

Abstract
Ecto-nucleoside triphosphate diphosphohydrolases (E-NTPDases, subtypes 1, 2, 3, 8 of NTPDases) dephosphorylate nucleoside tri- and diphosphates to the corresponding di- and monophosphates. In the present study we synthesized adenine and uracil nucleotide mimetics, in which the phosphate residues were replaced by phosphonic acid esters attached to the nucleoside at the 5′-position by amide linkers. Among the synthesized uridine derivatives, we identified the first potent and selective inhibitors of human NTPDase2. The most potent compound was 19a (PSB-6426), which was a competitive inhibitor of NTPDase2 exhibiting a Ki value of 8.2 μM and selectivity versus other NTPDases. It was inactive toward uracil nucleotide-activated P2Y2, P2Y4, and P2Y6 receptor subtypes. Compound 19a was chemically and metabolically highly stable. In contrast to the few known (unselective) NTPDase inhibitors, 19a is an uncharged molecule and may be perorally bioavailable. NTPDase2 inhibitors have potential as novel cardioprotective drugs for the treatment of stroke and for cancer therapy.
Introduction
Extracellular nucleotides, such as ATP, ADP, UTP, and UDP act as extracellular signaling molecules by activating purine and/ or pyrimidine P2 receptors.1,2 The activation of these receptors is controlled by various ecto-enzymes that dephosphorylate nucleotides.3–6 An important family of specific nucleoside tri-and diphosphate metabolizing enzymes is the ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase), whose members haveformerlyalsobeenknownasecto-ATPasesorecto-apyrases.3,4,7 By their membrane topology and specific catalytic properties, four of the eight members of the NTPDase family appear to be responsible for most of the hydrolysis of nucleotides at the cell surface, namely, NTPDase1 (CD39), NTPDase2 (CD39L1), NTPDase3 (CD39L3) and NTPDase8.4 These NTPDase members are cell surface proteins anchored in the plasma membrane with their active site orientated toward the extracellular space, while the remaining members (NTPDases 4, 5, 6, and 7) are mostly intracellular enzymes. NTPDase5 and 6 may be secreted into the extracellular space as soluble enzymes.3 NTPDases differentially catalyze the sequential hydrolysis of the β- and γ-phosphate of adenosine and uridine nucleoside di- and triphosphates,8 the physiological ligands of P2 receptors: NTPDase1 hydrolyzes nucleoside tri- and diphosphates similarly well, yielding the monophosphates (AMP, UMP) as products, while NTPDase2 has a clear preference for nucleoside triph-osphates and therefore yields ADP and UDP as the main products. NTPDase3 and 8 are functional intermediates between NTPDase1 and 2, showing an about 2- to 3-fold preference for nucleoside triphosphates.
The isolated action of NTPDase2 provides P2Y1, P2Y12, P2Y13 (ADP), and P2Y6 (UDP) receptor agonists9,10 while terminating the activation of P2X receptors (ATP), as well as P2Y2 (UTP, ATP), P2Y4 (UTP), and P2Y11 (ATP) receptors.11 NTPDase1 is the major physiological NTPDase member showing the broadest distribution and the highest expression levels in most tissues.4 The distribution of NTPDase2 appears to be more restricted. In the vasculature it is expressed in the adventitia of arteries and on subendothelial cells of veins.10 Upon vessel injury, large amounts of ATP are secreted in the extracellular environment which, when exposed to NTPDase2, are efficiently converted to platelet-activating ADP which activates P2Y1 and P2Y12 receptors on thrombocytes.10 In addition, ADP potently inhibits ecto-5′-nucleotidase,12–14 thereby preventing the extracellular formation of adenosine and impeding adenosine receptor-effected vasodilation and antithrombotic properties.15–17 El Omar et al. observed that patients with coronary artery disease had an altered ATP/ADP ratio of hydrolysis in favor of ATP hydrolysis.18 The ratio of ADP to ATP hydrolysis appears to play a vital role in the control of hemostasis, since even a small alteration of the balance may lead to higher platelet reactivity. A selective inhibitor for NTPDase2 may therefore exhibit cardioprotective properties.
In the central nervous system (CNSa) NTPDase2 is found on astrocytes.19 During cerebral ischemia, extracellular ATP concentrations rise. ATP is first dephosphorylated to ADP by astrocytic NTPDase2 and then dephosphorylated to AMP by other ectonucleotidases. The transiently accumulating ADP, a potent inhibitor of ecto-5′-nucleotidase,12 may delay further dephosphorylation of AMP to cytoprotective adenosine by ecto-5′-nucleotidase. Furthermore, activation of P2Y2 receptors by ATP may also have protective effects in glial cells.20 Thus, NTPDase2 inhibition may be useful for the treatment of ischemic conditions in brain, e.g., for the treatment of stroke. Recently, Zimmermann and colleagues identified NTPDase2 as a novel marker for certain progenitor cells in the adult and developing mouse brain, indicating a role in neurogenesis.21 High NTPDase2 expression was also found in human hepatoma22 and Walker 256 tumor cells23 and may be implicated in the stimulation of cancer progression.
NTPDase2 inhibitors could therefore modulate P1 and P2 receptor activation in a site- and event-specific manner. From a medicinal chemistry point of view this concept might be advantageous in comparison with the development of direct P2 receptor agonists because it has turned out to be difficult to develop metabolically stable and selective P2 agonists and it has been impossible to design uncharged, neutral molecules that can activate P2 receptors.24
So far, only a few NTPDase inhibitors have been described (Figure 1).25–32 However, reactive blue 2 (3), pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS, 4), and suramin (5) are also antagonists of many P2 receptors.32–35 Only the ATP analogue ARL67156 (FPL67156, N6-diethyl-β,γ-dibromomethylene-ATP, 1),36 the ATP derivative 8-thiobutyladenosine 5′-triphosphate (8-Bu-S-ATP, 2),26 and some polyoxometalates (POMs)31 have been shown to exhibit enzyme inhibitory potential without significantly affecting nucleotide receptors. However, these compounds are highly polar and cannot be perorally bioavailable. Furthermore, we have recently found that the frequently applied ARL67156 is a weak competitive inhibitor of NPP1 and, NTPDase1 and 3 and does not inhibit NPP3, NTPDase2, or NTPDase8 very well.30,37 Thus far, no selective inhibitors for NTPDase2 have been described.
Figure 1.

Structures of standard ectonucleotidase inhibitors.
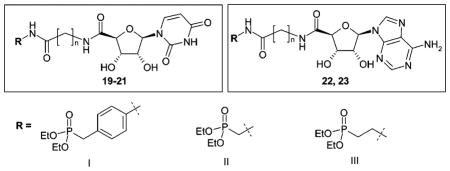
The present study was aimed at developing selective NTPD-ase inhibitors, which should not exhibit affinity for P2 receptors and, in contrast to phosphoric acid ester derivatives, were not to be dephosphorylated by ecto-nucleotidases or phosphatases. Furthermore, our goal was to obtain neutral molecules (not negatively charged at physiologic pH value of 7.4) in order to allow for peroral application. Our target structures were nucleotide mimetics 19–23 derived from ATP and UTP, the substrates of NTPDases, in which the triphosphate group was replaced by a phosphonic acid diester residue (I–III) linked via amide bonds and a linker group to the nucleoside core (Figure 2).
Figure 2.

Target structures: adenine and uracil nucleotide mimetics.
Syntheses
For the preparation of the target compounds 19–23 (Figure 2), a convergent synthetic strategy was applied (see Schemes 1, 2 and 3). The commercially available nucleosides adenosine (6a) and uridine (6b) were protected at the 2′- and 3′-hydroxyl groups (7a,b), and the 5′-hydroxyl group was subsequently oxidized to the corresponding 5′-carboxylic acid function (8a,b) (Scheme 1). Coupling of the nucleoside-5′-carboxylic acids 8a,b with amino-substituted phosphonic acid diethyl ester derivatives 16–18 (Scheme 2) followed by deprotection of the ribose moiety yielded the final products 19–23 (Scheme 3).
Scheme 1.

Synthesis of 2′,3′-Protected Nucleoside-5′-Carboxylic Acidsa
a Reagents and conditions: (a) p-methoxybenzaldehyde, ZnCl2, dry THF, room temp, 48 h; (b) TEMPO, bis(acetoxy)iodobenzene, H2O/MeCN = 1:1, room temp, 3 h.
Scheme 2.

Synthesis of ω-Aminoalkylcarboxamidoalkyl- and ω-Aminoalkylcarboxamido-p-benzyl-Substituted Phosphonic Acid Diethyl Estersa
a Reagents and conditions: (a) dry THF, N-methylmorpholine, ClCO2CH2CH(CH3)2, –25 °C to room temp, 3 h; (b) 4 N HCl in dry dioxane, room temp, 2h.
Scheme 3.

Synthesis of Nucleoside-5′-Carboxamides and Deprotection of the 2′,3′-Positiona
a Reagents and conditions: (a) (i) coupling reagent (PyBOP, HCTU, or HBTU; see Experimental Section), diisopropylethylamine, DMF, room temp, 12 h, (ii) silica gel flash chromatography, CH2Cl2/MeOH = 40:1; (b) (i) 3–5% TFA, CH2Cl2, H2O, 2 h, room temp, (ii) C-18 RP-HPLC, gradient of MeOH/H2O = 25:75 to 100:0.
Selective derivatization of nucleosides in the 5′-position usually requires protection of the 2′- and 3′-hydroxyl groups. Frequently, the nucleosides are treated with acetone to yield the 2′,3′-acetonides.38 In our hands, however, the hydrolytic cleavage of the acetonide group required concentrations of at least 50% of trifluoroacetic acid in water, which resulted in substantial degradation of the target compounds. We obtained a complex mixture of products, presumably because of hydrolysis of acid-sensitive phosphonic acid ester and nucleosidic bonds, especially in the case of the uridine derivatives. As an alternative, a protecting group that would be cleavable by catalytic hydrogenation such as a benzylidene group was considered. However, hydrogenolysis of this protective group with 10% Pd/C requires long reaction times. Johnson and Widlanski observed hydrogenation of the 5,6-double bond of the uracil ring under these conditions as an undesired major side reaction.39,40 Hydrogenolysis might also not be compatible with the benzylphosphonate moiety III (Figure 2). Therefore, we selected an alternative protecting group and reacted the nucleosides with p-methoxybenzaldehyde, yielding the acetal-protected compounds 7a,b in high yields (see Scheme 1).38 The p-methoxybenzylidene protecting group could be easily cleaved off later on under very mild conditions using 5% trifluoroacetic acid in dichloromethane. Under these conditions, no degradation of the nucleosides was observed (see Scheme 3).
Subsequent oxidation of the 5′-hydroxymethylene group of 7a,b to carboxylic acids 8a,b was performed in analogy to a published procedure using catalytic amounts of 2,2,6,6-tetramethyl-1-piperidinyl oxide (TEMPO) and stoichiometric amounts of bis(acetoxy)iodobenzene (Scheme 1).41 The 2′,3′-p-methoxybenzylidene protecting group turned out to be fully compatible with the reaction conditions, although it is somewhat bulkier and less stable than the 2′,3′-isopropylidene protecting group employed in the literature procedure.41 High yields of products 8a (72%) and 8b (78%) were obtained.
The triphosphate mimetic moieties 16–18 were synthesized by connecting amino-functionalized phosphonic acid diethyl ester derivatives via an amide bond with tert-butyloxycarbonyl-(Boc-) protected amino acids (see Scheme 2). For the condensation reaction, Wieland’s mixed anhydride method was employed using isobutyl chloroformate as an activating agent and N-methylmorpholine to deprotonate the carboxylic acid. The activation of the carboxylic acid was performed in dry tetrahydrofurane.42 The commercially available Boc-protected amino acids glycine (9a), β-alanine (9b), and γ-aminobutyric acid (9c) were thus coupled to p-aminobenzylphosphonic acid diethyl ester (10), aminomethylphosphonic acid diethyl ester (11), or 2-aminoethylphosphonic acid ester (12), respectively, yielding amides 13–15. Deprotection with hydrogen chloride in dry dioxane yielded products 16–18 (Scheme 2). Yields ranged between 60% and 90%, calculated over the two steps of amide coupling and Boc-deprotection.
The crucial step in the synthetic pathway was the formation of the nucleoside 5′-carboxylic acid amides (see Scheme 3). The carboxylic acid functions of nucleoside 5′-carboxylic acids are sterically hindered and therefore possess a very different reactivity than, for example, the carboxylic acid functions of amino acids. Initial attempts to react uridine-5′-carboxylic acid 8b with compounds 16a–c using standard coupling reagents, including carbodiimides (1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide hydrochloride, dicyclohexylcarbodiimide), or isobutyl chloroformate,42 were unsuccessful. Diphenylphosphorylazide, previously applied in the synthesis of uridine-5′-carboxylic acid amides, resulted in low yields.43 Therefore, we examined a series of coupling reagents based on 1-hydroxybenzotriazole, which have been described in the literature as powerful tools for amide synthesis.44 Among these, benzotriazol-1-yl-oxy-trispyrrolidinophosphonium hexafluorophosphate (PyBOP) had been successfully applied to the preparation of adenosine-5′-carboxamides.45,46 In addition to PyBOP, we also used 2-(1H-benzotriazolyl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 2-(6-chloro-1H-benzotriazolyl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU). The three reagents gave very different results depending on the starting compounds that were to be coupled. The best coupling reagent for each reaction was different mainly depending on the chain length of the alkylamine moiety; it did not depend on the nature of the nucleoside-5′-carboxylic acid (adenine or uracil derivative). If the alkyl chain consisted of only one methylene unit (n = 1: 16a, 17a, 18a), HCTU turned out to be the coupling reagent of choice. Longer alkyl chains consisting of two or three methylene groups (16b,c; 17b,c) required the use of HBTU or PyBOP (see Experimental Section). The amide formation had to be performed in the presence of a base. Three commonly used bases were compared for the synthesis of 19a and 19c: triethylamine, N-methylmorpholine, and diisopropylethylamine (DIPEA). Of these, only diisopropylethylamine resulted in satisfactory yields. The amide coupling reactions involving PyBOP were previously reported to proceed within 15 min;45 however, we found that stirring overnight gave the highest yields. The order of the addition of starting compounds and reagents to the solvent was important for the success of the reaction; the uridine-derived carboxylic acid 8b was codissolved with the appropriate coupling reagent and hydroxybenzotriazole in dimethylformamide, followed by the addition of diisopropylamine, which led to the activation of the carboxylic acid. Then the appropriate amine derivative was added. In contrast, the adenosine-derived carboxylic acid 8a had to be dissolved separately in a mixture of dimethylformamide and diisopropylamine, and a solution of the appropriate coupling reagent and hydroxybenzotriazole in dimethylformamide was subsequently added for activation, followed by the appropriate amine derivative (Scheme 3).
After deprotection of the ribose moiety using 5% trifluoroacetic acid in dichloromethane, the final products 19–23 were purified by reversed phase HPLC on a C-18 column and subsequently dried by lyophilization (see Scheme 3). Final products were isolated in yields ranging between 30% and 70%, calculated over the two steps of amide coupling and deprotection of the 2′,3′-position.
Biological Activity
The nucleotide mimetics were investigated as inhibitors of the four human NTPDase isoenzymes, NTPDases 1, 2, 3, 8. NTPDases were expressed in COS-7 cells or HEK 293 cells, and enzyme-containing membrane preparations were used. The assays were performed in reaction vials in the presence of ATP (400 μM) as a substrate. Product formation (AMP or ADP) was subsequently analyzed by capillary electrophoresis (CE) with UV detection.30 The recent development of easy and powerful CE-based nanoscale NTPDase inhibition assays in our group30 was crucial for the current identification and characterization of NTPDase2 inhibitors. It allowed high-throughput screening due to the direct and fast separation of the substrates from the reaction products within a few minutes without sample pre-treatment. Initial screening was performed at a concentration of 1 mM of test compounds. For active compounds, full concentration–inhibition curves were determined. For compound 19a, the type of NTPDase inhibition was investigated by determining enzyme kinetics in the presence of different concentrations of inhibitor, and a competitive mechanism of inhibition was found for NTPDase2. Ki values were calculated from IC50 values using the Cheng–Prusoff equation and the following Km values for the substrate ATP: NTPDase1, 17 μM; NTPDase2, 70 μM; NTPDase3, 75 μM; NTPDase8, 81 μM.8,47
In order to investigate the selectivity of the compounds on NTPDases versus P2 receptors, the effects of these compounds at the uracil nucleotide-activated P2 receptor subtypes P2Y2, P2Y4, and P2Y6 were also investigated. These subtypes were selected because the most potent NTPDase inhibitors of the present series were uracil rather than adenine nucleotide mimetics. Astrocytoma cell lines expressing the human P2Y2, the human P2Y4, or the rat P2Y6 receptor were used.48 Inhibition of agonist- (UTP for P2Y2, P2Y4; UDP for P2Y6) induced stimulation of intracellular calcium release of the phospholipase C coupled receptors was measured using the calcium fluorophore Fura-2 applied as its acetoxymethyl ester (Fura-2 AM).49–51 In this test system, test compounds with antagonistic as well as those with agonistic activity at the P2Y receptor subtypes can be detected, since preincubation with either agonists or antagonists will lead to an inhibition of ligand-induced receptor activation. Antagonists will block the receptors, while preincubation with agonists will lead to receptor desensitization.52 We have shown agonist-induced desensitization for all investigated receptors under the applied conditions (unpublished data). Selected compounds were also investigated directly for potentially agonistic activity.
Structure–Activity Relationships
NTPDase Inhibition
The investigated nucleotide mimetics can be divided into two structural groups: uracil derivatives (19–21) and adenine derivatives (22, 23). The compounds contain a 5′-amide linkage and a spacer unit of different length (one to three methylene units) connected via another amide linkage with a polar benzylphosphonate (I), methylenephosphonate (II), or ethylenephosphonate (III) moiety, respectively. Table 1 shows that adenine derivatives were generally less potent than the corresponding uracil derivatives (compare 19a/22a, 19b/22b, 20a/23a, 20b/23b, 20c/23c). This was surprising, since ATP is a better substrate of NTPDases than UTP.8 There was one exception, the benzylphosphonate derivatives 19c/22c containing a long (propylene) spacer. These compounds were weak, nonselective NTPDase inhibitors with Ki values ranging between 161 and 255 μM with only small differences between the uracil (19c) and the adenine derivative (22c). However, the adenine derivative, 22c, was inactive against NTPDase3 at the highest concentration tested (1 mM), whereas the uracil derivative, 19c, was inhibitory. The benzylphosphonates were the most potent NTPDase inhibitors of the present series. This was true for NTPDase1 and 2, while most of the investigated compounds were inactive or showed only low inhibitory activity with NTPDase3 and 8. The inhibitory potency was strongly dependent on the spacer length. The benzylphosphonates of the uracil series showed decreasing inhibitory potency with increasing spacer length, especially with NTPDase2. The most potent compound of the present series was 19a, a benzylphosphonate with a small methylene spacer, showing a Ki value for NTPDase2 of 8.2 μM. The compound is selective for NTPDase2 compared to NTPDase1, NTPDase3, and NTPDase8. The homologues with a longer spacer were 21-fold (ethylene, 19b) and 26-fold (propylene, 19c) less potent than 19a with NTPD-ase2. A comparison of 19a with analogues that contain the same spacer length but different phosphonate groups shows that the benzylphosphonate is superior to methylenephosphonate (20a) or ethylenephosphonate (21a) residues. Both analogues are 9-fold (20a) or 14-fold (21a) less potent at inhibiting NTPDase2. However, when the larger spacer (propylene) was combined with the smaller methylenephosphonate moiety (compound 20c), potency at NTPDase2 was enhanced (Ki = 29.2 μM). These results indicate that the distance between the terminal phosphonate moiety and the nucleoside plays an important role. Selected dose–response curves of potent inhibitors are shown in Figure 3. Compound 19a, the most potent inhibitor of the present series with a Ki value of 8.2 μM at NTPDase2 was further investigated with regard to the mechanism of enzyme inhibition. Since the compounds are substrate analogues, it is likely that they bind competitively to the nucleotide binding site in the NTPDases. Since they do not have phosphate ester bonds, they cannot be cleaved by the enzymes in contrast to the natural substrates. The kinetics of NTPDase2 were determined in the absence and in the prescence of 19a (5 and 10 μM). The Vmax was not affected by different concentrations of inhibitor, while the Km value increased with increasing inhibitor concentration, indicating a competitive mechanism of inhibition. The resulting Lineweaver–Burk plot is shown in Figure 4.
Table 1.
Inhibitory Potency of Synthesized Nucleotide Mimetics at Human NTPDases
| ||||||
|---|---|---|---|---|---|---|
| compd | R | n |
Ki ± SEM [μM] (or inhibition at 1 mM)
|
|||
| NTPDase1 | NTPDase2 | NTPDase3 | NTPDase8 | |||
| 19a PSB-6426 | I | 1 | (50%) | 8.2 ± 2.1 | (0%) | (0%) |
| 19b | I | 2 | 55.2 ± 2.6 | 173 ± 17.3 | (40%) | (0%) |
| 19c | I | 3 | 182 ± 24.3 | 210 ± 25.3 | (48%) | 242 ± 39.3 |
| 20a | II | 1 | 786 ± 33 | 71.7 ± 13.5 | (0%) | (0%) |
| 20b | II | 2 | (0%) | 167 ± 21.3 | (0%) | (0%) |
| 20c | II | 3 | (45%) | 29.2 ± 2.7 | (0%) | (0%) |
| 21a | III | 1 | (0%) | 116 ± 24.3 | (22%) | (0%) |
| 22a | I | 1 | (0%) | (0%) | (0%) | (0%) |
| 22b | I | 2 | 154 ± 37 | 1440 ± 64 | (0%) | (0%) |
| 22c | I | 3 | 161 ± 24 | 213 ± 34 | (0%) | 255 ± 25.3 |
| 23a | II | 1 | (0%) | (15%) | (25%) | (0%) |
| 23b | II | 2 | (0%) | (0%) | (0%) | (0%) |
| 23c | II | 3 | (0%) | (0%) | (0%) | (0%) |
| suramin | 300a | 65.4a | 12.7a | >100b | ||
| RB2 | 20.0a | 24.2a | 1.10a | >100b | ||
| PPADS | 46.0a | 44.2a | 3.0a | ndc | ||
| ARL67156 | 27.0a | (50%)a | 112.1a | >100d | ||
Figure 3.

Concentration–inhibition curves of selected inhibitors at NTPDase2. The enzyme assays were performed in a final volume of 100 μL using a substrate (ATP) concentration of 400 μM (Km = 70 μM). Product formation was determined by capillary electrophoresis as described in the Experimental Section. Data points represent the mean ± SEM from two separate experiments each performed in triplicate. Determined IC50 values were 42 μM (19a), 101 μM (20c), and 374 μM (20a). Calculated Ki values were 8.2 μM (19a), 29.2 μM (20c), and 71.7 μM (20a).
Figure 4.

Lineweaver–Burk plot for NTPDase2 kinetics in the absence and in the presence of two different concentrations of the inhibitor 19a (5 and 10 μM).
Interaction with P2Y Receptors
Most of the investigated nucleotide mimetics did not inhibit or only slightly inhibited the agonist-induced activation of P2Y2, P2Y4, and P2Y6 receptors at the tested concentration of 100 μM (see Supporting Information). Thus, the most potent NTPDase2 inhibitors of the present series, the uracil derivatives 19a and 20c, are selective for the enzyme versus these P2Y receptor subtypes, which are activated by uracil nucleotides: P2Y2 by UTP and ATP, P2Y4 by UTP, and P2Y6 by UDP. Three compounds (20a, 20c, 22a) were evaluated in a broader range of concentrations (0.1, 0.3, 1, 3, 10, 30, 100 μM) at P2Y2 receptors but did not show any concentration-dependent inhibition of UTP-induced calcium mobilization. Antagonists, but also agonists, would inhibit UTP-induced calcium mobilization in this assay, the latter due to desensitization of the receptors during the preincubation period. In addition, we showed that the selected compounds (19a, 19b, 20a, 20b, 22b, and 23a) do not or only weakly activate P2Y2 receptors (for details see Supporting Information). In contrast, known NTPDase2 inhibitors, such as RB2, suramin, and PPADS, not only are nonselective compared to other NTPDases but also are equally or more inhibitory for certain P2 receptor subtypes (see Supporting Information).
Investigation of Chemical and Metabolic Stability
Two selected phosphonates, the selective NTPDase2 inhibitor 19a and the moderately potent, nonselective NTPDase inhibitor 19c, were further investigated for their stability (i) under basic conditions in 0.001 N aqueous sodium hydroxide solution (pH 11),53 (ii) in artificial gastric fluid at 37 °C,54 and (iii) in rat liver microsomes55 in order to investigate potential metabolic degradation by liver enzymes. The experiments were performed essentially as described previously.53 Samples were incubated at 37 °C and subsequently analyzed by high performance liquid chromatography coupled to electrospray ionization mass spectroscopy (LC–MS).
Both uridine-derived benzylphosphonates, 19a and 19c, which contain different spacer lengths between the uridine-derived moiety and the benzylphosphonate partial structure, showed high stability in artificial gastric juice even after extended incubation periods of up to 24 h. No degradation products were detectable by LC–MS analysis. Similarly, both compounds were highly stable in sodium hydroxide solution at pH 11 (for details see Supporting Information), indicating that they may be stable in the alkaline milieu of the intestine.
The next step was to investigate whether the phosphonates were susceptible to liver metabolism. It has been described that short alkyl chain esters of phosphonates are metabolically quite stable.56,57 Incubation with rat liver microsomes showed that both compounds, 19a and 19c, were relatively stable toward liver enzymes. Only a very small percentage (<5%) was metabolized under the applied conditions (see Experimental Section), while >95% of the compounds were recovered unchanged. Both compounds were metabolized to a product with a molecular mass that was 162 u higher than that of the parent drug. An increase in the molecular mass of 162 u is atypical for a phase I reaction, such as oxidation, reduction, or hydrolysis. It points to a phase II reaction, i.e., a coupling reaction with an acid, such as glucuronic acid. Glucuronidation of the unsubstituted ribosyl hydroxyl groups of 19a and 19c is conceivable, but this would increase the molecular mass by 176 u. One possible metabolizing phase II reaction that would increase the molecular weight by 162 u would be coupling with glucose (Scheme 4).58 Several drugs, e.g., phenobarbital and other barbituric acid derivatives, have been reported to be conjugated to glucose in mammals, although this is usually a minor pathway.58
Scheme 4.

Proposed Metabolization of Compounds 19a and 19c by Rat Liver Homogenate
Candidate enzymes catalyzing this reaction could be phenol β-glucosyltransferase (EC 2.4.1.35), arylamine glucosyltransferase (EC 2.4.1.71), or nicotinate glucosyltransferase (EC 2.4.1.196).58
A second metabolite was detected after incubation of 19a with rat liver microsomes at 37 °C with a molecular mass of 243 u. This could be explained by a hydrolysis of the amide bond yielding the aniline derivative 10 (Scheme 5). The same metabolite was also detected after incubation of 19c. The expected metabolite 26 was not detectable, probably because of further degradation.
Scheme 5.

Amide Hydrolysis of Compound 19a
Conclusions
The synthesis of nucleotide mimetics derived from uridine 5′-carboxylic acid attached via amide bonds to a linker of variable length to various phosphonic acid diethyl ester moieties yielded the first potent and selective NTPDase2 inhibitors, uridine derivatives 19a and 20c. In contrast to nucleotides and standard NTPDase inhibitors the new compounds are uncharged at a physiological pH value and may therefore be perorally bioavailable. The compounds did not interact with the uracil nucleotide-activated P2 receptor subtypes P2Y2, P2Y4, or P2Y6. They showed high stability under conditions found in the gastrointestinal system and upon incubation with rat liver microsomes. Only a small fraction of 19a was metabolized, the main pathways presumably being amide hydrolysis and coupling to glucose as analyzed by LC–MS, while the phosphonic acid esters remained intact. Compound 19a may become a useful pharmacological tool for studying NTPDase2 and the potential of NTPDase2 inhibitors as novel drugs.
Experimental Section
Methods
Chemicals and solvents were obtained from various producers (Acros, Aldrich, Fluka, Merck, Sigma) and used without further purification unless otherwise noted. Dichloromethane was distilled prior to use. The reactions were monitored by thin layer chromatography (TLC) using aluminum sheets coated with silica gel 60 F254 (Merck). Column chromatography was carried out on silica gel 0.040–0.063 mm (Fluka) using a flash chromatography system (Büchi). Preparative HPLC was performed on a C18 column (250 mm × 20 mm, particle size 10 μm, Eurospher 100) using a mixture of MeOH and H2O as eluent at a flow rate of 20 mL/min. Mass spectra were recorded on an API 2000 mass spectrometer (electron spray ion source, Applied Biosystems, Darmstadt, Germany) coupled with an HPLC system (Agilent 1100) using a C18 column (particle size 3 μm, Phenomenex Luna). 1H, 31P, and 13C NMR spectra were recorded on a 500 MHz spectrometer (Bruker Avance). CDCl3, DMSO-d6, MeOD-d4, or D2O were used as solvents as indicated below. 31P NMR spectra were recorded using aqueous orthophosphoric acid (85%) as an external standard. Shifts are given in ppm relative to the external standard (31P NMR) or relative to the remaining protons of the deuterated solvents (1H, 13C). Elemental microanalyses were performed on a VarioEL apparatus. Yields and analytical data of all synthesized compounds are given in the Supporting Information.
2′,3′-p-Methoxybenzylideneadenosine (7a) and 2′,3′-p-Meth-oxybenzylideneuridine (7b)
Introduction of the p-methoxybenzylidene protecting group was performed in analogy to a described procedure.38 Adenosine (5.0 g) or uridine (5.0 g) and ZnCl2 (10.0 g) were suspended in a mixture of 10 mL of dry THF and 20 mL of p-methoxybenzaldehyde (anisaldehyde). The turbid mixture was stirred for 2 days, then the product was precipitated by the addition of 100 mL of diethyl ether, filtered off, and washed with water (3 × 50 mL) and diethyl ether (3 × 50 mL).
2′,3′-p-Methoxybenzylideneadenosine-5′-carboxylic Acid (8a) and 2′,3′-p-Methoxybenzylideneuridine-5′-carboxylic Acid (8b)
Oxidation was performed according to a published procedure41 starting from 6.0 mmol of 7a (2.31 g) or 7b (2.17 g).
General Procedures for the Synthesis of Amino-Substituted Phosphonic Acid Ester Derivatives 16a–c, 17a–c, and 18a. Preparation of Amides 13a–c, 14a–c, and 15a
In a dry vessel, N-tert-butyloxycarbonylglycine (10 mmol, 1750 mg), or N-tert-butyloxycarbonyl-β-alanine (10 mmol, 1890 mg), or N-tert-butyloxycarbonyl-γ-aminobutyric acid (10 mmol, 2030 mg) was dissolved in 10 mL of dry THF and cooled to –25 °C. N-Methylmorpholine (10 mmol, 1010 mg) and then isobutyl chloroformate (10 mmol, 1360 mg) were added under vigorous stirring. Immediately after the formation of a white precipitate (N-methyl-morpholine hydrochloride) a solution of p-aminobenzylphosphonic acid diethyl ester (11 mmol, 2673 mg) in dry THF (10 mL) or a solution of aminomethylphosphonic acid diethyl ester oxalate (11 mmol, 2830 mg) or aminoethylphosphonic acid diethyl ester oxalate (11 mmol, 2980 mg) in 11 mL of an aqueous 1 N NaOH solution, precooled on ice, was added. The resulting mixture was allowed to warm to room temperature. After 3 h, the volatiles were removed by rotary evaporation at 40 °C, the residue was dissolved in 10 mL of water, the pH was brought to 1–2 (with 10% aqueous NaHSO4 solution), and the solution was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with a saturated aqueous Na2CO3 solution (3 × 20 mL) and subsequently with water (3 × 20 mL), dried over Na2SO4, and evaporated to dryness. The products were directly used for the next step.
Deprotection: Removal of the tert-Butyloxycarbonyl (Boc) Protective Group
The Boc-protected amides 13a–c, 14a–c, or 15a (3–4 g, the whole amount of the Boc-protected amide except for a small sample for analytical purposes) were dissolved in 8 mL of a dry 4 N HCl solution in dioxane and stirred for 2 h at room temperature. The corresponding products 16a–c, 71a–c, and 18a were precipitated as hydrochlorides by the addition of 50 mL of diethyl ether, filtered off, and thoroughly washed with diethyl ether (3 × 50 mL).
p-(Aminomethylcarboxamido)benzylphosphonic Acid Diethyl Ester Hydrochloride (16a)
Yield over two steps, 3.0 g, 90%; mp 197 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.84 (s, 1H, CONH), 8.35 (br s, 3H, NH3+), 7.56 (d, 2H, 3J = 8.20 Hz, 2 × CHortho, benzylphosphonate), 7.13 (dd, 2H, 3J = 8.55 Hz and 4J = 2.55 Hz, 2 × CHmeta, benzylphosphonate), 4.02–3.94 (2q, 4H, 2 × O–CH2), 3.77 (br s, 2H, N–CH2, methylcarboxamide), 3.15 (d, 2H, 2JH,P = 21.45 Hz, CH2–P, benzylphosphonate), 1.22 (t, 6H, 2 × CH3). 13C NMR (125 MHz, DMSO-d6) δ 164.8 (C═O), 137.0 (Cpara, benzylphosphonate), 130.3 (2 × Cortho, benzylphosphonate), 127.6 (d, 2JC,P = 9.2 Hz, Cipso, benzylphosphonate), 119.2 (2 × Cmeta, benzylphosphonate), 61.5 (2 × OCH2), 41.1 (N–CH2, methylcarboxamide), 31.9 (d, 1JC,P = 137.8 Hz, CH2–P, benzylphosphonate), 16.4 and 16.3 (2 × CH3). 31P NMR (202 MHz, DMSO-d6) δ 26.9.
General Procedure for the Synthesis of Final Products 19a–c, 10a–c, 21a, 22a–c, and 23a–c. Synthesis of 2′,3′-p-Methoxybenzylideneadenosine-5′-carboxylic Acid Amides
Under an argon atmosphere, 2′,3′-p-methoxybenzylideneadenosine-5′-carboxylic acid 8a (1 mmol, 399 mg) was dissolved in a mixture of 2 mL of dry DMF and 0.4 mL of dry diisopropylethylamine at room temperature. The appropriate coupling reagent (see below) (1.1 mmol, HBTU 428 mg, HCTU 455 mg, PyBOP 570 mg) and 1-hydroxybenzotriazole (1.1 mmol, 149 mg) was dissolved in 2 mL of dry DMF, and after 1 min compound 16a, 16b, 16c, 17a, 17b, or 17c (2 mmol), dissolved in a mixture of dry DMF (2 mL) and diisopropylethylamine (0.5 mL), was added via a syringe to the solution. Vigorous stirring was continued for 24 h at room temperature. The volatiles were removed in vacuo at 40 °C, and the residue was purified by silica gel column chromatography using dichloromethane/methanol (40:1) as eluent. The solvent was removed by rotary evaporation at 40 °C, and the product was recrystallized from diethyl ether (20 mL).
Synthesis of 2′,3′-p-Methoxybenzylideneuridine-5′-carboxylic Acid Amides
Under an argon atmosphere 2′,3′-p-methoxybenzylideneuridine-5′-carboxylic acid 8b (1 mmol, 376 mg), the proper coupling reagent (see experimental part) (1.1 mmol, HBTU 428 mg, HCTU 455 mg, PyBOP 570 mg) and 1-hydroxybenzotriazole (1.1 mmol, 149 mg) were dissolved in 2 mL of dry DMF at room temperature. Then diisopropylethylamine (1.1 mmol, 143 mg) and, after 1 min, compound 16a, 16b, 16c, 17a, 17b, 17c, or 18a (2 mmol), dissolved in a mixture of dry DMF (2 mL) and diisopro-pylethylamine (0.5 mL) was sequentially added to the solution via a syringe. Reaction times and workup procedure were the same as for 2′,3′-p-methoxybenzylideneadenosine-5′-carboxylic acid amides.
The compounds were directly used in the following step.
Deprotection: Removal of the p-Methoxybenzylidene Protecting Group
Deprotection of the ribose moiety was performed by stirring 2′,3′-p-methoxybenzylidenenucleoside-5′-carboxylic acid amides (100 mg) in a mixture of dichloromethane (3 mL), trifluoroacetic acid (0.15 mL), and water (0.1 mL) at room temperature. After 2 h, the crude product was precipitated by the addition of diethyl ether (50 mL), and the suspension was stirred for 1 h. The product was collected by filtration, dissolved in 7 mL of a mixture of water/methanol (75:25), and purified on a C-18 column by RP-HPLC using a gradient of water/methanol from 75: 25 to 0:100. The final products were isolated by lyophilization. Yields were determined over two steps, after amide coupling and removal of the p-methoxybenzylidene group.
4-[2-((2S,3R,4S,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofurane-2-carboxamido)ethylamido-]benzylphosphonic Acid Diethyl Ester (19a)
Coupling reagent used in the synthesis was HCTU. Yield over two steps, 310 mg, 57%; mp 159 °C. 1H NMR (500 MHz, MeOD) δ 8.18 (d, 1H, 3J = 7.90 Hz, H-6), 7.58 (d, 2H, 3J = 8.85 Hz, 2 × CHortho, benzylphosphonate), 7.31 (dd, 2H, 3J = 8.80 Hz and 4J = 2.80 Hz, 2 × CHmeta, benzylphosphonate), 6.02 (d, 1H, 3J = 6.30 Hz, H-1′), 5.78 (d, 1H, 3J = 8.20 Hz, H-5), 4.51 (d, 1H, 3J = 3.20 Hz, H-4′), 4.47 (dd, 1H, 3J = 5.05 Hz and 3J = 5.95 Hz, H-2′), 4.41 (dd, 1H, 3J = 3.20 Hz and 3J = 5.05 Hz, H-3′), 4.19–4.01 (AB-system with A d and B d, partially overlapping with 2 × O-CH2, 2H, 2J = 16.35 Hz, N–CH2, ethylamide), 4.09–4.01 (2q, 4H, 2 × O–CH2), 3.25 (d, 2H, 2JH,P = 21.45 Hz, CH2–P, benzylphosphonate), 1.29 (t, 6H, 2 × CH3). 13C NMR (125 MHz, MeOD) δ 173.2 (C═O), 169.5 (C═O), 166.4 (C-4), 153.1 (C-2), 144.3 (C-6), 138.9 (Cpara, benzylphosphonate), 131.7 (2 × CHortho, benzylphosphonate), 128.7 (d, 2JC,P = 9.4 Hz, Cipso, benzylphosphonate), 121.5 (2 × CHmeta, benzylphosphonate), 103.5 (C-5), 92.2 (C-1′), 85.3 (C-4′), 74.9 (C-2′), 74.1 (C-3′), 64.1 and 64.0 (2 × O-CH2), 43.9 (N–CH2, ethylamide), 33.4 (d, 1JC,P = 137.6 Hz, CH2–P, benzylphosphonate), 17.0 and 16.9 (2 × CH3). 31P NMR (202 MHz, MeOD) δ 26.7. LC/ESI-MS: negative mode 539.3 ([M – H]−), positive mode 541.0 ([M + H]+). Anal. (C22H29N4O10P · 4.25H2O) C, H, N.
Biochemical Assays. Membrane Preparation Containing Expressed Human NTPDase2
The NTPDase2 cDNA cloned from human small cell lung carcinoma and inserted in a pcDNA3 vector was used to transfect human embryonic kidney (HEK293) cells. Stably transfected cells were obtained by geneticin selection as described.22 Membranes were prepared from stably transfected cells harvested from 10–15 10-cm plates by differential and sucrose gradient centrifugation as described.22
Cell Transfection with Human NTPDases 1, 3, 8 and Membrane Preparation
COS-7 cells were transfected in 10 cm plates using Lipofectamine (Invitrogen), as previously described.59 Briefly, 80–90% confluent cells were incubated for 5 h at 37 °C in Dulbecco’s modified Eagle medium (DMEM) in the absence of fetal bovine serum (FBS) with 6 μg of the appropriate plasmid DNA and 24 μL of Lipofectamine reagent. The reaction was stopped by the addition of an equal volume of DMEM containing 20% FBS, and the cells were harvested 40–72 h later. For the preparation of protein extracts, transfected cells were washed three times with Tris-saline buffer at 4 °C, collected by scraping in the harvesting buffer (95 mM NaCl, 0.1 mM phenylmethanesulfonyl fluoride (PMSF) and 45 mM Tris at pH 7.5), and washed twice by 300g centrifugation for 10 min at 4 °C. Cells were resuspended in the harvesting buffer containing 10 mg/mL aprotinin and sonicated. Nucleus and cellular debris were discarded by centrifugation at 300g for 10 min at 4 °C, and the supernatant (crude protein extract) was aliquoted and stored at –80 °C until used for activity assays. The protein concentration was estimated by the Bradford microplate assay using bovine serum albumin as a standard.60
Capillary Electrophoresis (CE) Instrumentation
All experiments were carried out using a P/ACE MDQ capillary electrophoresis system (Beckman Instruments, Fullerton, CA) equipped with a UV detection system coupled with a diode array detector (DAD). Data collection and peak area analysis were performed by the P/ACE MDQ software 32 KARAT obtained from Beckman Coulter. The capillary and sample storing unit temperature was kept constant at 25 °C. The electrophoretic separations were carried out using an eCAP polyacrylamide-coated fused-silica capillary [(30 cm (20 cm effective length) × 50 μm internal diameter (i.d.) × 360 μm outside diameter (o.d.), obtained from CS-Chromatographie (Langerwehe, Germany)]. The separation was performed using an applied current of –60 μA and a data acquisition rate of 8 Hz. Analytes were detected using direct UV absorbance at 210 nm. The capillary was conditioned by rinsing with water for 2 min and subsequently with buffer (phosphate 50 mM, pH 6.5) for 1 min. Sample injections were made at the cathodic side of the capillary.
Determination of NTPDase Inhibition
Enzyme inhibition assays were carried out at 37 °C in a final volume of 100 μL. The reaction mixture contained 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 10 mM Hepes, pH 7.4, and 400 μM ATP. Different concentrations of inhibitors dissolved in water or containing 1% DMSO in cases of low water solubility when a stock solution could not be prepared in water (10 μL) were added, and the reaction was initiated by the addition of 10 μL of the appropriately diluted membrane preparations containing human NTPDase1, NTPDase2, NTPDase3, or NTPDase8. The mixture was incubated for 10 min, and the reaction was terminated by heating at 99 °C for 5 min. Aliquots of the reaction mixture (50 μL) were then diluted 10-fold with water containing UMP (final concentration 20 μM) as an internal standard, transferred to mini-CE vials, and injected into the CE instrument under the conditions described above. Each analysis was repeated three times (triplicates) in two separate experiments. Control experiments were performed using membrane preparations of cells transfected with the empty plasmid (pcDNA3). The Cheng–Prusoff equation was used to calculate Ki values from the IC50 values, determined by the nonlinear curve-fitting program PRISM 3.0 (GraphPad, San Diego, CA), assuming a competitive inhibition mechanism. Km values were as follows: 17 μM (NTPDase1), 70 μM (NTPDase2), 75 μM (NTPDase3), and 81 μM (NTPDase8).8,47
For the determination of the mechanism of inhibition, the enzymatic reaction of NTPDase2 was monitored as described above using different concentrations of ATP as a substrate (12.5, 25, 50, 125, 250, and 1000 μM) in the absence and presence of the inhibitor 19a (0, 5, and 10 μM). The curves were analyzed by PRISM 3.0 and transformed to obtain a Lineweaver–Burk plot.
Stability and Metabolism Studies. Preparation of Solutions and Rat Liver Microsomes
The intestinal environment was simulated with 0.001 N aqueous sodium hydroxide solution (pH 11). Simulated gastric acid was prepared as previously described:53,54 pepsin (3.20 g), NaCl (2.0 g), and HCl (1 M, 80 mL) were mixed with distilled water to obtain 1000 mL, and the resulting solution was stored at 4 °C. Rat liver microsomes were prepared as described with slight modifications:53,55,61 fresh rat liver (6.5 g) was homogenized in 30 mL of freshly prepared Dulbecco’s phosphate buffered saline (DPBS) consisting of 132.5 mg of CaCl2 · 2H2O, 100 mg of MgCl2 · 6H2O, 200 mg of KCl, 200 mg of KH2PO4, 8000 mg of NaCl, and 1500 mg of Na2HPO4 in a total volume of 1000 mL, pH 7.2, and centrifuged at 9000g for 30 min at 4 °C. The supernatant, which contained the soluble microsomes, was carefully decanted and stored at –80 °C until used. The protein concentration was 18 mg/mL as determined by the method of Bradford.60
LC–MS Analyses
HPLC was performed on a C18 column (50 mm × 2 mm, particle size 3 μm, Phenomenex Luna) using a mixture of H2O (solvent A) and MeOH (solvent B) containing 20 mM of NH4OAc as eluent at a flow rate of 250 μL/min. Mass spectra were recorded on an API 2000 mass spectrometer (electron spray ion source, Applied Biosystems, Darmstadt, Germany) coupled with an HPLC system (Agilent 1100, Böblingen, Germany). Data were collected and analyzed by Analyst Software, version 1.3.1. The separation was carried out at room temperature by gradient elution. The elution was started with a mixture of solvent A and solvent B (90:10, v/v) up to 15 min. Subsequently only solvent B was used up to 30 min. The limit of detection (LOD), defined as the lowest analyte concentration with a signal-to-noise (S/N) ratio of 3, was determined for compounds 19a and 19c to be approximately 0.1 μg/mL.
Stability in Basic Solution and in Artificial Gastric Acid
Stock solutions (2 mM) of 19a and 19c in H2O containing 20 mM NH4OAc were prepared, and 10 μL aliquots were incubated for 2 or 24 h with 990 μL of either 0.001 N aqueous NaOH solution or simulated gastric acid at 37 °C and subsequently analyzed by LC–MS. No degradation products were detectable by sensitive LC–MS analysis.
Metabolism by Rat Liver Microsomes
Compounds 19a and 19c were incubated with rat liver microsomes (6 mg of protein per vial) at a concentration of 100 μM in a final volume of 1 mL. The solution contained an NADPH regenerative system consisting of NADP (0.57 mM), NADH (0.57 mM), isocitrate (6.4 mM), isocitrate dehydrogenase (0.57 mM), and MgCl2 (23.4 mM) (pH 7.2).62 The samples were incubated for 2 h at 37 °C in a water bath and subsequently heated at 99 °C for 3 min to stop the enzymatic reaction. After centrifugation at 14000g, the supernatant was analyzed by LC–MS.
Supplementary Material
Acknowledgments
Karen Schmeling and Sonja Sternberg are acknowledged for skillful technical assistance. We are grateful to Dr. Gary Weisman, University of Missouri-Columbia, and Dr. Petra Hillmann, University of Bonn, for providing recombinant astrocytoma cell lines expressing P2Y receptor subtypes. J.I. is grateful for a STIBET scholarship by the Deutscher Akademischer Austauschdienst (DAAD). A.F.K. was supported by the California Metabolic Research Foundation. J.S. was supported by grants from the Canadian Institutes of Health Research (CIHR) and was the recipient of a New Investigator award from the CIHR.
Footnotes
Preliminary results were presented at the 8th International Symposium on Adenosine and Adenine Nucleotides in Ferrara, 2006 (Abstract published in Purinergic Signalling 2006, 2, 170).
Abbreviations: Boc, tert-butyloxycarbonyl; 8-Bu-S-ATP, 8-thiobutyl-adenosine 5′-triphosphate; CE, capillary electrophoresis; CDCl3, deuterated chloroform; CNS, central nervous system; DIPEA, diisopropylethylamine; DMEM, Dulbecco’s modified Eagle medium; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; DMSO-d6, deuterated dimethyl sulfoxide; D2O, deuterated water; DPBS, Dulbecco’s phosphate-buffered saline; FBS, fetal bovine serum; EI, electron impact; ESI, electrospray ionization; HBTU, 2-(1H-benzotriazolyl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HCTU, 2-(6-chloro-1H-benzotriazolyl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HEPES, N-[2-hydroxyethyl]piperazine-N′-2-ethansulfonic acid; HPLC, high performance liquid chromatography; HRMS, high resolution mass spectrometry; LC–MS, liquid chromatography–mass spectrometry; LOD, limit of detection; MeOD-d4, deuterated methanol; MeOH, methanol; NMR, nuclear magnetic resonance; NPP, nucleoside pyrophosphatase; (E)-NTPDase, (ecto)-nucleoside 5′-triphosphate diphosphohydrolase; PMSF, phenylmethanesulfonyl fluoride; POM, polyoxometalate; PPADS, pyridox-alphosphate-6-azophenyl-2′,4′-disulfonic acid; PyBOP, benzotriazol-1-yl-oxytrispyrrolidinophosphonium hexafluorophosphate; RB-2, reactive blue 2; SAR(s), structure–activity relationship(s); S/N, signal-to-noise; TEMPO, 2,2,6,6-tetramethyl-1-piperidinyl oxide; THF, tetrahydrofurane; TLC, thin layer chromatography.
Supporting Information Available: Elemental analyses, yields, and analytical data for final products; yields and analytical data for 7a and 7b; procedures and test results at P2Y2, P2Y4, and P2Y6 receptors, and HPLC chromatograms and MS spectra of stability and metabolism studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]; (b) Burnstock G. Purine and pyrimidine receptors. Cell Mol Life Sci. 2007;64:1471–1483. doi: 10.1007/s00018-007-6497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbracchio MP, Burnstock G, Boeynaems J, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zimmermann H. Ectonucleotidases: some recent developments and a note on nomenclature. Drug Dev Res. 2001;52:44–56. [Google Scholar]
- 4.Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: Structure–function relationships and pathophysiological significance. Purinergic Signalling. 2006;2:409–430. doi: 10.1007/s11302-006-9003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73) Purinergic Signalling. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmermann H. Two novel families of ectonucleotidases: molecular structures, catalytic properties and a search for function. Trends Pharmacol Sci. 1999;20:231–236. doi: 10.1016/s0165-6147(99)01293-6. [DOI] [PubMed] [Google Scholar]
- 8.Kukulski F, Lévesque SA, Lavoie EG, Lecka J, Bigonnesse F, Knowles AF, Robson SC, Kirley TL, Sévigny J. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3, and 8. Purinergic Signalling. 2005;1:193–204. doi: 10.1007/s11302-005-6217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarado-Castillo C, Harden TK, Boyer JL. Regulation of P2Y1 receptor-mediated signaling by the ectonucleoside triphosphate diphosphohydrolase isozymes NTPDase1 and NTPDase2. Mol Pharmacol. 2005;67:114–122. doi: 10.1124/mol.104.006908. [DOI] [PubMed] [Google Scholar]
- 10.Sévigny J, Sundberg C, Braun N, Guckelberger O, Csizmadia E, Qawi I, Imai M, Zimmermann H, Robson SC. Differential catalytic properties and vascular topography of murine nucleoside triphosphate diphosphohydrolase 1 (NTPDase1) and NTPDase2 have implications for thromboregulation. Blood. 2002;99:2801–2809. doi: 10.1182/blood.v99.8.2801. [DOI] [PubMed] [Google Scholar]
- 11.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 12.Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285(Part 2):345–365. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal J, Jirovsky D, Lee SY, Zimmermann H, Müller CE. Capillary electrophoresis-based nanoscale assays for monitoring ecto-5′-nucleotidase activity and inhibition in preparations of recombinant enzyme and melanoma cell membranes. Anal Biochem. 2008;373:129–140. doi: 10.1016/j.ab.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 14.Sträter N. Ecto-5′-nucleotidase: structure function relationships. Purinergic Signalling. 2006;2:343–350. doi: 10.1007/s11302-006-9000-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shryock JC, Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am J Cardiol. 1997;79:2–10. doi: 10.1016/s0002-9149(97)00256-7. [DOI] [PubMed] [Google Scholar]
- 16.Koszalka P, Ozüyaman B, Huo Y, Zernecke A, Flögel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sévigny J, Gear A, Weber AA, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H, Gödecke A, Schrader J. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- 17.Kawashima Y, Nagasawa T, Ninomiya H. Contribution of ecto-5′-nucleotidase to the inhibition of platelet aggregation by human endothelial cells. Blood. 2000;96:2157–2162. [PubMed] [Google Scholar]
- 18.El Omar MM, Islam N, Broekman MJ, Drosopoulos JH, Roa DC, Lorin JD, Sedlis SP, Olson KE, Pulte ED, Marcus AJ. The ratio of ADP- to ATP-ectonucleotidase activity is reduced in patients with coronary artery disease. Thromb Res. 2005;116:199–206. doi: 10.1016/j.thromres.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 19.Wink MR, Braganhol E, Tamajusuku ASK, Lenz G, Zerbini LF, Libermann TA, Sévigny J, Battastini AMO, Robson SC. Nucleoside triphosphate diphosphohydrolase-2 (NTPDase2/ CD39L1) is the dominant ectonucleotidase expressed by rat astrocytes. Neuroscience. 2006;138:421–432. doi: 10.1016/j.neuroscience.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 20.Chorna NE, Santiago-Perez LI, Erb L, Seye CI, Neary JT, Sun GY, Weisman GA, Gonzalez FA. P2Y2 receptors activate neuroprotective mechanisms in astrocytic cells. J Neurochem. 2004;91:119–132. doi: 10.1111/j.1471-4159.2004.02699.x. [DOI] [PubMed] [Google Scholar]
- 21.Langer D, Ikehara Y, Takebayashi H, Hawkes R, Zimmermann H. The ectonucleotidases alkaline phosphatase and nucleoside triphosphate diphosphohydrolase 2 are associated with subsets of progenitor cell populations in the mouse embryonic, postnatal and adult neurogenic zones. Neuroscience. 2007;150:863–879. doi: 10.1016/j.neuroscience.2007.07.064. [DOI] [PubMed] [Google Scholar]
- 22.Knowles AF, Chiang W-C. Enzymatic and transcriptional regulation of human ecto-ATPase/E-NTPDase 2. Arch Biochem Biophys. 2003;418:217–227. doi: 10.1016/j.abb.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Buffon A, Ribeiro VB, Wink MR, Casali EA, Sarkis JJ. Nucleotide metabolizing ecto-enzymes in Walker 256 tumor cells: molecular identification, kinetic characterization and biochemical properties. Life Sci. 2007;80:950–958. doi: 10.1016/j.lfs.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson KA, Jarvis MF, Williams M. Purine and pyrimidine (P2) receptors as drug targets. J Med Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gendron FP, Halbfinger E, Fischer B, Beaudoin AR. Inhibitors of NTPDase: key players in the metabolism of extracellular purines. Adv Exp Med Biol. 2000;486:119–123. doi: 10.1007/0-306-46843-3_24. [DOI] [PubMed] [Google Scholar]
- 26.Gendron FP, Halbfinger E, Fischer B, Duval M, D’Orleans-Juste P, Beaudoin AR. Novel inhibitors of nucleoside triphosphate diphosphohydrolases: chemical synthesis and biochemical and pharmacological characterizations. J Med Chem. 2000;43:2239–2247. doi: 10.1021/jm000020b. [DOI] [PubMed] [Google Scholar]
- 27.Gendron FP, Benrezzak O, Krugh BW, Kong Q, Weisman GA, Beaudoin AR. Purine signaling and potential new therapeutic approach: possible outcomes of NTPDase inhibition. Curr Drug Targets. 2002;3:229–245. doi: 10.2174/1389450023347713. [DOI] [PubMed] [Google Scholar]
- 28.Cechin SR, Schetinger MR, Zanatta N, Madruga CC, Pacholski IL, Flores DC, Bonacorso HG, Martins MAP, Morsch VM. Inhibitory effect of novel pyrimidines on ATP and ADP hydrolysis in synaptosomes from rat cerebral cortex. Chem Res Toxicol. 2003;16:1433–1439. doi: 10.1021/tx034102p. [DOI] [PubMed] [Google Scholar]
- 29.Joseph SM, Pifer MA, Przybylski RJ, Dubyak GR. Methylene ATP analogs as modulators of extracellular ATP metabolism and accumulation. Br J Pharmacol. 2004;142:1002–1014. doi: 10.1038/sj.bjp.0705865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iqbal J, Vollmayer P, Braun N, Zimmermann H, Müller CE. A capillary electrophoresis method for the characterization of ecto-nucleoside triphosphate diphosphohydrolases (NTPDases) and the analysis of inhibitors by in-capillary enzymatic microreaction. Purinergic Signalling. 2005;1:349–358. doi: 10.1007/s11302-005-8076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller CE, Iqbal J, Baqi Y, Zimmermann H, Rollich A, Stephan H. Polyoxometalates—a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorg Med Chem Lett. 2006;16:5943–5947. doi: 10.1016/j.bmcl.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Munkonda MN, Kauffenstein G, Kukulski F, Lévesque SA, Legendre C, Pelletier J, Lavoie EG, Lecka J, Sévigny J. Inhibition of human and mouse plasma membrane bound NTPDases by P2 receptor antagonists. Biochem Pharmacol. 2007;74:1524–1534. doi: 10.1016/j.bcp.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Tuluc F, Bültmann R, Glänzel M, Frahm AW, Starke K. P2-receptor antagonists: IV. Blockade of P2-receptor subtypes and ecto-nucleotidases by compounds related to reactive blue 2. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:111–120. doi: 10.1007/pl00005144. [DOI] [PubMed] [Google Scholar]
- 34.Bültmann R, Wittenburg H, Pause B, Kurz G, Nickel P. Blockade of P2-purinoceptor subtypes and ecto-nucleotidases by compounds related to suramin. Naunyn Schmiedeberg’s Arch Pharmacol. 1996;354:498–504. doi: 10.1007/BF00168442. [DOI] [PubMed] [Google Scholar]
- 35.Chen BC, Lee CM, Lin WW. Inhibition of ecto-ATPase by PPADS, suramin and reactive blue in endothelial cells, C6 glioma cells and RAW 264.7 macrophages. Br J Pharmacol. 1996;119:1628–1634. doi: 10.1111/j.1476-5381.1996.tb16082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crack BE, Pollard CE, Beukers MW, Roberts SM, Hunt SF, Ingall AH, McKechnie KC, Jzerman IAP, Leff P. Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br J Pharmacol. 1995;114:475–481. doi: 10.1111/j.1476-5381.1995.tb13251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lévesque SA, Lavoie EG, Lecka J, Bigonnesse F, Sévigny J. Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol. 2007;152:141–150. doi: 10.1038/sj.bjp.0707361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith M, Rammler DH, Goldberg IH, Khorana HG. PolynucleotidesXIV Specific synthesis of the C3′–C5′ internucleotide linkage. Synthesis of uridylyl-(3′ → 5′)-uridine and uridylyl-(3′ → 5′)-adenosine. J Am Chem Soc. 1962;84:430–440. [Google Scholar]
- 39.Watkins BE, Kiely JS, Rapoport H. Synthesis of oligodeoxyribonucleotides using N-(benzyloxycarbonyl)-blocked nucleosides. J Am Chem Soc. 1982;104:5702–5708. [Google Scholar]
- 40.Johnson DC, Widlanski TS. Facile deprotection of O-Cbz-protected nucleosides by hydrogenolysis: an alternative to O-benzyl ether-protected nucleosides. Org Lett. 2004;6:4643–4646. doi: 10.1021/ol048426w. [DOI] [PubMed] [Google Scholar]
- 41.Epp JB, Widlanski TS. Facile preparation of nucleoside-5′-carboxylic acids. J Org Chem. 1999;64:293–295. doi: 10.1021/jo981316g. [DOI] [PubMed] [Google Scholar]
- 42.Wieland T. Use of anhydrides of N-acylated amino acids and derivatives of inorganic acids. Liebigs Ann Chem. 1951;572:190–194. [Google Scholar]
- 43.Montgomery JA, Thomas HJ, Brockman RW, Wheeler GP. Potential inhibitors of nucleotide biosynthesis. 1. Nitrosoureidonucleosides. J Med Chem. 1981;24:184–189. doi: 10.1021/jm00134a011. [DOI] [PubMed] [Google Scholar]
- 44.Sabatino G, Mulinacci B, Alcaro MC, Chelli M, Rovero P, Papini AM. Assessment of new 6-Cl-HOBt based coupling reagents for peptide synthesis. Part 1: Coupling efficiency study. Lett Pept Sci. 2003;9:119–123. [Google Scholar]
- 45.Uri A, Järlebark L, von Kügelgen I, Schönberg T, Unden A, Heilbronn E. A new class of compounds, peptide derivatives of adenosine 5′-carboxylic acid, includes inhibitors of ATP receptor-mediated responses. Bioorg Med Chem. 1994;2:1099–1105. doi: 10.1016/s0968-0896(00)82060-x. [DOI] [PubMed] [Google Scholar]
- 46.Sak K, Uri A, Enkvist E, Raidaru G, Subbi J, Kelve M, Jarv J. Adenosine-derived non-phosphate antagonists for P2Y1 purinoceptors. Biochem Biophys Res Commun. 2000;272:327–331. doi: 10.1006/bbrc.2000.2775. [DOI] [PubMed] [Google Scholar]
- 47.Fausther M, Lecka J, Kukulski F, Lévesque SA, Pelletier J, Zimmermann H, Dranoff JA, Sévigny J. Cloning, purification, and identification of the liver canalicular ecto-ATPase as NTPDase8. Am J Physiol. 2007;292:G785–G795. doi: 10.1152/ajpgi.00293.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Tayeb A, Qi A, Müller CE. Synthesis and structure–activity relationships of uracil nucleotide derivatives and analogues as agonists at human P2Y2, P2Y4, and P2Y6 receptors. J Med Chem. 2006;49:7076–7087. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 49.Kassack MU, Höfgen B, Lehmann J, Eckstein N, Quillan JM, Sadee W. Functional screening of G protein-coupled receptors by measuring intracellular calcium with a fluorescence microplate reader. J Biomol Screening. 2002;7:233–246. doi: 10.1177/108705710200700307. [DOI] [PubMed] [Google Scholar]
- 50.Kaulich M, Streicher F, Mayer R, Müller I, Müller CE. Flavonoids—novel lead compounds for the development of P2Y2 receptor antagonists. Drug Dev Res. 2003;59:72–81. [Google Scholar]
- 51.Weyler S, Baqi Y, Hillmann P, Kaulich M, Hunder AM, Müller IA, Müller CE. Combinatorial synthesis of anilinoanthraquinone derivatives and evaluation as non-nucleotide-derived P2Y2 receptor antagonists. Bioorg Med Chem Lett. 2008;18:223–227. doi: 10.1016/j.bmcl.2007.10.082. [DOI] [PubMed] [Google Scholar]
- 52.Brunschweiger A, Müller CE. P2 receptors activated by uracil nucleotides—an update. Curr Med Chem. 2006;13:289–312. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 53.Yan L, Müller CE. Preparation, properties, reactions, and adenosine receptor affinities of sulfophenylxanthine nitrophenyl esters: toward the development of sulfonic acid prodrugs with peroral bioavailability. J Med Chem. 2004;47:1031–1043. doi: 10.1021/jm0310030. [DOI] [PubMed] [Google Scholar]
- 54.Guven KC, Ozaydin F. Stability of sodium cyclamate in simulated gastric and intestinal media. Pharmazie. 1981;36:297. [PubMed] [Google Scholar]
- 55.Oliyai R, Shaw JP, Sueoka-Lennen CM, Cundy KC, Arimilli MN, Jones RJ, Lee WA. Aryl ester prodrugs of cyclic HPMPC I: Physicochemical characterization and in vitro biological stability. Pharm Res. 1999;16:1687–1693. doi: 10.1023/a:1018945713623. [DOI] [PubMed] [Google Scholar]
- 56.Krise JP, Stella VJ. Prodrugs of phosphates, phosphonates, and phosphinates. Adv Drug Delivery Rev. 1996;19:287–310. [Google Scholar]
- 57.Serafinowska HT, Ashton RJ, Bailey S, Harnden MR, Jackson SM, Sutton D. Synthesis and in vivo evaluation of prodrugs of 9-[2-(phosphonomethoxy)ethoxy]adenine. J Med Chem. 1995;38:1372–1379. doi: 10.1021/jm00008a015. [DOI] [PubMed] [Google Scholar]
- 58.Tang BK. Drug glucosidation. Pharmacol Ther. 1990;46:53–56. doi: 10.1016/0163-7258(90)90034-y. [DOI] [PubMed] [Google Scholar]
- 59.Kaczmarek E, Koziak K, Sévigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- 60.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 61.Fletcher SR, Burkamp F, Blurton P, Cheng SK, Clarkson R, O’Connor D, Spinks D, Tudge M, van Niel MB, Patel S, Chapman K, Marwood R, Shepheard S, Bentley G, Cook GP, Bristow LJ, Castro JL, Hutson PH, MacLeod AM. 4-(Phenylsulfonyl)piperidines: novel, selective, and bioavailable 5-HT(2A) receptor antagonists. J Med Chem. 2002;45:492–503. doi: 10.1021/jm011030v. [DOI] [PubMed] [Google Scholar]
- 62.De Graaf IA, Van Meijeren CE, Pektas F, Koster HJ. Comparison of in vitro preparations for semi-quantitative prediction of in vivo drug metabolism. Drug Metab Dispos. 2002;30:1129–1136. doi: 10.1124/dmd.30.10.1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.