

Abstract
Porous organic cages present many opportunities in functional materials chemistry, but the synthetic challenges for these molecular solids are somewhat different from those faced in the areas of metal–organic frameworks, covalent–organic frameworks, or porous polymer networks. Here, we highlight the practical methods that we have developed for the design, synthesis, and characterization of imine porous organic cages using CC1 and CC3 as examples. The key points are transferable to other cages, and this perspective should serve as a practical guide to researchers who are new to this field.
Introduction
Porous organic cages (POCs) are a unique class of microporous material composed of discrete molecules with intrinsic, guest accessible cavities (Figure 1a).1−5 To be porous in the solid state, these cavities must be connected by a 1-, 2-, or 3-dimensional pore network (Figure 1b). Without this connectivity, the intrinsic cavities are isolated and inaccessible to guest molecules.6 The cages must also remain shape-persistent upon addition and removal of guests, such as solvent, because collapse of the intrinsic cavity would disrupt the pore network.7 The intrinsic porosity inside the cages may also be augmented by extrinsic voids between cages.8 The combination of these two requirements (porous crystal packing and shape-persistence), coupled with the synthetic challenge of forming a cage in the first place, makes POCs easy to design “on paper” but somewhat harder to realize in the laboratory.
Figure 1.
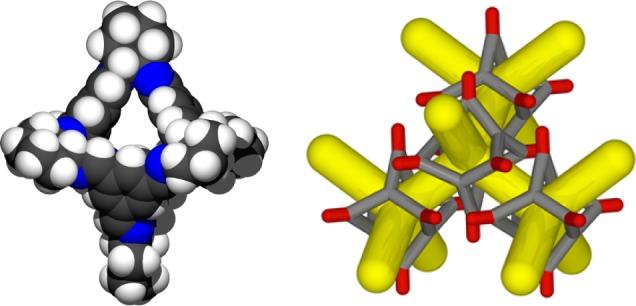
(a) Space filling representation of the porous organic imine cage CC3-R, taken from its single crystal structure (gray, carbon; white, hydrogen; blue, nitrogen). The cage is shape-persistent and has an intrinsic cavity that is accessible via four windows. (b) Schematic representation of crystalline CC3α: each cage packs window-to-window with four adjacent cages to form a 3-D pore network. The intrinsic cage cavities are connected by extrinsic voids between the cage windows (gray, core cage structure; red, cyclohexane groups located on the cage vertices; yellow, 3-D pore network).
POCs can pack either in a crystalline or an amorphous fashion.9,10 The cage packing has a dramatic effect on porosity, and different crystalline polymorphs of the same molecule can have quite different physical properties.11 POCs share some similarities with metal–organic frameworks (MOFs),12 covalent organic frameworks (COFs),13 and porous polymer networks,14 but because of their discrete molecular nature, they are usually solution processable.15 This processability allows POCs to be used in applications that would be more challenging with insoluble porous solids.16−21 To give one example, we have used soluble POCs to prepare liquids with molecular porosity.22 The molecular nature of POCs also gives options for characterization (e.g., solution NMR, HPLC) and purification (e.g., recrystallization, preparative chromatography) that are unavailable for insoluble, extended frameworks.
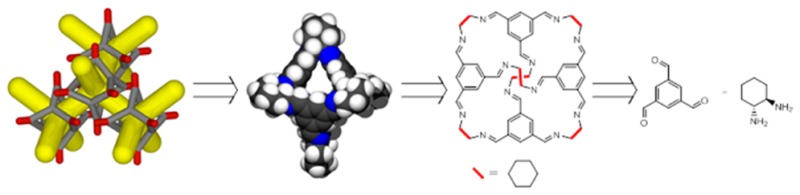
In the Cooper group, we have primarily focused on the synthesis of imine POCs, but the following discussion is also relevant to cages formed using other bond forming reactions (e.g., boronate esters).23 Our aim is to highlight more general experimental strategies by using CC1 and CC3 as detailed worked examples (Scheme 1).
Scheme 1. Synthesis of CC1 and CC3-R Formed by the Reaction of 1,3,5-Triformylbenzene with Ethylendiamine or 1R,2R-1,2-Diaminocyclohexane, Respectively, with CC3-S formed from 1S,2S-1,2-Diaminocyclohexane.
Design and Synthesis of New POCs
There are a number of challenges involved in the design and synthesis of POCs; this is especially true for completely new molecules, but even the preparation of structural variants of known POCs can present unforeseen difficulties. First, suitable cage precursors must be chosen. As a minimum requirement, the precursors must have the correct geometry to form a cage. We have found that even subtle changes to bond angles in the precursors can have a dramatic effect on the outcome of the reaction by changing the size and stoichiometry of the cage product,7,24,25 as also found for metal–organic cages.26 Moreover, essentially any cage precursor combination can, in principle, form an amorphous polymer network instead of a cage. Sometimes, a small change in one of the precursors or the use of unsuitable reaction conditions can “flip” the system from being cage-forming to being polymer-forming. High-dilution synthesis coupled with dynamic covalent chemistry is a common strategy to avoid this, but this will only succeed if the target cage is the thermodynamic product.27,28 The synthesis of imine cages from amine and aldehyde precursors is an example of this.29 Dynamic covalent routes allow the thermodynamic cage product to emerge from the various kinetic products that are initially formed in the reaction, and this can lead in many cases to high synthetic yields; for example, the yield of CC3 in batch syntheses usually exceeds 80%.9 We have synthesized up to 30 g of CC3 in a single batch (Figure 2a), and there is no reason to think that larger scale syntheses are not possible.
Figure 2.

(a) Large scale batch synthesis (>10 g) of CC3 yields a pure polycrystalline material. (b) Large, millimeter-sized single crystals suitable for single crystal X-ray diffraction can also be isolated directly from reaction mixtures.
Obviously, it would be a major advantage to have methods to design appropriate precursors for POC synthesis without resorting to trial and error. In collaboration with the Jelfs group, we are exploring methods to compute the size and shape of cages formed from a given set of starting materials, and their likelihood of remaining shape-persistent, before attempting any synthesis in the lab. We are doing this by calculating the relative energies of candidate structures.30,31 For example, if the candidate cage structure is too strained, then it is unlikely to form. In time, we believe this will be a powerful and generalizable method for in silico POC design. For the moment, though, some intuitive design strategies exist. For example, if a cage is too flexible, then it will often undergo a structural rearrangement upon desolvation, resulting in collapse of the intrinsic cavity and loss of porosity. This can be due to the cumulative effect of small degrees of freedom in multiple “rigid” bonds as well as (more obviously) the inclusion of freely rotatable or highly flexible linkers. As a result, the design of large shape-persistent cages (>2–3 nm diameter) is generally more difficult than for smaller POCs, and there are fewer examples of large cages in the literature.23
Once potential cage precursors have been identified, then suitable synthesis, purification, and isolation conditions must be developed. Parameters that can affect the outcome of the cage-forming reaction include concentration, temperature, and solvent and catalyst choice. Whether or not water (or other condensate) is removed during the reaction can also be important as can the order and speed of the reactant addition. Selection of the reaction parameters should be informed by the properties of the reactants, such as their reactivity and solubility. For instance, the wrong solvent choice can lead to premature precipitation of intermediates from the reaction mixture before the target cage is formed. Once kinetic products precipitate, they may not be able to equilibrate into the desired cage, even if it is the thermodynamic product. Changing the reaction solvent or using a suitable cosolvent can address this. The addition of solubilizing groups (e.g., alkyl chains) is another strategy, although for POCs, this bears the potential disadvantage that these groups may diminish the porosity in the solid state or decrease the propensity of the cage to crystallize, if that is the goal.32,33 A different strategy to improve cage solubility is to decrease the symmetry in the cage, for example, by using mixed linkers,22 but this may strongly inhibit crystallization.10 It may also be necessary to add a catalyst, such as an acid, to enhance the reversibility of the dynamic covalent bond forming chemistry, although in some cases this can also direct the reaction toward other products, such as interlocked catenanes.34
With sensible precursor selection and optimization of the reaction conditions, it is often possible to obtain the desired cage molecule in good yield. Also, if there is sufficient preorganization in the cage precursors, dynamic covalent chemistry may not always be required.35−37 With luck, the cage may crystallize directly from the reaction mixture in a porous phase that remains stable to direct desolvation (e.g., CC3), but this is relatively uncommon. Even if a cage does crystallize directly from the reaction mixture, it may not be easy to determine whether insoluble oligomers are also present as side products, especially if the cage itself is poorly soluble. In addition, amorphous cage or side products might not be revealed by powder X-ray diffraction (PXRD); we have found that a significant proportion of amorphous material can be present before there is a noticeable change in the baseline of the PXRD. Hence, even cages that appear to be chemically pure and phase pure (e.g., by solution NMR and by PXRD) might still be contaminated, for example, with a small quantity of insoluble amorphous polymer that is invisible to these techniques. Moreover, crystals that grow directly and in some cases rapidly from a reaction mixture might be of lower quality and possess a greater number of defects than crystals grown in a more controlled process. This is an important consideration because crystal quality can strongly affect porosity.9,38
Quite often, the desired cage product can remain in solution, perhaps with some insoluble polymeric byproduct. In such cases, one should take care not to mistake this precipitate for the cage product and discard the supernatant! Indeed, even if the cage product crystallizes from the reaction solvent, the supernatant may contain a significant quantity of cage, which can be recovered.8 A further complication, often overlooked, is that even a 100% yield of the cage does not automatically mean that it is the thermodynamic product. Provided that there are no steric clashes, the global thermodynamic product may instead be a catenated cage where two (or conceivably more) independent cages become mechanically interlocked.34,39,40 In such cases, the discrete cage product is a local minimum on the reaction energy surface. However, because catenation often requires extended reaction times and conditions that promote reversibility, such as higher temperatures or the use of a catalyst, then the noncatenated cage can often be isolated even if it is not the overall thermodynamic product.34,39,41 More generally, minor side-products, whether catenanes or other species, need not be fatal to success: as discussed in the Methods section, various chromatographic, precipitation, and crystallization methods can be used to isolate cages as chemically pure single components. Finally, solvent choice can play a part in determining the size and shape of the cage. For example, Warmuth et al. demonstrated that three different cages could be formed from the same starting materials simply by changing the reaction solvent.42 Recently, we also found that other imine cages can equilibrate to form new cage products on prolonged standing in certain crystallization solvents, suggesting that a kinetic cage product is formed initially.
POC Isolation and Characterization
Once pure cage material has been isolated, we would typically screen a range of conditions to afford either crystalline or amorphous phases of the cage, as required. Usually, crystalline phases will be isolated as solvates, although in rare instances the crystallization solvent may be excluded from the crystal, particularly if the cage cavity is small and the solvent is large. Crystals may also spontaneously and rapidly lose solvent at ambient temperature in air while remaining crystalline, particularly if the solvent is volatile and weakly interacting (see Figure S19 in ref (1)): in such cases, it might appear that the cage is crystallizing “solvent free” unless the material is characterized immediately upon isolation from the crystallization solvent. More commonly, the crystalline solvated phase must be carefully desolvated, or “activated”, to isolate a porous solid. As observed for MOFs and COFs, activation is generally more challenging for POCs with low density (high pore volumes) and for solvates where the solvent has a high boiling point, particularly if the solvent strongly interacts with the POC framework, for example, by hydrogen bonding. In such cases, solvent exchange for a less polar, more volatile solvent prior to solvent removal may be necessary.23 PXRD in combination with electron microscopy can be used to establish whether the cage has changed phase or become amorphous after desolvation. Also, if gas adsorption is to be performed, we strongly advocate repeating these characterization techniques after the adsorption measurement to ensure that the cage has not changed phase.
Not all applications require crystallinity; for some purposes, amorphous POCs may be advantageous. Techniques to isolate amorphous phases, or defective crystalline materials, include chemical scrambling,10,22 freeze-drying,9 rapid precipitation,9 spin-coating,17 electrospray,18 and (conceivably) mechanical grinding.
Throughout each stage of synthesis, isolation, and properties evaluation, analytical methods should be used to ensure that the purity and structure of the cage material is unchanged. For example, HPLC and solution NMR are simple methods for ensuring chemical stability, and PXRD and electron microscopy, as discussed above, can be used for phase identification at any stage in the process. All of these methods require only small (<20 mg) quantities of material.
Methods
Synthesis
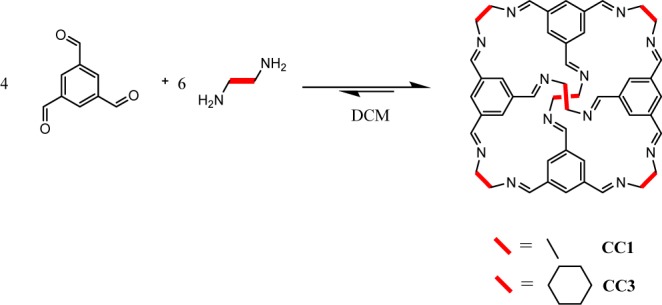
Our most studied POCs, CC1 and CC3, are synthesized by a [4 + 6] cycloimination reaction in which four molecules of the aldehyde 1,3,5-triformylbenzene (TFB) react with six molecules of either ethylenediamine (EDA, CC1) or 1R,2R-1,2-diaminocyclohexane (CHDA, CC3-R) (Scheme 1). To date, we have published two separate high yielding procedures for the synthesis of CC1 and CC3: a batch synthesis that affords multigram quantities of poorly crystalline CC1 or crystalline CC3 within a week and a continuous flow synthesis that affords milligram quantities of amorphous CC1 and semicrystalline CC3 within minutes and gram quantities within hours.
Batch Synthesis
In the batch synthesis of CC3, a solution of homochiral trans-CHDA in DCM is layered onto a suspension of TFB in DCM containing a catalytic amount of TFA. The concentration is relatively high compared with standard cage or macrocycle reactions, which avoids excessive solvent volumes. However, the slow dissolution of the poorly soluble TFB over a number of days, along with the crystallization of CC3 from the supersaturated reaction mixture, is essentially equivalent to running the reaction at high dilution with the slow addition of the TFB. Similar approaches that use solubility tuning of the reagents should be possible with other cages. The rigidity and homochirality of CHDA ensure that the intermediates are at least partially preconfigured to form the CC3 cage, thus reducing the formation of misaligned kinetic oligomers in comparison with more flexible reactants. Indeed, in this system, no oligomeric side-products are observed to precipitate from the concentrated reaction mixture. The role of the acid catalyst is to increase the reversibility of the imine reaction and to afford the thermodynamic cage product within a reasonable time frame. The use of a catalyst is particularly suited to the synthesis of cages that use less reactive starting materials. Heating may also be employed either to increase reversibility or in cases where the reactants or the cage product has poor solubility.
The isostructural POC, CC1, formed by the batch reaction of TFB with EDA (Scheme 1), is synthesized under different conditions. This reaction is run at low temperature and high dilution with slow addition of TFB to the diamine. These conditions are required to mitigate the higher reactivity and inherent flexibility of this diamine, which can result in the formation of insoluble oligomeric products at higher concentrations and higher reaction temperatures. Hence, this route is more reminiscent of classical macrocycle syntheses. To synthesize CC1, a solution of TFB in DCM is added dropwise over 48 h to a solution of the EDA in DCM at 0 °C. The use of a syringe or peristaltic pump rather than an addition funnel ensures more accurate control of addition rates and better reproducibility. After warming the reaction mixture to room temperature and stirring for a further 24 h, the starting materials show complete conversion to CC1 with no soluble byproducts observed. The high reactivity of the starting materials means that an acid catalyst is not required even at high dilution. In fact, when the reaction is run in acetonitrile, the presence of an acid catalyst affords catenated CC1, which is the true thermodynamic product.34CC3, on the other hand, is unable to catenate due to the presence of the bulky cyclohexane groups on the cage vertices. Catenated cages are easily spotted because they often display a marked change in their 1H NMR spectra due to a decrease in symmetry and a pronounced through-space anisotropic effect caused by close contact between the interlocked cages. They can also be identified, of course, by techniques such as mass spectrometry, HPLC, or diffusion-ordered NMR spectroscopy (DOSY).34,39
The formation of CC1 and CC3 does not appear to be sensitive to the water liberated during the reaction, either in batch or continuous flow conditions. However, we have encountered other cages that require the removal of water via a Dean–Stark apparatus (a reverse Dean–Stark apparatus can be used for chlorinated solvents) or the addition of a desiccant to ensure good conversion of the reactants to the cage product.8 This requirement is often associated with the low reactivity of one of the starting materials due to steric or electronic effects.
As a rule of thumb for the synthesis of imine cages under batch reactions, reactive starting materials require high dilution (unless solubility is controlled), low temperatures, and slow addition of one of the precursors (again, unless solubility can act as a control). Less reactive starting materials tend to require the use of an acid catalyst, higher temperatures, more concentrated reaction mixtures, and the removal of water (e.g., with a Dean–Stark trap) to improve conversion. The order of addition is also important. Generally reactions proceed more cleanly when the aldehyde is added to an excess of diamine; the excess amine in the reaction helps to promote reversibility and should minimize oligomer formation.
Flow Synthesis
The synthesis of CC1 and CC3 has also been successfully transferred to a Vaportec R-series continuous flow reactor as a proof of concept.43 By using flow systems, the reaction temperature can be increased above the boiling point of the solvent. Coupled with the highly efficient reagent mixing characteristic of flow systems, this allows the rate of reaction to be increased and the reaction time to be reduced, in some cases from days to minutes. The short reaction time allows multiple combinations of starting materials to be assessed rapidly across a range of conditions. Concentration, stoichiometry, reaction time, flow rate, and temperature are all easily varied. Hence, flow synthesis is a desirable method to screen for new POCs.
The synthesis of CC3 under flow conditions afforded full conversion of TFB to cage in just 10 min at 100 °C, whereas the batch synthesis takes a number of days. A stoichiometry of 4 TFBs to 6.5 diamines was used in the reaction (ideal stoichiometry is 4:6) to mitigate fluctuations in the pump performance. The excess diamine was well tolerated and ensured complete conversion of TFB to the cage, whereas the use of excess aldehyde resulted in incomplete cage formation. The same effect has been noted before in our group in the batch synthesis of imine cages, where the use of a small excess of amine can result in cleaner and more reproducible reactions. The effect of temperature and reaction time on the flow synthesis was also studied with lower temperatures or shorter reaction times affording incomplete conversion to the cage product. Higher temperatures resulted in the precipitation of oligomeric material, whereas a longer reaction time had no effect on the outcome of the reaction. Surprisingly, we also found that CC1 could be synthesized using the same reaction conditions despite significant differences between the batch synthesis conditions for each cage (above), and the reduced thermal and hydrolytic stability of CC1. Unfortunately, due to the poor solubility of TFB in DCM, we were unable to test the effect of more concentrated reaction mixtures on the outcome of either reaction using this flow system. However, the flow synthesis of CC1 is still three times more concentrated than its batch synthesis, significantly reducing solvent volumes. For both CC1 and CC3, the reaction stream exiting the reactor afforded a solution of the cage contaminated with only the excess diamines, which are easily removed by antisolvent reprecipitation.
The importance of directly monitoring cage reactions should be emphasized. We have found several examples of cages that form in solution in good yields but are sensitive to the isolation procedure and can readily decompose into an insoluble polymeric material upon solvent removal.44 One simple monitoring technique is to dilute a sample of the reaction mixture with a suitable deuterated solvent for analysis by 1H NMR. Although the signals of interest may be weak and partially obscured by the nondeuterated reaction solvent, such spectra often provide sufficient information to determine whether the aldehyde has been consumed and how cleanly the reaction has progressed.44 If the target cage is symmetrical, then the 1H NMR spectrum is usually relatively simple. A more complex 1H NMR would potentially indicate the formation of oligomeric products, incomplete conversion to the cage, or potentially catenated products. Reactions can also be monitored directly by LC–MS, which can give information on the purity and composition of the reaction mixture. A final technique that can be used directly on reaction mixtures is DOSY. To do this, however, the reaction should be performed in deuterated solvents. With careful calibration, DOSY can be used to determine the size and hence stoichiometry of the cage that is formed, if any.23,45
For reaction mixtures that form a suspension (e.g., batch synthesis of CC3), it is good practice to analyze a sample of both the solid and the supernatant to determine what quantity, if any, of cage has formed and its purity in each phase. Depending on the composition of the solid and the supernatant, it is often easier to redissolve the solid and combine it with the supernatant to purify the material as a single batch. Obviously, if the quantity of cage in the supernatant (or in the precipitated solid) is insignificant, then that phase can be discarded. If the cage is mainly present in the solid phase, and even if it gives a PXRD pattern consistent with cage formation, this does not guarantee that it has crystallized “phase pure”: it is still good practice to dissolve some of this material and to check that it is not contaminated with oligomeric materials or other side products. POCs are organic molecules, and they can be characterized by the standard range of organic chemistry techniques. Also, as discussed above, crystallization of the cage from the reaction solvent may not produce “good” crystals, although in several cases we have obtained excellent quality millimeter-sized crystals directly from reaction mixtures (Figure 2b) that were suitable for single crystal X-ray diffraction.9 In many cases, though, it is advisible to grow crystals of purified material in a subsequent step, although this may produce a different polymorph to that which precipitated from the reaction mixture, especially if a different crystallization solvent is used. In this respect, soluble POCs differ significantly from insoluble MOFs and COFs where the crystals must, by necessity, be used “as synthesized”.
Purification and Isolation
The first priority when developing an isolation procedure to yield a porous crystal (or an amorphous POC solid) is to check that you are starting with chemically pure material. If the chemical purity of the cage is poor, then it can make the subsequent isolation of crystalline phases more difficult. In the batch synthesis of CC3, the cage crystallizes directly from the reaction mixture in good yield and excellent chemical purity, though this is not always the case for all POCs. Pure CC3 is isolated by filtration of the reaction mixture, followed by a displacement wash with 95:5 EtOH/DCM to remove any surface impurities. CC1, on the other hand, remains in solution when the reaction is complete. The reaction mixture is first filtered to remove any traces of polymeric material that have formed during the reaction, and this filtered solution is evaporated to dryness at <20 °C; higher solvent evaporation temperatures can result in polymerization of the cage. Indeed, because of the extended evaporation time as a result of the large reaction volume, some cage polymerization can still occur, even at low temperatures. Hence, after reaching dryness, the residue is redissolved in a small volume of volatile solvent, usually DCM, refiltered to remove any insoluble polymeric material, and this much smaller volume of solvent evaporated at <20 °C to afford CC1 as a chemically pure solid. More generally, dissolution and filtration is a good method to remove any contamination with insoluble oligomeric side-products.
In the flow synthesis of CC1 and CC3, both products exit the reactor as a DCM solution that also contains excess diamine. CC3 is precipitated directly from the reaction mixture by mixing it with excess hexane. However, because of the higher solubility of CC1, the reaction mixture must first be evaporated; the material is then redissolved in a small amount of solvent, filtered to remove any polymeric material formed during the evaporation, and precipitated by addition to hexane. In both cases, the product is then isolated by filtration to yield the cage in excellent purity and yield; the excess diamine remains in the filtrate. A number of factors affect the choice of antisolvent: it must be miscible with the cage solution and induce precipitation of the cage in good yield and purity. Comparing the HPLC of the initial cage solution with the supernatant of the precipitated suspension allows rapid screening of solvent/antisolvent combinations; in a successful purification, the filtrate will be heavily enriched in impurities as the cage will have precipitated. A related technique that we often use to isolate cages is to swap the solvent, often DCM or chloroform, to a higher boiling antisolvent. The cage solution is usually diluted with the antisolvent, which may result in a cloudy solution/suspension, and then the original solvent is slowly evaporated to leave a suspension of the cage. The cage can then be isolated by filtration while more soluble impurities (e.g., unreacted monomers) remain in solution. Alkanes, such as hexane, and acetone have proven to be particularly effective antisolvents for this technique. We routinely employ this technique to isolate soluble cages directly from their reaction mixtures, and in some instances, it has led directly to the formation of a crystalline porous phase. Recrystallization has also been used to purify cages. As a starting point, common crystallization solvents can be screened on a small scale with heating to establish the solubility of the cage. On cooling, the recovery and purity of any cage precipitate can again be assessed by HPLC analysis of the supernatant. For recovery to be increased, an antisolvent may be added to the hot cage solution. Cages that are thermally unstable may also be purified by adopting the ambient temperature crystallization techniques outlined below. The correct solvent choice for any of these methods is based on a combination of experience and (substantial) trial and error.
If pure cage cannot be obtained using any of the methods discussed, then chromatography may be used for purification. We have had the most success with preparative HPLC and have been able to separate desired cage products from soluble impurities, usually cage fragments or oligomers, isostructural cages, and catenated cages. In our hands, purification usually involves injecting a solution of the crude cage onto a C8 reverse phase column then eluting with methanol, the presence of imine bonds in the cages precludes the use of water-containing gradients. Unsurprisingly, we have found huge differences in performance between column manufacturers; hence, the suitability of a column should first be assessed using an analytical system. Other chromatographic techniques, such as size exclusion chromatography, have also been investigated, but these have so far met with limited success in our experience. The purification of POCs by vacuum sublimation, the organic chemist’s “last resort”, is a further possibility, although the vapor pressure of these relatively large macrocyclic molecules may often be too low for this to be viable.
Crystallization, Cocrystallization, and Amorphization
Unlike insoluble MOFs and COFs, the porosity in POCs can be modified by postsynthetic crystallization steps. As such, isolation of a cage from a reaction mixture that is initially found to be nonporous might not preclude porosity when recrystallized or amorphized.
CC3 crystallizes from the batch reaction mixture in a single desolvatable phase, CC3α, where the intrinsic cage cavities are connected via extrinsic cavities to afford a 3-dimensional pore network (Figure 1b). However, as mentioned above, the porosity is influenced by the quality of the crystals that are formed. We have found that the apparent Brunauer–Emmett–Teller surface area (SABET) of highly crystalline CC3α is, reproducibly, 409 ± 8 m2 g–1, whereas standard “as-made” CC3α has been reported with SABET values of 592 and 624 m2 g–1.1,9 This is thought to be due to defects, such as missing cages, in the crystal packing, which add to the porosity in the material. The only discernible difference between the PXRD patterns of the highly crystalline and “as made” samples is a slight broadening in the diffraction peaks for the latter.9 A polymorph screen with CC3 identified a second crystalline phase, CC3β, which crystallizes from DCM/diethyl ether and has an SABET of 555 m2 g–1 when desolvated.46 Hence, different levels of porosity can be obtained for the CC3 molecule depending on the method of crystallization.
Deliberate amorphization of CC3 via freeze-drying of CC3 from a solution of DCM affords material with an increased SABET of 859 ± 63 m2 g–1.9 This increase in SABET upon amorphization observed for CC3 is not a general phenomenon; more commonly, amorphization results in a decrease or a complete loss of porosity, as found for the isostructural cage CC1.47CC3 increases its porosity in the amorphous state because the bulky cyclohexane groups on the cage vertices prevent efficient packing (CC1 lacks these bulky groups).
CC1 is isolated from the batch and flow reaction mixtures as a poorly crystalline solid, but a screen of different reaction and crystallization solvents has led to the discovery of a number of crystalline solvates. CC1α, CC1β, CC1γ, and CC1δ were isolated from ethyl acetate, DCM, o-xylene, and dioxane, respectively.36 Upon desolvation, CC1α and CC1β undergo subtle structural changes to yield the related desolvated phases, CC1α′ and CC1β′. Each polymorph was observed to possess different porous properties, including different gas selectivities. CC1α′ and CC1β′ are formally nonporous to nitrogen, whereas CC1γ, when desolvated, has an SABET of 550 m2 g–1. CC1δ was found to be unstable to desolvation and afforded a mixture of unidentified phases. Interestingly, it was also found that nonporous and porous phases of CC1 could be interconverted in the solid state simply via exposure to solvent vapor.11
When conducting polymorph screens for POCs, there are two main challenges. The first is to identify new crystalline phases, initially as solvates, and to obtain crystals of sufficient quality to allow them to be solved by SCXRD. Once crystalline phases have been identified, the next step is to isolate sufficient phase-pure material to allow characterization of physical properties. This can be more difficult than isolating the chemically pure cage in the first place. Bulk samples should be analyzed by PXRD to ensure that they are phase pure and that they match the simulated PXRD from the solvated SCXRD. Electron microscopy can also be used to ensure sample homogeneity as secondary crystalline or amorphous phases should be visible.
We routinely use a number of techniques to screen for crystalline phases of organic cages. Single or polycrystalline phases can be obtained by slow evaporation of single or mixed solvent cage solutions under a nitrogen flow; this may be achieved using a desiccator with a gas inlet on the lid. The solutions should be checked regularly for the appearance of suitable crystals for SCXRD during the evaporation. After solvent evaporation, the bulk material should also be analyzed by PXRD: a common pitfall here is to select a “nice” single crystal that is, in fact, not representative of the bulk solid. Vial-in-vial diffusion of an antisolvent into the cage solution or slow diffusion of an antisolvent layered onto the cage solution can also yield single crystal or polycrystalline materials. These materials will most likely be isolated as solvates unless the material spontaneously undergoes desolvation upon removal from the solvent. Rapid desolvation of single crystals upon removal from the crystallization solvent may also be accompanied by a loss of singularity, although the material may still be polycrystalline. In practice, it is often better to keep the crystal in the crystallization solvent until it can be analyzed by SCXRD. Particularly sensitive crystals may be stabilized by encapsulation in a protective oil before analysis.44 One should also be wary of predicting properties from the solvated SCXRD structures unless the material can be desolvated without a significant change in crystal packing.6
Because POCs can be recrystallized after synthesis, it is also possible to produce cocrystals in a modular way containing two9,48 or more49 chemically distinct cages. Clearly, the main technique for proving such structures is single crystal X-ray diffraction. However, because it is possible to form cocrystals with significant positional disorder,49 one may also need to exploit the solubility of POCs, for example, by using HPLC and/or NMR to prove that the chemical composition of the cocrystal is the same as that suggested by the X-ray structure refinement. This is another key difference between POCs and MOFs, COFs, and porous organic polymers: their chemical composition can be determined accurately using solution phase analyses without resorting to techniques such as acid digestion or other chemical decomposition.
Activation
Removal of solvent from POC solvates can have a number of effects. In an ideal case, the solvent can be removed with no or little change in the crystal packing, such that the PXRD of the activated material matches the pattern calculated from the solvated single crystal structure. Sometimes, desolvation leads to a phase change where a new crystalline phase (or a mixed phase) forms. Alternatively, and particularly with large and/or flexible cages or highly solvated structures, desolvation can lead to complete amorphization. Lastly, and less widely recognized, desolvation can sometimes be accompanied by chemical decomposition of some or all of the cage material.44
Various activation methods for porous materials have been used by our group and by others, including vacuum drying,1 supercritical drying,50 and solvent exchange.51 Thermogravimetric analysis of the solvated material is a useful guide to the desolvation temperature required while also providing information on the thermal stability of the cage.1 The best method of activation is dictated by the stability of the crystal packing in the solvate, the boiling point of the crystallization solvent, and the strength of the interaction between the solvent and the cage. The diamondoid window-to-window packing observed for CC3α is a particularly stable “self-reinforcing” packing motif, and CC3α survives rapid desolvation at 80 °C in a vacuum oven with no phase change or loss of crystallinity. However, not all cages, or indeed polymorphs of the same cage, possess such a strong packing motif, and abrupt desolvation can lead to mixed phases or partial amorphization. Such materials will have different properties to their phase pure constituents, and their gas sorption isotherms may not agree, for example, with isotherms predicted from idealized SCXRD structures.
Careful solvent exchange, via one or more solvents, to a lower boiling weakly interacting solvent is the most successful method so far for gentler desolvation of POCs,23,51 but it is likely that techniques such as supercritical drying, as used successfully for MOFs, should also be applicable.50
Ideally, one should also try and obtain a desolvated single crystal structure to provide an accurate structure of the “activated” POC. In practice, this can be difficult to achieve, particularly if the crystal structure is heavily solvated. Solvent removal can lead to cracking of the crystals, and single-crystal-to-single-crystal transformations are substantially rarer than crystal-to-crystal transformations. Quite often, though, the solvent can be removed while the crystal is still mounted in the diffractometer by slowly heating under a flow of nitrogen. CC3α, for example, can be readily desolvated in situ by heating to 117 °C.
POCs should be analyzed postactivation by 1H NMR to ensure that no chemical decomposition has occurred and also that the material has been fully desolvated: here, again, there is an analytical advantage over MOFs and COFs in that the whole sample can be dissolved and any entrained guests released without chemically decomposing the POC. It should be noted that POCs often readily adsorb atmospheric moisture, which could affect any porosity measurements.52 This can also affect elemental analyses significantly: a POC can adsorb several weight percent of water, depending on ambient humidity, which is not an issue typically encountered with the elemental analysis of dense, nonporous organic compounds. Therefore, even “activated” samples should be reactivated in situ prior to gas sorption measurements. PXRD and, ideally, electron microscopy analysis should also be used to ensure that no phase change or amorphization of the sample has occurred when removing the solvent. These tests should be repeated after gas sorption measurements or exposure to other guests, such as solvent vapors, to ensure that the POC is still chemically and phase pure. Finally, all analysis and characterization should be carried out on a single batch of material to ensure that the measured physical properties match those predicted from the POC structure.
Characterization
Throughout the POC discovery process, a number of analytical tests are required to ensure that chemical and phase purity are maintained. Again, POCs are molecules, and it is possible to analyze more than simple phase purity by PXRD, which should be the minimal analysis requirement. Without these various analytical tests, it may be impossible to correlate physical properties with structure: for example, one may end up trying to correlate a gas sorption isotherm calculated from a single crystal X-ray structure with a physical measurement derived from a sample that has become partially amorphous, changed phase, or chemically decomposed. Figure 3 provides an overview of our standard cage discovery workflow and the minimum analytical requirements recommended for each stage of the discovery process.
Figure 3.

Overview of the cage discovery process and the chemical and physical analysis needed at each stage.
Summary
We hope that this perspective raises awareness of the pitfalls that may be encountered along the road to discovering new POCs while at the same time suggesting solutions to these problems. The field of POCs is wide open for development, but progress requires a combination of techniques developed by organic and supramolecular chemists, coupled with methods borrowed from the world of extended framework solids. In this respect, it is a mistake to think of this area as “organic chemistry” or as “materials chemistry”: it is both. Problems occur when either the synthetic chemistry (e.g., insufficient purification) or the physical chemistry (e.g., wrong activation conditions) are neglected. However, by approaching POCs in a systematic way, it is possible to discover truly remarkable materials. For example, there are now organic cage molecules in the literature with surface areas exceeding 3,500 m2 g–1 that begin to rival the most porous MOFs and COFs.23
Experimental Section
Synthesis of CC3-R in a Batch Reaction
DCM (100 mL) was added slowly onto solid 1,3,5-triformylbenzene (5.00 g, 30.9 mmol) without stirring at room temperature. Trifluoroacetic acid (100 μL) was added directly to this suspension as a catalyst for imine bond formation. Finally, a solution of 1R,2R-1,2-diaminocyclohexane (5.00 g, 44.6 mmol) in DCM (100 mL) was layered onto the suspension. The unmixed reaction was covered and left to stand at ambient temperature. Over 5 days, all of the solid 1,3,5-triformylbenzene was consumed, and octahedral crystals of CC3-R grew on the sides of the vessel. The white, crystalline product was removed by filtration and washed with 95:5 EtOH/DCM (25 mL). The solid was dried to constant weight at 80 °C in a vacuum oven, resulting in a yield of 6.5 g (83%).
Synthesis of CC3-R in a Continuous Flow Reaction
The Vaportec reactor was assembled using the R-2+/R-2 Pump Modules with the R-4 Reactor Module. The reaction was run using the conditions outlined in Figure 4. Once the system had reached steady state, the suspension was collected for 110 min. The suspension was isolated by filtration and dried to constant weight in a vacuum oven at 60 °C to afford CC3-R as a white crystalline powder (0.918 g, 95%).
Figure 4.

Schematic of flow reactor setup showing the optimized parameters for the synthesis of CC3-R. Flow system setup: system solvent, DCM; reagent A, 0.083 M 1R,2R-1,2-diaminocyclohexane in DCM (0.948 g/100 mL of DCM); reagent B, 0.083 M 1,3,5-triformylbenzene in DCM (1.34 g/100 mL of DCM); reagent C, hexane; flow rate A, 0.62 mL/min; flow rate B, 0.38 mL/min; flow rate C, 4 mL/min; reactor volume, 10 mL; reactor temperature, 100 °C; back pressure regulator, 8 bar.
Synthesis of CC1 in a Batch Reaction
A solution of 1,3,5 triformylbenzene (3.75 g, 23.1 mmol) in DCM (1150 mL) was added dropwise over 48 h (∼0.3 mL/min) via pressure equalizing dropping funnel (or a syringe or peristaltic pump) to a solution of ethylenediamine (2.08 g, 34.7 mmol) in DCM (850 mL) at 0 °C. After complete addition, the reaction was allowed to stir for another 24 h at room temperature. The solution was then filtered through filter paper. The solvent was removed from the filtrate via rotary evaporation (temperature of the water bath maintained below 20 °C); the crude product was redissolved in CHCl3 (100 mL), and the solution was refiltered. The residue was washed with CHCl3 (50 mL), and the combined organic filtrate was concentrated under vacuum on a rotary evaporator (temperature of the water bath maintained below 20 °C) to give the product as a beige powder with a yield of 4.25 g (94%).
Synthesis of CC1 in a Continuous Flow Reaction
The Vaportec reactor was assembled using the R-2+ Pump Module with the R-4 Reactor Module. The reaction was run using the conditions outlined in Figure 5. Once the system had reached steady state, the reaction mixture was collected for 53 min. The solution was evaporated to dryness at 20 °C, and the residue was redissolved in the minimum amount of DCM, filtered, and poured into an approximately equal volume of hexane to afford a white suspension. The solid was collected by filtration, then dried to constant weight in a vacuum oven at 50 °C to afford CC1 as a beige powder with a yield of 0.310 g (93%).
Figure 5.

Schematic of flow reactor setup showing the parameters for the synthesis of CC1. Flow system setup: System solvent: DCM; reagent A, 0.083 M ethylenediamine in 1:3 MeOH/DCM (0.499 g/(25 mL of MeOH + 75 mL of DCM)); reagent B, 0.083 M 1,3,5-triformylbenzene in DCM (1.34 g/100 mL of DCM); flow rate A, 0.62 mL/min; flow rate B, 0.38 mL/min; reactor volume, 10 mL; reactor temperature, 100 °C; back pressure regulator, 8 bar.
Acknowledgments
We thank Dr. Rebecca L. Greenaway, Dr. Tom Hasell, and Dr. Samantha Y. Chong for helpful comments and discussion.
Glossary
Abbreviations
- CHDA
1,2-diaminocyclohexane
- DCM
dichloromethane
- DOSY
diffusion-ordered spectroscopy
- EDA
ethylenediamine
- HPLC
high-performance liquid chromatography
- IR
infrared
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- POC
porous organic cage
- PXRD
powder X-ray diffraction
- SABET
apparent Brunauer–Emmett–Teller surface area
- TFB
1,3,5-triformylbenzene
- TGA
thermogravimetric analysis
- SCXRD
single-crystal X-ray diffraction
We thank the European Research Council under FP7 (RobOT, ERC Grant Agreement No. 321156).
The authors declare no competing financial interest.
References
- Tozawa T.; Jones J. T. a; Swamy S. I.; Jiang S.; Adams D. J.; Shakespeare S.; Clowes R.; Bradshaw D.; Hasell T.; Chong S. Y.; Tang C.; Thompson S.; Parker J.; Trewin A.; Bacsa J.; Slawin A. M. Z.; Steiner A.; Cooper A. I. Porous Organic Cages. Nat. Mater. 2009, 8, 973–978. 10.1038/nmat2545. [DOI] [PubMed] [Google Scholar]
- Holst J. R.; Trewin A.; Cooper A. I. Porous Organic Molecules. Nat. Chem. 2010, 2, 915–920. 10.1038/nchem.873. [DOI] [PubMed] [Google Scholar]
- Mastalerz M. One-Pot Synthesis of a Shape-Persistent Endo-Functionalised Nano-Sized Adamantoid Compound. Chem. Commun. 2008, 4756–4758. 10.1039/b808990f. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Mastalerz M. Organic Cage Compounds--from Shape-Persistency to Function. Chem. Soc. Rev. 2014, 43, 1934–1947. 10.1039/C3CS60358J. [DOI] [PubMed] [Google Scholar]
- Evans J. D.; Sumby C. J.; Doonan C. J. Synthesis and Applications of Porous Organic Cages. Chem. Lett. 2015, 44, 582–588. 10.1246/cl.150021. [DOI] [Google Scholar]
- Barbour L. J. Crystal Porosity and the Burden of Proof. Chem. Commun. 2006, 1163–1168. 10.1039/b515612m. [DOI] [PubMed] [Google Scholar]
- Jelfs K. E.; Wu X.; Schmidtmann M.; Jones J. T. A.; Warren J. E.; Adams D. J.; Cooper A. I. Large Self-Assembled Chiral Organic Cages: Synthesis, Structure, and Shape Persistence. Angew. Chem., Int. Ed. 2011, 50, 10653–10656. 10.1002/anie.201105104. [DOI] [PubMed] [Google Scholar]
- Bojdys M. J.; Briggs M. E.; Jones J. T. A.; Adams D. J.; Chong S. Y.; Schmidtmann M.; Cooper A. I.; Chong S. Y. Supramolecular Engineering of Intrinsic and Extrinsic Porosity in Covalent Organic Cages. J. Am. Chem. Soc. 2011, 133, 16566–16571. 10.1021/ja2056374. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Chong S. Y.; Jelfs K. E.; Adams D. J.; Cooper A. I. Porous Organic Cage Nanocrystals by Solution Mixing. J. Am. Chem. Soc. 2012, 134, 588–598. 10.1021/ja209156v. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Jones J. T. a; Hasell T.; Blythe C. E.; Adams D. J.; Trewin A.; Cooper A. I. Porous Organic Molecular Solids by Dynamic Covalent Scrambling. Nat. Commun. 2011, 2, 207. 10.1038/ncomms1207. [DOI] [PubMed] [Google Scholar]
- Jones J. T. A.; Holden D.; Mitra T.; Hasell T.; Adams D. J.; Jelfs K. E.; Trewin A.; Willock D. J.; Day G. M.; Bacsa J.; Steiner A.; Cooper A. I. On-off Porosity Switching in a Molecular Organic Solid. Angew. Chem., Int. Ed. 2011, 50, 749–753. 10.1002/anie.201006030. [DOI] [PubMed] [Google Scholar]
- Butova V. V.; Soldatov M. A.; Guda A. A.; Lomachenko K. A.; Lamberti C. Metal-Organic Frameworks: Structure, Properties, Synthesis and Characterization. Russ. Chem. Rev. 2016, 85, 280–307. 10.1070/RCR4554. [DOI] [Google Scholar]
- Ding S.-Y.; Wang W. Covalent Organic Frameworks (COFs): From Design to Applications. Chem. Soc. Rev. 2013, 42, 548–568. 10.1039/C2CS35072F. [DOI] [PubMed] [Google Scholar]
- Dawson R.; Cooper A. I.; Adams D. J. Nanoporous Organic Polymer Networks. Prog. Polym. Sci. 2012, 37, 530–563. 10.1016/j.progpolymsci.2011.09.002. [DOI] [Google Scholar]
- Slater A. G.; Cooper A. I. Function-Led Design of New Porous Materials. Science 2015, 348, aaa8075. 10.1126/science.aaa8075. [DOI] [PubMed] [Google Scholar]
- Kewley A.; Stephenson A.; Chen L.; Briggs M. E.; Hasell T.; Cooper A. I. Porous Organic Cages for Gas Chromatography Separations. Chem. Mater. 2015, 27, 3207–3210. 10.1021/acs.chemmater.5b01112. [DOI] [Google Scholar]
- Song Q.; Jiang S.; Hasell T.; Liu M.; Sun S.; Cheetham A. K.; Sivaniah E.; Cooper A. I. Porous Organic Cage Thin Films and Molecular-Sieving Membranes. Adv. Mater. 2016, 28, 2629–2637. 10.1002/adma.201505688. [DOI] [PubMed] [Google Scholar]
- Brutschy M.; Schneider M. W.; Mastalerz M.; Waldvogel S. R. Direct Gravimetric Sensing of GBL by a Molecular Recognition Process in Organic Cage Compounds. Chem. Commun. 2013, 49, 8398–8400. 10.1039/c3cc43829e. [DOI] [PubMed] [Google Scholar]
- Bushell A. F.; Budd P. M.; Attfield M. P.; Jones J. T. A.; Hasell T.; Cooper A. I.; Bernardo P.; Bazzarelli F.; Clarizia G.; Jansen J. C. Nanoporous Organic Polymer/cage Composite Membranes. Angew. Chem., Int. Ed. 2013, 52, 1253–1256. 10.1002/anie.201206339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasell T.; Zhang H.; Cooper A. I. Solution-Processable Molecular Cage Micropores for Hierarchically Porous Materials. Adv. Mater. 2012, 24, 5732–5737. 10.1002/adma.201202000. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Chen L.; Briggs M. E.; Hasell T.; Cooper A. I. Functional Porous Composites by Blending with Solution-Processable Molecular Pores. Chem. Commun. 2016, 52, 6895–6898. 10.1039/C6CC01034B. [DOI] [PubMed] [Google Scholar]
- Giri N.; Del Pópolo M. G.; Melaugh G.; Greenaway R. L.; Rätzke K.; Koschine T.; Pison L.; Gomes M. F. C.; Cooper A. I.; James S. L. Liquids with Permanent Porosity. Nature 2015, 527, 216–220. 10.1038/nature16072. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. A Permanent Mesoporous Organic Cage with an Exceptionally High Surface Area. Angew. Chem., Int. Ed. 2014, 53, 1516–1520. 10.1002/anie.201308924. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Yu C.; Zhang C.; Long H.; Azarnoush S.; Jin Y.; Zhang W. Dynamic Covalent Synthesis of Aryleneethynylene Cages through Alkyne Metathesis: Dimer, Tetramer, or Interlocked Complex?. Chem. Sci. 2016, 7, 3370–3376. 10.1039/C5SC04977F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Jin A.; McCaffrey R.; Long H.; Zhang W. Design Strategies for Shape-Persistent Covalent Organic Polyhedrons (COPs) through Imine Condensation/Metathesis. J. Org. Chem. 2012, 77, 7392–7400. 10.1021/jo3011683. [DOI] [PubMed] [Google Scholar]
- Sun Q.-F.; Iwasa J.; Ogawa D.; Ishido Y.; Sato S.; Ozeki T.; Sei Y.; Yamaguchi K.; Fujita M. Self-Assembled M24L48 Polyhedra and Their Sharp Structural Switch upon Subtle Ligand Variation. Science 2010, 328, 1144–1147. 10.1126/science.1188605. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Yu C.; Denman R. J.; Zhang W. Recent Advances in Dynamic Covalent Chemistry. Chem. Soc. Rev. 2013, 42, 6634–6654. 10.1039/c3cs60044k. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Wang Q.; Taynton P.; Zhang W. Dynamic Covalent Chemistry Approaches toward Macrocycles, Molecular Cages, and Polymers. Acc. Chem. Res. 2014, 47, 1575–1586. 10.1021/ar500037v. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Zhu Y.; Zhang W. Development of Organic Porous Materials through Schiff-Base Chemistry. CrystEngComm 2013, 15, 1484–1499. 10.1039/C2CE26394G. [DOI] [Google Scholar]
- Jelfs K. E.; Eden E. G. B.; Culshaw J. L.; Shakespeare S.; Pyzer-Knapp E. O.; Thompson H. P. G.; Bacsa J.; Day G. M.; Adams D. J.; Cooper A. I. In Silico Design of Supramolecules from Their Precursors: Odd-Even Effects in Cage-Forming Reactions. J. Am. Chem. Soc. 2013, 135, 9307–9310. 10.1021/ja404253j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelfs K. E.; Cooper A. I. Molecular Simulations to Understand and to Design Porous Organic Molecules. Curr. Opin. Solid State Mater. Sci. 2013, 17, 19–30. 10.1016/j.cossms.2012.12.001. [DOI] [Google Scholar]
- Giri N.; Davidson C. E.; Melaugh G.; Del Pópolo M. G.; Jones J. T. a.; Hasell T.; Cooper A. I.; Horton P. N.; Hursthouse M. B.; James S. L. Alkylated Organic Cages: From Porous Crystals to Neat Liquids. Chem. Sci. 2012, 3, 2153. 10.1039/c2sc01007k. [DOI] [Google Scholar]
- Xu D.; Warmuth R. Edge-Directed Dynamic Covalent Synthesis of a Chiral Nanocube. J. Am. Chem. Soc. 2008, 130, 7520–7521. 10.1021/ja800803c. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Wu X.; Jones J. T. A.; Bacsa J.; Steiner A.; Mitra T.; Trewin A.; Adams D. J.; Cooper A. I. Triply Interlocked Covalent Organic Cages. Nat. Chem. 2010, 2, 750–755. 10.1038/nchem.739. [DOI] [PubMed] [Google Scholar]
- Avellaneda A.; Valente P.; Burgun A.; Evans J. D.; Markwell-Heys A. W.; Rankine D.; Nielsen D. J.; Hill M. R.; Sumby C. J.; Doonan C. J. Kinetically Controlled Porosity in a Robust Organic Cage Material. Angew. Chem., Int. Ed. 2013, 52, 3746–3749. 10.1002/anie.201209922. [DOI] [PubMed] [Google Scholar]
- Kitchin M.; Konstas K.; Sumby C. J.; Czyz M. L.; Valente P.; Hill M. R.; Polyzos A.; Doonan C. J. Continuous Flow Synthesis of a Carbon-Based Molecular Cage Macrocycle via a Three-Fold Homocoupling Reaction. Chem. Commun. 2015, 51, 14231–14234. 10.1039/C5CC05181A. [DOI] [PubMed] [Google Scholar]
- Burgun A.; Valente P.; Evans J. D.; Huang D. M.; Sumby C. J.; Doonan C. J. Endohedrally Functionalised Porous Organic Cages. Chem. Commun. 2016, 52, 8850. 10.1039/C6CC04423A. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Culshaw J. L.; Chong S. Y.; Schmidtmann M.; Little M. A.; Jelfs K. E.; Pyzer-Knapp E. O.; Shepherd H.; Adams D. J.; Day G. M.; Cooper A. I. Controlling the Crystallization of Porous Organic Cages: Molecular Analogs of Isoreticular Frameworks Using Shape-Specific Directing Solvents. J. Am. Chem. Soc. 2014, 136, 1438–1448. 10.1021/ja409594s. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. A Shape-Persistent Quadruply Interlocked Giant Cage Catenane with Two Distinct Pores in the Solid State. Angew. Chem., Int. Ed. 2014, 53, 5126–5130. 10.1002/anie.201400285. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Yu C.; Long H.; Du Y.; Jin Y.; Zhang W. Solution-Phase Dynamic Assembly of Permanently Interlocked Aryleneethynylene Cages through Alkyne Metathesis. Angew. Chem., Int. Ed. 2015, 54, 7550–7554. 10.1002/anie.201501679. [DOI] [PubMed] [Google Scholar]
- Lydon D. P.; Campbell N. L.; Adams D. J.; Cooper A. I. Scalable Synthesis for Porous Organic Cages. Synth. Commun. 2011, 41, 2146–2151. 10.1080/00397911.2010.499487. [DOI] [Google Scholar]
- Liu X.; Warmuth R. Solvent Effects in Thermodynamically Controlled Multicomponent Nanocage Syntheses. J. Am. Chem. Soc. 2006, 128, 14120–14127. 10.1021/ja0644733. [DOI] [PubMed] [Google Scholar]
- Briggs M. E.; Slater A. G.; Lunt N.; Jiang S.; Little M. A.; Greenaway R. L.; Hasell T.; Battilocchio C.; Ley S. V.; Cooper A. I. Dynamic Flow Synthesis of Porous Organic Cages. Chem. Commun. 2015, 51, 17390–17393. 10.1039/C5CC07447A. [DOI] [PubMed] [Google Scholar]
- Briggs M. E.; Jelfs K. E.; Chong S. Y.; Lester C.; Schmidtmann M.; Adams D. J.; Cooper A. I. Shape Prediction for Supramolecular Organic Nanostructures: [4 + 4] Macrocyclic Tetrapods. Cryst. Growth Des. 2013, 13, 4993–5000. 10.1021/cg401171v. [DOI] [Google Scholar]
- Avram L.; Cohen Y. Diffusion NMR of Molecular Cages and Capsules. Chem. Soc. Rev. 2015, 44, 586–602. 10.1039/C4CS00197D. [DOI] [PubMed] [Google Scholar]
- Little M. a; Chong S. Y.; Schmidtmann M.; Hasell T.; Cooper A. I. Guest Control of Structure in Porous Organic Cages. Chem. Commun. 2014, 50, 9465–9468. 10.1039/C4CC04158E. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Jelfs K. E.; Holden D.; Hasell T.; Chong S. Y.; Haranczyk M.; Trewin A.; Cooper A. I. Molecular Dynamics Simulations of Gas Selectivity in Amorphous Porous Molecular Solids. J. Am. Chem. Soc. 2013, 135, 17818–17830. 10.1021/ja407374k. [DOI] [PubMed] [Google Scholar]
- Jones J. T. A.; Hasell T.; Wu X.; Bacsa J.; Jelfs K. E.; Schmidtmann M.; Chong S. Y.; Adams D. J.; Trewin A.; Schiffman F.; Cora F.; Slater B.; Steiner A.; Day G. M.; Cooper A. I. Modular and Predictable Assembly of Porous Organic Molecular Crystals. Nature 2011, 474, 367–371. 10.1038/nature10125. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Chong S. Y.; Schmidtmann M.; Adams D. J.; Cooper A. I. Porous Organic Alloys. Angew. Chem., Int. Ed. 2012, 51, 7154–7157. 10.1002/anie.201202849. [DOI] [PubMed] [Google Scholar]
- Mondloch J. E.; Karagiaridi O.; Farha O. K.; Hupp J. T. Activation of Metal–organic Framework Materials. CrystEngComm 2013, 15, 9258. 10.1039/c3ce41232f. [DOI] [Google Scholar]
- Mastalerz M.; Oppel I. M. Rational Construction of an Extrinsic Porous Molecular Crystal with an Extraordinary High Specific Surface Area. Angew. Chem., Int. Ed. 2012, 51, 5252–5255. 10.1002/anie.201201174. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Schmidtmann M.; Stone C. A.; Smith M. W.; Cooper A. I. Reversible Water Uptake by a Stable Imine-Based Porous Organic Cage. Chem. Commun. 2012, 48, 4689–4691. 10.1039/c2cc31212c. [DOI] [PubMed] [Google Scholar]