer’s disease 2010 [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P. The A4 Study: Stopping AD Before Symptoms Begin? Sci Translat Med. 2014;6:228fs213. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R, Godin O, Crivello F, Maillard P, Mazoyer B, Tzourio C, Dufouil C. Longitudinal neuroimaging correlates of subjective memory impairment: 4-year prospective community study. The British Journal of Psychiatry. 2011;198:199–205. doi: 10.1192/bjp.bp.110.078683. [DOI] [PubMed] [Google Scholar]
- Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007;24:118–124. doi: 10.1159/000105017. [DOI] [PubMed] [Google Scholar]
- van der Flier WM, et al. Memory complaints in patients with normal cognition are associated with smaller hippocampal volumes. J Neurol. 2004;251:671–675. doi: 10.1007/s00415-004-0390-7. [DOI] [PubMed] [Google Scholar]
- van Harten AC, Visser PJ, Pijnenburg YAL, Teunissen CE, Blankenstein MA, Scheltens P, van der Flier WM. Cerebrospinal fluid Aβ42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimer’s & Dementia. 2012 doi: 10.1016/j.jalz.2012.08.004. doi: http://dx.doi.org/10.1016/j.jalz.2012.08.004. [DOI] [PubMed]
- van Oijen M, de Jong FJ, Hofman A, Koudstaal PJ, Breteler MM. Subjective memory complaints, education, and risk of Alzheimer’s disease. Alzheimers Dement. 2007;3:92–97. doi: 10.1016/j.jalz.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Visser PJ, et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. The Lancet Neurology. 2009;8:619–627. doi: 10.1016/S1474-4422(09)70139-5. [DOI] [PubMed] [Google Scholar]
- Wang L, van Belle G, Crane PK, Kukull WA, Bowen JD, McCormick WC, Larson EB. Subjective memory deterioration and future dementia in people aged 65 and older. J Am Geriatr Soc. 2004;52:2045–2051. doi: 10.1111/j.1532-5415.2004.52568.x. doi:JGS52568 [pii] 10.1111/j.1532-5415.2004.52568.x. [DOI] [PubMed] [Google Scholar]
- Wolfsgruber S, et al. Subjective cognitive decline is related to CSF biomarkers of AD in patients with MCI. Neurology. 2015;84:1261–1268. doi: 10.1212/wnl.0000000000001399. [DOI] [PubMed] [Google Scholar]
- Zwan MD, et al. Subjective Memory Complaints in APOEɛ4 Carriers are Associated with High Amyloid-β Burden. Journal of Alzheimer’s Disease. 2015;49:1115–1122. doi: 10.3233/JAD-150446. [DOI] [PubMed] [Google Scholar]