

Abstract

Human aldo-keto reductases (AKRs) are NAD(P)H-dependent oxidoreductases that convert aldehydes and ketones to primary and secondary alcohols for subsequent conjugation reactions and can be referred to as “phase 1” enzymes. Among all the human genes regulated by the Keap1/Nrf2 pathway, they are consistently the most overexpressed in response to Nrf2 activators. Although these enzymes play clear cytoprotective roles and deal effectively with carbonyl stress, their upregulation by the Keap1/Nrf2 pathway also has a potential dark-side, which can lead to chemotherapeutic drug resistance and the metabolic activation of lung carcinogens (e.g., polycyclic aromatic hydrocarbons). They also play determinant roles in 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone metabolism to R- and S-4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanol. The overexpression of AKR genes as components of the “smoking gene” battery raises the issue as to whether this is part of a smoking stress response or acquired susceptibility to lung cancer. Human AKR genes also regulate retinoid, prostaglandin, and steroid hormone metabolism and can regulate the local concentrations of ligands available for nuclear receptors (NRs). The prospect exists that signaling through the Keap1/Nrf2 system can also effect NR signaling, but this has remained largely unexplored. We present the case that chemoprevention through the Keap1/Nrf2 system may be context dependent and that the Nrf2 “dose-response curve” for electrophilic and redox balance may not be monotonic.
1. INTRODUCTION
Aldo-keto reductases (AKRs) belong to a protein superfamily that mainly contains monomeric cytosolic oxidoreductases that catalyze the NAD(P)H-dependent reduction of a large number of endogenous and exogenous carbonyl substrates, e.g., aldehydes and ketones to produce primary and secondary alcohols, respectively, Figure 1.1–4 In addition, these enzymes can also perform double-bond reduction, e.g., AKR1D1 (steroid 5β-reductase), or nitroreduction, e.g., AKR1C3.5,6 The alcohol products are often conjugated by sulfonation catalyzed by sulfotransferases (SULTs) or by glucuronidation catalyzed by uridine diphosphate glucuronsyl transferases (UGTs). As such, AKRs are phase 1 enzymes. Because of their ability to detoxify reactive aldehydes and ketones, it is not surprising to find that these enzymes can be cytoprotective, are evolutionary conserved, and are found in archaebacteria, prokaryotes, and eukaroytes. The AKR genes encoding these enzymes can be considered stress response genes because they are regulated by oxidative, electrophilic,7–10 and osmotic stress11 and heat shock.12 Thus, these primordial genes are regulated by primordial signals.13
Figure 1.
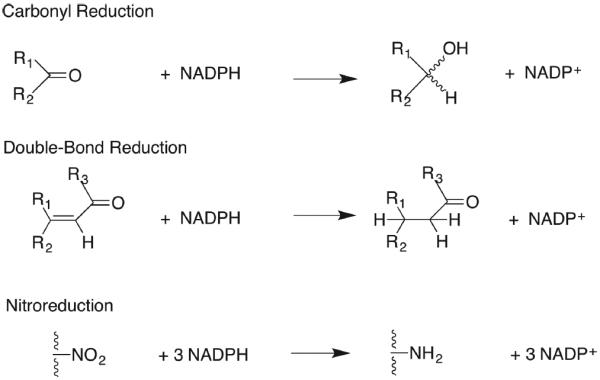
Reduction reactions catalyzed by human AKRs; steroid double-bond reduction is catalyzed by AKR1D1, and nitroredution is catalyzed by AKR1C3.
In humans, there are 15 different AKRs that play key roles in the metabolism of sugar and lipid aldehydes, e.g., advanced glycation end products14,15 and 4-hydroxy-2-nonenal (4-HNE), respectively;8,16 chemical carcinogens, e.g., aflatoxin dialdehyde;17 polycyclic aromatic trans-dihydrodiols;18,19 nicotine-derived nitrosaminoketones (NNK);20,21 as well as cancer chemotherapeutic agents.22–25 The human AKR genes are involved in the metabolism of prostaglandins26–29 and natural and synthetic steroid hormones,30–34 all of which contain carbonyl functionalities (Table 1).
Table 1.
Human Aldo-Keto Reductases
| gene | chromosomal localization | nonsystematic name | physiologic reaction(s) | associated disease | specific inhibitors |
|---|---|---|---|---|---|
| AKR1A1 | 1p33-p32 | aldehyde reductase; dihydrodiol dehydrogenase (DD) 3 | glyceraldehyde ➔ glycerol; mevaldic acid➔mevalonic acid | ||
| AKR1B1 | 7q35 | aldose reductase | glucose ➔ sorbitol | diabetic complications (caratogenesis; retinopathy; nephropathy) | sorbinil, tolrestat, epalrestat, ranirestsat fidarestat |
| AKR1B10 | 7q33 | aldose reductase “small intestine like aldose reductase”'; retinaldehyde reductase | retinal ➔ retinol | NSCLC | oleanolic acid |
| hepatocarcinogenesis | |||||
| AKR1B15 | 7q33 | aldose reductase | 3-keto-acyl CoA reduction | ||
| AKR1C1 | 10p15-10p14 | 3(20α)-hydroxysteroid dehydrogenase; DD1 | progesterone ➔20α-hydroxprogesterone | preterm birth; endometriosis | salicylate 3-bromophenyl salicylate |
| AKR1C2 | 10p15-10p14 | type 3 3α-hydroxysteroid dehydrogenase; DD2 | DHT➔ 3α-androstanediol; | androgen insufficiency | ursodeoxycholate |
| DHP➔allopregnanolone | prementsural syndrome | ||||
| AKR1C3 | 10p15-10p14 | type 5 17β-hydroxysteroid dehydrogenase; prostaglandin F synthase; DDx | Δ4-AD➔ Testosterone | prostate cancer; breast cancer | indomethacin 6-medroxyprogesterone acetate |
| 5α-androstane-3,17-dione➔ DHT | prostate cancer | ||||
| estrone➔17β-estradiol | breast cancer | ||||
| PGH2➔ PGF2α; PGD2➔11β-PGF2α | acute myeloid leukemia | ||||
| AKR1C4 | 10p15-10p14 | Type 1 3α-hydroxysteroid dehydrogenase; chlordecone reductase; DD4 | 5β-cholestan-7α-ol-3-one ➔ 5β-cholestane-3α,7α-diol; 5β-cholestane-7α, 12α-diol-3-one ➔ 5β-cholestane-3α,7α,12α-triol; | phenolphthalein | |
| AKR1D1 | 7q32-7q33 | Steroid 5β-reductase | Δ4-cholesten-3-one-➔5β-cholestan-3-one | bile acid deficiency | |
| AKR1E1 | 10p15 | 1,5-anhydro-D-fructose reductase | |||
| AKR6A3 | 3q26.1 | K+ voltage gated channel β-subunit 1 | NADPH dependent channel opening | aberrant redox regulation of Kv channels and CV disease | |
| AKR6A5 | 1p36.3 | K+ voltage gated channel β-subunit 2 | |||
| AKR6A9 | 17p13.1 | K+ voltage gated channel β-subunit 3 | |||
| AKR7A2 | 1p35.1-p36.23 | afllatoxin aldehyde reductase | succinic semialdehyde➔γ–hydroxybutyrate | neuromodulation and SSDH deficiency | |
| AKR7A3 | 1p35.1-p36.23 | aflatoxin aldehyde reductase | aflatoxin dialdehyde➔aflatoxin bis-alcohol | hepatocarcinogenesis |
The NF-E2 p45-related factor (also known as nuclear factor (erythroid-derived 2)-like 2 (Nrf2)–Kelch-like ECH-associated protein 1 (Keap1) system35,36) induces many of the human AKR genes to respond to oxidative and electrophilic stress.7,9,10,37 In this perspective, I will discuss the roles of these enzymes and present the case that, depending on the context, upregulation of the AKR genes by the Keap1/Nrf2 system can represent a “dark-side” to Nrf2 activation.
2. THE KEAP1/NRF2 SYSTEM
Levels of the Cap'n'Collar basic leucine zipper transcription factor Nrf2 are kept in check by binding to the cytosolic protein Keap1.36,38 Keap1 targets Nrf2 for ubiquintination and proteasomal degradation by acting as a substrate adaptor protein for the cullin 3-dependent ubiquitin ligase.39 Keap1 is decorated with cysteine residues, which permits the protein to sense reactive oxygen species, electrophiles, Michael acceptors, and heavy metals.40 Once the cysteine residues are oxidized and form adducts with electrophiles or complexes with metals, dimeric Keap1 undergoes a conformational change to bind Nrf2 more tightly. This permits newly translated Nrf2 to evade Keap1-mediated ubiquintination and translocate to the nucleus.41 Keap1 is not the only repressor of Nrf2. Glycogen synthase kinase 3 also creates a phosphodegron on Nrf2 to reduce endogenous Nrf2 protein.42,43 Once Nrf2 is translocated to the nucleus, it forms heterodimers with small Maf.35 The heterodimer binds to the antioxidant response element (ARE), also known as the electrophilic response element (EpRE), in responsive genes,44,45 which belong to the ARE-gene battery. Human genes in this battery include those involved in xenobiotic detoxication (e.g., NADPH:quinone oxidoreductase (NQO1)), detoxication of reactive oxygen species (e.g., SOD1, CAT), heme detoxication (e.g., heme oxygenase), and glutathione synthesis (e.g., γ-glutamyl cysteine ligase light chain).39,46 The human AKR genes AKR1B1, AKR1B10, AKR1C1, AKR1C2, AKR1C3, AKR7A2, and AKR7A3 are all regulated by the Keap1/Nrf2 system.7,9,10,47–52 Although many studies measure NQO1 induction as a read out of Nrf2 activation, often the level of induction of NQO1 is a quite modest 2–3 fold by comparison to the induction seen in the AKR genes, which can be 1–2 orders of magnitude greater.52,53 In fact, the AKR gene response can be one of the most robust signatures of Nrf2 activation in humans, yet less attention is placed on the regulation of these genes and its consequences.
3. EVIDENCE THAT HUMAN AKR GENES ARE REGULATED BY ARES
Ciaccio and Tew were the first to report the induction of an AKR gene in human colon (HT29) cells that were resistant to ethacrynic acid and induced by dimethyl maleate, t-butyl hydroquinone, and hydroquinone, which are classical Nrf2 activators.54 The gene regulated was identified as a dihydrodiol dehydrogenase (DD) and was ultimately identified as AKR1C1.9,55 Subsequent studies revealed that exposure of HepG2 cells to benzo[a]pyrene and other polycyclic aromatic compounds (bifunctional inducers), and electrophilic Michael acceptors, phenolic antioxidants (monofunctional inducers), and reactive oxygen species (ROS) led to a 3–10 fold increase in AKR1C mRNA.7 In this context, bifunctional inducers bind to the aryl hydrocarbon receptor (AhR) and are metabolically activated to electrophiles and/or ROS to subsequently activate the Keap1/Nrf2 pathway. By contrast, monofunctional inducers act only as direct acting electrophiles or produce ROS to activate the same pathway. The distinction between bifunctional and monofunctional inducers was originally made by Prochaska and Talalay.56 However, this terminology is no longer used because there are Nrf2 activators that are neither electrophilic nor ROS producing.
In HepG2 cells, induction of AKR1C mRNA by compounds that activate AhR and Nrf2 (e.g., benzo[a]pyrene, β-naphthoflavone) was delayed with respect to the induction of CYP1A1 mRNA, indicating their need to undergo metabolism. By contrast and AKR1C mRNA was not induced by the nonmetabolizable AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). These data suggest that, in contrast to CYP1A1, induction of AKR1C member(s) by polycyclic aromatic hydrocarbons (PAHs) is mediated indirectly via an ARE by their electrophilic metabolites rather than a xenobiotic response element regulated by the AhR. RNase protection assays identified AKR1C1 (DD1) mRNA as the transcript, which was upregulated by β-naphthoflavone and Nrf2 activators (ethacrynic acid and tert-butyl hydroquinone) and ROS in both human hepatoma (HepG2) and colon carcinoma (HT29) cells.7 Subsequently, siRNA against Keap1 and R-sulforaphane treatment of human HaCAT keratinocytes resulted in induction of AKR1C1/2.57 Promoter analysis of the AKR1C1 and AKR1C2 genes located a distal consensus ARE regulated by Nrf2.10 Concurrently, studies on AKR7A1 identified this enzyme as an ethoxyquin-inducible aldehyde reductase in rat liver that was induced by Nrf2.58,59 Human aflatoxin dialdehyde reductases AKR7A2 and AKR7A3 protect against products of lipid peroxidation and aflatoxin dialdehyde in cell lines, respectively.50,60 Each of the genes for these enzymes contained AREs. Knockdown of Nrf2 in HepG2 cells led to a reduction of AKR7A3 mRNA and AKR7A3 protein and increased sensitivity to acetaminophen-induced cytotoxicity.47 Analysis of the human AKR gene promoters identified the existence of multiple AREs in 13/15 human genes, and many have subsequently been found to be functional, Table 2.46
Table 2.
Human AKR Genes Regulated by Nrf2
| gene name | number of AREs based on Nrf2 consensus sequence | positional matrix human LD < 6 | induced by Nrf2 |
|---|---|---|---|
| AKR1A1 | 6 | 5 | X49 |
| AKR1B1 | 1 | 3 | X49 |
| AKR1B10 | 4 | 2 | X49,52,53 |
| AKR1B15 | 4 | 5 | |
| AKR1C1 | 10 | 16 | X7,49,52–54 |
| AKR1C2 | 15 | 24 | X10,49,52,53 |
| AKR1C3 | 4 | 6 | X49,53 |
| AKR1C4 | 2 | 2 | |
| AKR1D1 | 11 | 27 | |
| AKR1E2 | 6 | 3 | |
| AKR6A5 | 26 | 8 | |
| AKR7A2 | 9 | 10 | X51 |
| AKR7A3 | 4 | 4 | X47 |
Agyeman et al. conducted transcriptomic and proteomic analysis of gene expression in MCF10A cells treated with either R-sulforaphane as an Nrf2 activator or with si-RNA for Keap1 so that Nrf2 would be constitutively active. Transcriptomic studies were performed by microarray and quantitative proteomic analysis was performed by stable-isotope labeling of amino acids in cell culture (SILAC).53 Transcriptomic analysis showed that AKR1B10, AKR1C1/2, and AKR1C3 were highly induced by R-sulforaphane where the fold changes were 302-, 15-, and 27-fold, respectively; by contrast, NQO1 was induced 4.4-fold. SILAC analysis showed that AKR1C1/2 and AKR1C3 proteins were highly induced by R-sulforaphane where the changes were 31 and 39, respectively; by contrast, NQO1 was induced 3.7-fold. Thus, the expression of the AKR1 genes was consistently more robust than NQO1, and this is now considered an important biomarker of the antioxidant response in humans.
4. CHANGES IN HUMAN AKR GENE EXPRESSION WITH DISEASE STATE
Changes in AKR1 gene expression have been observed in a number of disease states, especially those related to tobacco carcinogenesis. Most notable was the overexpression of AKR1C1/2 in non-small cell lung carcinoma (NSCLC), where a dramatic increase in AKR1C1 expression was observed in tumor versus adjacent normal tissue in 317/381 NSCLC patients using differential display;61 AKR1B10 and AKR1C1 were found to be two of the seven most overexpressed of the 30,000 genes displayed on an Affymetrix microarray in NSCLC;62 CYP1A1, CYP1B1, AKR1B10, AKR1C1, and AKR1C3 were also increased 15–30 fold in expression in oral squamous carcinoma and induced by cigarette smoke condensate in oral dysplasic cells.63 In addition, AKR1B10, AKR1C1, and ALDH3A1 were three of the ten most overexpressed genes in tobacco-exposed bronchial epithelial cells,64 and CYP1A1, CYP1B1, AKR1C1, NQO1, and ALDH3A1 were part of a gene battery upregulated in buccal oral specimens of smokers.65 It was also found that CYP1B1, AKR1B10, AKR1C1, and AKR1C2 were the most upregulated genes in bronchial epithelial cell brushes of smokers and were downregulated in smokers who quit.66,67 The consistent finding is that, in either smoking-related cancer or upon smoke exposure, AKR1B10 and AKR1C1 are consistently overexpressed. Interestingly, A549 cells, which are derived from a human lung adenocarcinoma patient, constitutively express Nrf2 due to a somatic mutation in Keap1 and, as a result, have high expression of AKR1C1–AKR1C3.68
Dysfunction of the Keap1/Nrf2 system has been observed in NSCLC patients where a high frequency of mutations (19% in 50 cases) were observed in a region of Keap1 that would attenuate its repressive effects on Nrf2.68 Keap1 can also become hypermethylated to reduce its expression in lung cancer. Methylation of the Keap1 promoter was observed in 22/47 NSCLC patients.69 Thus, AKR1 genes are clearly upregulated as part of the stress response to tobacco smoke, and at some point in the oncogenic process, Keap1 becomes either mutated or is epigenetically silenced, leading to high constitutive expression of Nrf2 and hence induction of AKR1 genes. This raises the question as to whether the high overexpression of AKR1 genes is a protective stress response or whether it contributes to disease pathogenesis. To address this question, it is important to consider the roles of these enzymes and in what context their overexpression is protective or harmful or may contribute to oncogenesis. It is known that AKR1B10 prevents retinoic acid signaling by its all-trans-retinaldehyde reductase activity70,71 and that AKR1C genes are implicated in the metabolism of tobacco carcinogens,18–20,72 see sections 8 and 9.
5. AKRS AND DETOXICATION OF LIPID PEROXIDES AND REACTIVE ALDEHYDES
4-HNE and 4-oxo-2-nonenal are by products of lipid peroxidation, which result from the free radical attack of polyunsaturated fatty acids and are a hallmark of oxidative stress.73 These reactive lipid aldehydes are bifunctional electrophiles because they contain an α,β-unsaturated carbonyl and are therefore Michael acceptors, and through their aldehyde group, they can form Schiff bases. Thus, they have the potential to act as cross-linking agents with proteins and form DNA adducts that are mutagenic.74 In considering this bifunctionality, Michael addition is favored due to the presence of the α,β-unsaturated carbonyl, but once this occurs, the aldehyde is able to react with lysine residues.
There are redundant mechanisms that can detoxify these reactive aldehydes. AKRs involved in this process are AKR1B1, AKR1B10, AKR1C1, AKR1C2, and AKR7A2.8,16,50,75,76 Evidence for their critical role comes from si-RNA studies and the use of chemical probes (isoform-selective AKR inhibitors) to show that, in the absence of an AKR, cells become sensitized to the cytotoxic effects of these lipid peroxidation byproducts. Which AKRs are the most important is often determined by differences in substrate preference and their cellular distribution. 4-HNE can be rapidly conjugated by glutathione, and 4-HNE-glutathionyl conjugates are the preferred substrates for AKR1B1.16 By contrast, AKR1B10, AKR1C1, and AKR7A2 reduce 4-HNE to 4-hydroxy-1,2-nonenol, eliminating the bifunctionality of 4-HNE in a single step. AKR1B10, AKR1C1, and AKR7A2 play this cytoprotective role in the small intestine and colon,75 lung,8 and neuronal cells,77 respectively.
6. AKRS AND CHEMOTHERAPEUTIC DRUG RESISTANCE
Overexpression of AKR genes in cancer cells that are resistant to cancer chemotherapeutic agents is commonplace. This is observed with cisplatin,22,23 anthracyclins (doxorubicin and duanorubciin),24 mitomycin,78 Temozolomide,79 cyclophosphosphamide,80–82 and oracin,25 see Table 3. This overexpression could be associated with a counter-response to the stress generated by these cytotoxic agents or it could be causative in the resistance phenotype. Simpkins was the first to demonstrate that ovarian cancer cells grown in the presence of cisplatin become resistance to the drug and that this resistance is associated with the overexpression of AKR1C1. Stable transfection of AKR1C1 into nonresistant ovarian cancer cells endowed the cancer chemotherapeutic drug resistant phenotype, showing that it was causative.22,23 Felsted et al.83 was among the first to show that rat daunorubicin reductase was aldehyde reductase and that this enzyme likely increased the cardiotoxicity of the agent.
Table 3.
Human AKRs and Chemotherapeutic Drug Resistance
AKRs also reduce the cytotoxicity of cyclophosphamide, Table 3. Cyclophosphamide is activated by P450-mediated hydroxylation to yield 4-hydroxy-cyclophosphamide, the immediate precursor of aldophosphamide, which gives rise to phosphoramide mustard and the reactive aldehyde acrolein. AKR1B1 catalyzes the reduction of acrolein to allyl alcohol (Km = 80 μM, kcat = 87 min−1). Acrolein also causes a time-dependent 7–20-fold increase in the activity of AKR1B1.81 In medulloblastoma, AKR1B10 is implicated in drug resistance by also reducing aldophosphamide.80
AKR1B10 is recognized as a diagnostic tumor marker in lung cancer and in the proliferation of cancer cells.57 AKR1B10 is also overexpressed in mitomycin-resistant colon cancer cell lines (HT29 cells), where it is implicated in protecting against ROS and the production of lipid peroxidation by products generated by mitomycin treatment.78 The drug resistance phenotype could be reversed by the AKR1B10 selective inhibitor (Z)-2-(4-methoxyphenylimino)-7-hydroxy-N-(pyridin-2-yl)-2H-chromene-3-carboxamide and si-RNA.78
AKR1C1–3 were significantly increased in both U373 and T98G human glioblastoma cells (at both the mRNA and protein levels) after long-term treatment with the alkylating agent Temozolomide.79 How overexpression mediates resistance to this agent is unknown, but AKR1C1–3 can all stimulate a growth proliferative phenotype in these different cell models.
Oracin is a DNA intercalating anticancer agent. AKR1C1–4 mediate the carbonyl reduction of 6-[2-(2-hydroxyethyl)-aminoethyl]-5,11-dioxo-5,6-dihydro-11H-indeno[1,2-c]-isoquinoline (oracin) to its inactive metabolite dihydrooracin, leading to drug resistance.25
Resistance to enzalutamide is mediated by AKR1C3 through an entirely different mechanism. Enzalutamide is a potent androgen-receptor antagonist used in androgen deprivation therapy in castrate-resistant prostate cancer. Median survival seen with this agent is 3–4 months before drug resistance emerges. This resistance phenotype is caused by overexpression of AKR1C3, which through its 17β-hydroxysteroid dehydrogenase activity can produce potent ligands for the androgen receptor to surmount the effect of this AR antagonist.88
In these instances, human AKRs appear to mediate drug resistance, and inhibitors of either the AKRs or the Keap1/Nrf2 system would be desirable. Thus, Nrf2 activation in terms of cancer chemotherapeutic drug resistance represents a “dark-side” to Nrf2 activation.
7. AKRS AND CANCER CHEMOTHERAPEUTIC DRUG ACTIVATION
PR104A is a new anticancer drug developed for the treatment of solid hypoxic tumors. Recently, PR104 was found to be effective under aerobic conditions, increasing its broad anticancer spectrum.5 The drug contains a nitro group, which is metabolically activated by nitroreduction to the hydroxylamino compound, which is then activated to a DNA damaging species by either acetylation or sulfonation, Figure 2. In comparison with NQO1, AKR1C3 was found to be the superior nitroreductase for this reaction, Figure 1.5 Induction of AKR1C3 with R-sulforaphane led to enhanced bioactivation of the pro-drug and antiproliferative effects.5,48 Thus, with cancer chemotherapeutic drug response, whether induction of AKRs is deleterious or beneficial may be drug and tumor dependent.
Figure 2.

Bioactivation of PR104A by AKR1C3. Steps involve the sequential formation of the nitroso and hydroxylamino products.
8. NRF2-REGULATED GENES AND CARCINOGEN METABOLISM
8.1. Aflatoxin
Many of the human AKR genes are implicated in carcinogen metabolism. The hepatocarcinogen aflatoxin is activated by P4503A4 to aflatoxin epoxide, which undergoes hydrolysis and ring opening to form the dialdehyde. Ethoxyquin-inducible aflatoxin dialdehyde reductase (AKR7A1) was originally described by Ellis et al. in rat liver,59 where the enzyme metabolizes aflatoxin dialdehyde to the mono- and dialcohols, preventing the dialdehyde from forming Schiff bases with proteins. This activity has also been assigned to human AKR7A3.60 Using liver specific AKR7A1 transgenic Sprague–Dawley rats, two lines were developed, AKR7A1Tg2 and AKR7A1Tg5. Both overexpressed AKR7A1 to different degrees, and rates of formation of aflatoxinB1 alcohols were increased. However, neither Tg line protected against acute aflatoxin-induced bile acid proliferation, suggesting that upregulation of AKR7A genes is insufficient to protect against protein adduct formation from the dialdehyde and attenuate aflatoxin B1 tumorigenicity.89 These data suggest that different outcomes in cell-based experiments and whole animals can exist when considering the protective effects of AKR7A3 and perhaps other AKRs as well. Thus, using in vitro experiments to predict in vivo acquired phenotypes resulting from Nrf2 activation need to be interpreted with caution.
8.2. Tobacco Carcinogens
Human AKRs play important roles in the metabolism of two major classes of tobacco carcinogens, polycyclic aromatic hydrocarbons (PAH) and 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK), which may account for their overexpression as part of the “smoking gene” battery.
AKR1A1, AKR1C1–4, and AKR1B10 have all been shown to play a role in the metabolic activation of PAH trans-dihydrodiols via their dihydrodiol dehydrogenase activity.18,19,90 In this reaction trans-dihydrodiols are oxidized in the presence of NAD(P)+ to a ketol, which then spontaneously rearranges to a catechol. The catechol undergoes two sequential one-electron oxidation reactions in air to form an o-semiquinone anion radical in route to electrophilic o-quinones and reactive oxygen species. In the presence of NADPH, NQO1 and AKR enzymes themselves reduce the o-quinones back to the catechol, establishing an enzymatic futile redox cycle thereby amplifying the production of ROS. For benzo[a]pyrene, the sequence involves the oxidation of benzo[a]pyrene-7,8-trans-dihydrodiol to form 7,8-dihydroxybenzo[a]pyrene and its autoxidation to benzo[a]pyrene-7,8-dione (BPQ), Figure 3.91–93 Subtle differences exist in PAH trans-dihydrodiol specificity and stereochemical preference of AKRs. AKR1A1 has the highest catalytic preference for (−)7R,8R-trans-dihydrodiol of benzo[a]pyrene, which is the metabolically favored isomer.19 By contrast, AKR1C1–4 oxidize both the (−)-R,R and (+)-S,S isomers but prefer bay-region methylated trans-dihydrodiols, which are more tumorigenic than their unmethylated counterparts.18 AKR1B10 has lower specific activities and has a preference for the minor stereoisomers.90
Figure 3.

Role of AKRs in human lung cancer: function of AKRs induced by the smoking gene battery. AKR1C1–3 are involved in the metabolic activation of PAH; AKR1C1 and AKR1C2 show a preference for forming S-NNAL, which can be retained in lung tissues for bioactivation, and AKR1B10 prevents retinoic acid signaling. Acting together, these reactions could contribute to lung cancer initiation and promotion.
The AKR1C1–3 enzymes appear to be the most important in the oxidization of PAH trans-dihydrodiols in human lung cells, where they are inducible by the ARE and overexpressed in A549 lung adenocarcinoma cells.94 In A549 cells, the complete sequence for the activation of benzo[a]pyrene-7,8-trans-dihydrodiol oxidation was observed, resulting in the production of BPQ, ROS, and elevated amounts of mutagenic 8-oxo-dGuo.94 The production of BPQ and ROS suggests that AKR1C genes regulate their own expression via AREs, generating a positive feedback loop that exacerbates the carcinogenicity of PAH tobacco carcinogens, i.e., this would mean that PAH trans-dihydrodiols enhance their own genotoxicity by inducing AKR1C genes, Figure 3. This feed-forward loop could be attenuated if the intermediate catechol could be either O-methylated or conjugated by sulfotransferases or glucuronosyl transferases. In A459 cells treated with B[a]P-7,8-trans-dihydrodiol, both ROS and 8-oxo-dGuo were increased in the presence of a catechol-O-methyl transferase (COMT) inhibitor, showing that some interception of the catechol can occur.94 To determine the extent by which phase 2-conjugating enzymes can protect against the redox cycling of o-quinones, recombinant forms of COMT, SULTs, and UGTs expressed in lung cells were examined for their ability to form catechol conjugates versus the preference of o-quinones to redox cycle.95–97 The specific activities for NQO1 and AKRs to redox cycle BPQ were much greater than those observed for the phase 2 enzymes to conjugate 7,8-dihydroxybenzo[a]-pyrene, Table 4.98 In fact, conversion of the trans-dihydrodiols to the o-quinones catalyzed by AKRs was 100–1,000-fold slower than their subsequent reduction of the o-quinone to produce the catechol. Thus, the redox cycling observed with the o-quinones overwhelms the activity of the phase 2-conjugating enzymes. These data support the notion that enzymatic redox cycling of PAH o-quinones occurs at an alarming rate, leading to ROS and Nrf2 activation.
Table 4.
Enzymatic PAH Catechol Conjugation Relative to PAH o-Quinone Redox Cycling
| recombinant enzyme−1 | specific activity (nmol min−1 mg−1) 10 μM B[a]P-7.8-dionea |
|---|---|
| NQO1 + NADPH | 1070b |
| AKR7A2 + NADPH | 1270b |
| AKR1C1 + NADPH | 64b |
| COMT + S-adenosyl-L-methionine | 55c |
| SULT1A1 + phosphoadenosine phosphosulfate (PAPS) | 0.8c |
| UGT1A3 + UDP-glucuronic acid | >1.0d |
For catechol conjugation reactions catalyzed by COMT, SULT, or UGT, B[a]P-7,8-dione was reduced to the catechol with NADPH under anaerobic conditions.
Mean of six replicates with SD < 10%.
From Michaelis–Menten plots with homogeneous recombinant enzyme.
From commercial microsomes expressing recombinant UGT.
The tobacco carcinogen NNK is metabolically activated by P4502A6 and P4502A13 (in the aerodigestive tract)99 by α-methyl or α-methylene group hydroxylation. α-Methyl hydroxylation results in the formation of formaldehyde and a pyridyloxylbutyl (POB) diazohydroxide, which leads to formaldehyde and POB-DNA adducts,100,101 respectively. α-Methylene hydroxylation can lead to the formation of an α-methyl-diazohydroxide and a pyridyloxybutylaldehyde. The former collapses to form an α methyl carbonium ion that can form O6-methyl-guanine DNA adducts.102 Metabolic activation of NNK can be prevented if it is reduced to R-4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanol (R-NNAL) and S-NNAL provided the alcohols are glucuronidated by UGTs and eliminated. In the smoking gene battery, AKR1C1–3 reduce NNK to S-NNAL, whereas human lung microsomes predominately produce R-NNAL.72 S-NNAL is stereoselectively retained in rat lung and has a higher tumorigencity than that of R-NNAL.103,104 S-NNAL can still undergo activation by α-methyl and α-methylene hydroxylation. Thus, induction of AKR1C–3 could prevent the formation of R-NNAL and its subsequent elimination as S-NNAL–O-glucuronide and exacerbate DNA-adduct formation. It should be noted that, in the UGT reaction mechanism, there is proposed inversion of stereochemistry at the anomeric carbon.105
8.3. Nitroarenes
Nitroarenes are nitrated PAHs, which are unique to diesel exhaust emissions. Diesel exhaust has been recently classified as a Group 1 carcinogen by IARC.106 Nitroarenes are metabolically activated by six-electron reduction to form the corresponding amines. The sequence involves formation of nitroso, hydroxylamino, and amine intermediates.107,108 Following formation of the hydroxylamino, intermediate acetylation by NAT or sulfonation by SULTs109 introduces a good leaving group so that a nitrenium ion can form, which will result in DNA adducts, Figure 4. This sequence is observed with 3-nitrobenzanthrone (3-NBA), which is the most potent mutagen identified in the Ames test, and with 6-nitrochrysene, which is the most potent tumorigen in a newborn mouse model for lung cancer.110,111 Enzymes implicated in the metabolic activation of nitroarenes are NQO1 and NADPH:P450 oxidoreductase.108,109 Using 32P-post-labeling, it was found that the number of DNA adducts derived from NBA112 was significantly reduced in the presence of dicoumarol, an NQO1 inhibitor.109 The metabolic activation of nitroarenes to electrophiles and their back oxidation in air in the presence of heavy metals to produce reactive oxygen species113 indicates that nitroarenes may also induce their own genotoxicity by induction of NQO1 via the ARE.109,114
Figure 4.

Nrf2-regulated genes and carcinogen metabolism in humans: Role of NQO1 in the metabolic activation of nitroarenes. NQO1 plays a pivotal role in the metabolic activation of 3-NBA, yet it is the signature gene measured to demonstrate Nrf2 activation.
In these examples, the induction of Nrf2-regulated genes in the context of air pollutants could be deleterious, leading to an enhancement of their genotoxicity; this may be observed with PAH, NNK, and nitroarenes and represents another “dark-side” of Nrf2 activation.
9. NRF2 REGULATION OF AKR GENES AND NUCLEAR RECEPTOR SIGNALING
Human AKR genes metabolize retinoids, prostaglandins, and steroid hormones and play a pivotal role in the prereceptor regulation of nuclear receptors (NR) by regulating the amount of ligand that can bind to these receptors, Table 5. Thus, the prospect exists that the same AKR genes that are regulated by the Keap1/Nrf2 pathway may also influence nuclear receptor action.
Table 5.
Nuclear Receptor Regulation by Human AKRs
| human AKR | nuclear receptor ligand regulation | nuclear receptor target | change in gene expression |
|---|---|---|---|
| AKR1B10 | all-trans-retinaldehyde → all-trans-retinol | RAR | decrease |
| AKR1B15 | 9-cis-retinaldhehyde → cis-retinol | RXR | decrease |
| AKR1C1 | progesterone → 20α-hydroxyprogesterone | PR | decrease |
| AKR1C2 | dihydrotestosterone → 3α-androstanediol | AR | decrease |
| AKR1C3 | Δ4-androstene-3,17-dione → testosterone; 5α-androstane-3,17-dione → dihydrotestosterone | AR | increase |
| AKR1C3 | estrone → 17β-estradiol | ER | increase |
| AKR1C3 | 11β-PGD2 → PGF2α | PPARγ | decrease |
| AKR1D1 | bile-acid biosynthesis | FXR | increase |
AKR1B10 is part of the “smoking gene battery”,62,71,115 and this enzyme is a highly efficient retinal reductase that reduces all-trans-retinaldehyde to all-trans-retinol and thus prevents the formation of retinoic acid, Figure 4.70,116 Retinoic acid binds to the retinoic acid receptor (RAR), which signals cells to differentiate and is thus antiproliferative. The attenuation of this signaling pathway by AKR1B10 suggests that its regulation by the Keap1/Nrf2 system provides a pro-proliferative signal. In NSCLC, where AKR1B10 is highly constitutively expressed due to a Keap1 mutation, or with Keap1 silencing, this could lead to the promotion of lung cancer.
AKR1C3 is also known as prostaglandin (PG) F2α synthase, converts PGH2 to PGF2α, and also possesses 11-ketoprostaglandin reductase activity, where it converts PGD2 to 11β-PGF2α.28,29 Both PGF2α and 11β-PGF2α bind to the FP1 receptor, which activates MAPK pathways to stimulate cell growth and inactivate PPARγ by phosphorylation. Additionally, conversion of PGD2 to 11β-PGF2α prevents the formation of PGJ2 ligands (15-deoxy-Δ12,14-prostaglandin J2; 15d-PGJ2) for the peroxisome proliferator-activated receptor (PPARγ).117 By forming heterodimer complexes with RXR, cell differentiation can proceed. Thus, AKR1C3 promotes cell proliferation by forming FP1 ligands and by depriving PPARγ of its ligands.26,118 This mechanism has been shown to be an important signaling mechanism in acute myeloid leukemia, and AKR1C3 inhibitors are being sought for their antineoplastic effects.119
AKR1C1-AKR1C3 display NADPH dependent 3-, 17- and 20-ketosteroid activity.32 Thus, AKR1C1 is the primary 20-ketosteroid reductase in humans responsible for the conversion of progesterone to 20α-hydroxyprogesterone and terminates progesterone action at the progesterone receptor.120 Progestin therapy is used for the treatment of precancerous endometrial conditions (e.g., atypical hyperplasia or endometrial intraepithelial neoplasia), but a portion of patients are resistant to progestin therapy. It has been shown that Nrf2-driven AKR1C1 expression mediates this progestin resistance.121
AKR1C2 is the primary 3-ketosteroid reductase responsible for the conversion of 5α-dihydrotestosterone to 5α-androstan-3α,17β-diol and can terminate androgen action at the androgen receptor.33 By contrast, AKR1C3 is also a major peripheral 17-ketosteroid reductase, is responsible for the formation of potent androgens in the prostate, and is a source of testosterone, the substrate for aromatase, in the breast.26,31,122 Extensive drug discovery programs exist for the development of AKR1C3 inhibitors for hormone-dependent malignancies.123–130 The fact that each of these genes are regulated by the Keap1/Nrf2 pathway suggests that Nrf2 activators may have unintended consequences for steroid hormone receptor occupancy and hence steroid hormone action in both physiology and hormone-dependent malignancies.
10. “U”-SHAPED DOSE RESPONSE CURVE FOR NRF2 IN THE CONTEXT OF ELECTROPHILIC AND OXIDATIVE STRESS
Induction of AKR genes is clearly cytoprotective against the formation of lipid peroxidation by products and ROS.7,50,51,75,76 However, this induction can also mediate cancer chemotherapeutic drug resistance, regulate NR action, and may exacerbate hormonal or chemical carcinogenesis based on context. There is no question that Nrf2 activators can be chemopreventive in rodent tumor models, where studies have been extensive.131–133 There is also evidence that the principle of chemoprevention can be translated into humans, e.g., the prevention of hepatocarcinogenesis initiated by aflatoxin.134 However, the animal studies on which the clinical studies are based were performed in species that lack orthologues of the human AKR1C genes. As indicated above, there are scenarios in which induction of the AKR1B10 and AKR1C genes may contribute to tumor initiation and promotion. The ability of Nrf2 activation to have both beneficial and harmful consequences suggests that context is important and begs the question as to the shape of the Nrf2-dose response curve, the exposure paradigm and whether epigenetic regulation of the Nrf2 system influences the outcome.
We propose that, even for its cytoprotective effects, the shape of the dose response curve for Nrf2 activation is “U”-shaped and would mirror that seen for either an essential nutrient or trace metal, Figure 5. This hormetic dose–response curve for Nrf2 has also been proposed by other investigators to account for the role of Keap1/Nrf2 in oncogenesis.135,136 Here, the dose is Nrf2 expression, and the response is oxidative or electrophilic stress. In the absence of Nfr2 expression, exposure to electrophilic or redox active compounds would increase cellular stress and would be cytotoxic; as Nrf2 is activated by increased exposure to electrophiles or redox-active compounds, redox balance is addressed, and redox homeostasis is achieved; however, at high levels of exposure to electrophiles and redox active compounds, the Keap1/Nrf2 system induces AKR genes that may exacerbate the stress, especially when the compounds are lung carcinogens.
Figure 5.
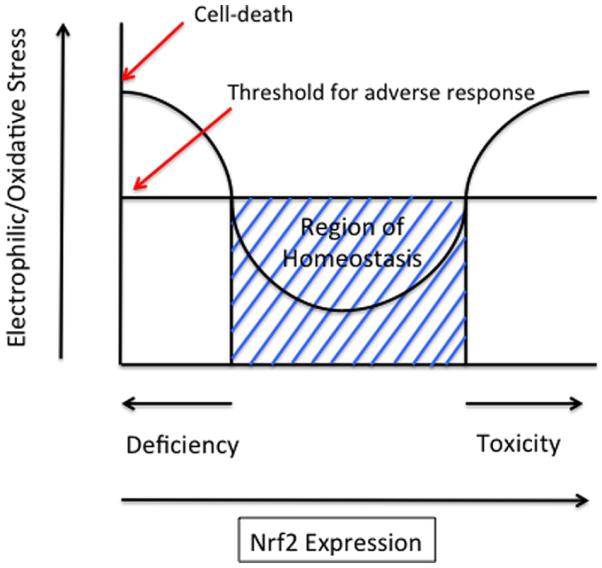
“U”-shaped dose response for Nrf2. This hermetic dose–response curve is based on data from refs 135, 136, 140, and 141.
Many of the in vitro experimental systems that have been used to activate the Keap1/Nrf2 pathway use single acute exposures. However, real world exposure to Nrf2 activator inducers are likely to be to low dose and chronic. Important in vitro an in vivo experiments in which repeated single doses of Nrf2 activator are given with concurrent measurement of Nrf2 response were conducted using the chemoprevention agent Oltipraz, where the Nrf2 response was reported as induction of glutathione-S-transferase (GST) levels in rats.137 Daily, twice weekly, and even weekly administration produced a prolonged response of GST activity. Thus, intermittent exposure to an Nrf2 inducer can have a prolonged effect. In other studies, long-term dietary administration of Oltipraz led to an induction of AKR7A1 that was less pronounced than that of shorter exposures.138 Both studies indicate that adaptive responses to Nrf2 expression can occur after exposure to inducers. Low dose chronic exposures may also lead to permanent epigenetic changes in gene expression of the Cul3/Nrf2/Keap1 system. The system is known to be epigenetically regulated by DNA methylation, histone modification, and microRNAs.139 These are important questions that need to be addressed when considering the pharmacokinetic/pharmacodyamic correlates of chemopreventive reagents targeting the Keap1/Nrf2 system.
Acknowledgments
Funding This work was supported by P30-ES013508 awarded to T.M.P.
ABBREVIATIONS
- AhR
aryl hydrocarbon receptor
- AKR
aldo-keto reductase
- ARE
antioxidant response element
- B[a]P
benzo[a]pyrene
- BPQ
benzo[a]pyrene-7,8-dione
- CAT
catalase
- COMT
catechol-O-methyl transferase
- Keap1
Kelch-like ECH associate protein
- P450
cytochrome P450
- PAH
polycyclic aromatic hydrocarbons
- PPARγ
peroxisome proliferator-activated receptor γ
- 3-NBA
3-nitrobenzanthrone
- NNK
4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanol
- NR
nuclear receptor
- NSCLC
non-small cell lung carcinoma
- NQO1
NAD(P)H-quinone oxiodreductase
- Nrf2
NF-E2 p45-related factor (also known as nuclear factor (erythroid-derived 2)-like 2)
- RAR
retinoic acid receptor
- ROS
reactive oxygen species
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- SOD1
superoxide dismutase
- SULTs
sulfotransferases
- UGTs
uridine diphosphate glucuronsyl transferases
Biography
Trevor M. Penning, Ph.D., is the Thelma Brown and Henry Charles Molinoff Professor of Pharmacology and Director of the Center of Excellence in Environmental Toxicology at the Perelman School of Medicine at the University of Pennsylvania (UPenn). He obtained his Ph.D. in Biochemistry with Professor M. Akhtar F.R.S. at Southampton University, UK and conducted postdoctoral training with Professor Paul Talalay at Johns Hopkins University School of Medicine. He joined the faculty at UPenn in 1982. He is internationally recognized for his research on how hormones and chemicals cause cancer. He is a member of The Johns Hopkins Society of Scholars and Fellow of the American Chemical Society.
Footnotes
ORCID Trevor M. Penning: 0000-0002-3937-1066
The author declares no competing financial interest.
REFERENCES
- (1).Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem.-Biol. Interact. 2003;143–144:621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- (2).Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem. Pharmacol. 1997;54:639–647. doi: 10.1016/s0006-2952(97)84253-0. [DOI] [PubMed] [Google Scholar]
- (3).Mindnich RD, Penning TM. Aldo-keto reductase (AKR) superfamily: genomics and annotation. Hum. Genomics. 2009;3:362–370. doi: 10.1186/1479-7364-3-4-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jin Y, Penning TM. Aldo-Keo Reductases and bioactivation/detoxication. In: Cho AK, Blaschke TF, Insel PA, editors. Annu. Rev. Pharmacol. Toxicol. Annual Reviews; Palo Alto CA: 2007. pp. 263–292. [Google Scholar]
- (5).Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Doñate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70:1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- (6).Chen M, Drury JE, Penning TM. Substrate specificity and inhibitor analyses of human steroid 5β-reductase (AKR1D1) Steroids. 2011;76:484–490. doi: 10.1016/j.steroids.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999;59:607–614. [PubMed] [Google Scholar]
- (8).Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species-and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J. Biol. Chem. 2001;276:2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- (9).Ciaccio PJ, Jaiswal AK, Tew K. Regulation of human dihydrodiol dehydrogenase by Michael acceptor xenobiotics. J. Biol. Chem. 1994;269:15558–15562. [PubMed] [Google Scholar]
- (10).Lou HD, Ji Q, Stolz A. Induction of AKR1C2 by phase II inducers: identification of a distal consensus antioxidant response element regulated by NRF2. Mol. Pharmacol. 2006;69:1662–1672. doi: 10.1124/mol.105.019794. [DOI] [PubMed] [Google Scholar]
- (11).Ko BC, Ruepp B, Bohren KM, Gabbay KH, Chung SS. Identification and characterization of multiple osmotic response sequences in the human aldose reductase gene. J. Biol. Chem. 1997;272:16431–16437. doi: 10.1074/jbc.272.26.16431. [DOI] [PubMed] [Google Scholar]
- (12).Chang Q, Harter T, Griest T, Murthy BSN, Petrash JM. Aldo-Keto Reductases in the stress response of the budding yeast Saccharomyces cerevisiae. In: Penning TM, Petrash M, editors. Aldo-Keto Reductases and Toxicant Metabolism. ACS; Washington DC: 2004. pp. 213–224. [Google Scholar]
- (13).Penning TM, Drury JE. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007;464:241–250. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vander Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Reduction of trioses by NADPH-dependent aldo-keto reductases. Aldose reductase, methylglyoxal, and diabetic complications. J. Biol. Chem. 1992;267:4364–4369. [PubMed] [Google Scholar]
- (15).Vander Jagt DL, Hunsaker LA, Young BS, Brown WM. Aldo-keto reductase-catalyzed detoxication of endogenus aldehydes associated with diabetic complications. In: Penning TM, Petrash M, editors. Aldo-Keto Reductases and Toxicant Metabolism. ACS; Washington DC: 2004. pp. 23–36. [Google Scholar]
- (16).Ramana KV, Dixit BL, Srivastava S, Balendiran GK, Srivastava SK, Bhatnagar A. Selective recognition of glutathiolated aldehydes by aldose reductase. Biochemistry. 2000;39:12172–12180. doi: 10.1021/bi000796e. [DOI] [PubMed] [Google Scholar]
- (17).O'Connor T, Ireland LS, Harrison DJ, Hayes JD. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem. J. 1999;343(Pt 2):487–504. [PMC free article] [PubMed] [Google Scholar]
- (18).Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J. Biol. Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- (19).Palackal NT, Burczynski ME, Harvey RG, Penning TM. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: Potential role in polycyclic aromatic hydrocarbon activation. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- (20).Atalla A, Breyer-Pfaff U, Maser E. Purification and characterization of oxidoreductases-catalyzing carbonyl reduction of the tobacco-specific nitrosamine 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) in human liver cytosol. Xenobiotica. 2000;30:755–769. doi: 10.1080/00498250050119826. [DOI] [PubMed] [Google Scholar]
- (21).Atalla A, Maser E. Characterization of enzymes participating in carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) in human placenta. Chem.-Biol. Interact. 2001;130–132:737–748. doi: 10.1016/s0009-2797(00)00304-5. [DOI] [PubMed] [Google Scholar]
- (22).Chen J, Adikari M, Pallai R, Parekh HK, Simpkins H. Dihydrodiol dehydrogenases regulate the generation of reactive oxygen species and the development of cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother. Pharmacol. 2008;61:979–987. doi: 10.1007/s00280-007-0554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Deng HB, Parekh HK, Chow KC, Simpkins H. Increased expression of dihydrodiol dehydrogenase induces resistance to cisplatin in human ovarian carcinoma cells. J. Biol. Chem. 2002;2002:15035–15043. doi: 10.1074/jbc.M112028200. [DOI] [PubMed] [Google Scholar]
- (24).Hofman J, Malcekova B, Skarka A, Novotna E, Wsol V. Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014;278:238–248. doi: 10.1016/j.taap.2014.04.027. [DOI] [PubMed] [Google Scholar]
- (25).Novotna R, Wsol V, Xiong G, Maser E. Inactivation of the anticancer drugs doxorubicin and oracin by aldoketo reductase (AKR) 1C3. Toxicol. Lett. 2008;181:1–6. doi: 10.1016/j.toxlet.2008.06.858. [DOI] [PubMed] [Google Scholar]
- (26).Byrns MC, Duan L, Lee SH, Blair IA, Penning TM. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J. Steroid Biochem. Mol. Biol. 2010;118:177–187. doi: 10.1016/j.jsbmb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lacroix Pépin N, Chapdelaine P, Rodriguez Y, Tremblay JP, Fortier MA. Generation of human endometrial knockout cell lines with the CRISPR/Cas9 system confirms the prostaglandin F2α synthase activity of aldo-ketoreductase 1B. Mol. Hum. Reprod. 2014;20:650–663. doi: 10.1093/molehr/gau023. [DOI] [PubMed] [Google Scholar]
- (28).Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J. Biochem. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- (29).Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, Watanabe K. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 1999;462:335–340. doi: 10.1016/s0014-5793(99)01551-3. [DOI] [PubMed] [Google Scholar]
- (30).Jin Y, Duan L, Chen M, Penning TM, Kloosterboer HJ. Metabolism of the synthetic progestogen norethynodrel by human ketosteroid reductases of the aldo-keto reductase super-family. J. Steroid Biochem. Mol. Biol. 2012;121:546–555. doi: 10.1016/j.jsbmb.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β–HSD activity and cellular distribution. Mol. Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- (32).Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3α-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- (34).Steckelbroeck S, Jin Y, Oyesanmi B, Kloosterboer HJ, Penning TM. Tibolone is metabolized by the 3α/3β-hydroxysteroid dehydrogenase activities of the four human isozymes of the aldo-keto reductase 1C subfamily: inversion of stereospecificity with a delta5(10)-3-ketosteroid. Mol. Pharmacol. 2004;66:1702–1711. doi: 10.1124/mol.104.004515. [DOI] [PubMed] [Google Scholar]
- (35).Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response element. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- (36).Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Talalay P, Dinkova-Kostova AT. Role of nicotinamide quinone oxidoreductase 1 (NQO1) in protection against toxicity of electrophiles and reactive oxygen intermediates. Methods Enzymol. 2004;382:355–364. doi: 10.1016/S0076-6879(04)82019-6. [DOI] [PubMed] [Google Scholar]
- (38).Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- (39).Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of keap-1 in cellular protective responses. Chem. Res. Toxicol. 2005;18:1779–1795. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- (40).Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Baird L, Lleres D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. U. S. A. 2013;110:15259–15264. doi: 10.1073/pnas.1305687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rada P, Rojo A, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobón-Velasco JC, Devijver H, García-Mayoral MF, Van Leuven F, Hayes JD, Bertho G, Cuadrado A. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2011;31:1121–1133. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991;1991:11632–11639. [PubMed] [Google Scholar]
- (45).Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 1990;265:16648–16653. [PubMed] [Google Scholar]
- (46).Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radical Biol. Med. 2015;88:108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ahmed MM, Wang T, Luo Y, Ye S, Wu Q, Guo Z, Roebuck BD, Sutter TR, Yang JY. Aldo-keto reductase-7A protects liver cells and tissues from acetaminophen-induced oxidative stress and hepatotoxicity. Hepatology. 2011;54:1322–1332. doi: 10.1002/hep.24493. [DOI] [PubMed] [Google Scholar]
- (48).Erzinger MM, Bovet C, Hecht KM, Senger S, Winiker P, Sobotzki N, Cristea S, Beerenwinkel N, Shay JW, Marra G, Wollscheid B, Sturla SJ. Sulforaphane preconditioning sensitizes human colon cancer cells towards the boreductive anticancer prodrug PR-104A. PLoS One. 2016;11:e0150219. doi: 10.1371/journal.pone.0150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jung KA, Choi BH, Nam CW, Song M, Kim ST, Lee JY, Kwak MK. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol. Lett. 2013;218:39–49. doi: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- (50).Li D, Ferrari M, Ellis EM. Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem.-Biol. Interact. 2012;195:25–34. doi: 10.1016/j.cbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- (51).Li D, Ma S, Ellis EM. Nrf2-mediated adaptive response to methyl glyoxal in HepG2 cells involves the induction of AKR7A2. Chem.-Biol. Interact. 2015;234:366–371. doi: 10.1016/j.cbi.2014.10.019. [DOI] [PubMed] [Google Scholar]
- (52).MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis. 2009;30:1571–1580. doi: 10.1093/carcin/bgp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, Kensler TW. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012;132:175–187. doi: 10.1007/s10549-011-1536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ciaccio PJ, Stuart JE, Tew KD. Overproduction of a 37.5-kDa cytosolic protein structurally related to prostaglandin F synthase in ethacrynic acid-resistant human colon cells. Mol. Pharmacol. 1993;43:845–853. [PubMed] [Google Scholar]
- (55).Ciaccio PJ, Tew K. cDNA and deduced amino acid sequences of a human colon dihydrodiol dehydrogenase. Biochim. Biophys. Acta, Bioenerg. 1994;1186:129–132. doi: 10.1016/0005-2728(94)90144-9. [DOI] [PubMed] [Google Scholar]
- (56).Prochaska HJ, Talalay P. Regulatory mechanisms of monofunctional and bifunctional anticarcinogenic enzyme inducers in murine liver. Cancer Res. 1988;48:4776–4782. [PubMed] [Google Scholar]
- (57).Devling TW, Lindsay CD, McLellan LI, McMahon M, Hayes JD. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. U. S. A. 2005;102:7280–7285. doi: 10.1073/pnas.0501475102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ellis EM, Slattery CM, Hayes JD. Characterization of the rat aflatoxin B1 aldehyde reductase gene, AKR7A1. Structure and chromosomal localization of AKR7A1 as well as identification of antioxidant response elements in the gene promoter. Carcinogenesis. 2003;24:727–737. doi: 10.1093/carcin/bgg016. [DOI] [PubMed] [Google Scholar]
- (59).Ellis EM, Judah DJ, Neal GE, Hayes JD. An ethoxyquin-inducible aldehyde reductase from rat liver that metabolizes aflatoxin B1 defines a subfamily of aldo-keto reductases. Proc. Natl. Acad. Sci. U. S. A. 1993;90:10350–10354. doi: 10.1073/pnas.90.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bodreddigari S, Jones LK, Egner PA, Groopman JD, Sutter CH, Roebuck BD, Guengerich FP, Kensler TW, Sutter TR. Protection against aflatoxin B1-induced cytotoxicity by expression of the cloned aflatoxin B1-aldehyde reductases rat AKR7A1 and human AKR7A3. Chem. Res. Toxicol. 2008;21:1134–1142. doi: 10.1021/tx7004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hsu N-Y, Ho H-C, Chow K-C, Lin T-Y, Shih C-S, Wang L-S, Tsai C-M. Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res. 2001;61:2727–2731. [PubMed] [Google Scholar]
- (62).Fukumoto S-I, Yamauchi N, Moriguchi H, Hippo Y, Watanbe A, Shibahara J, Taniguichi H, Ishikawa S, Ito H, Yamamato S, Iwanari H, Horonaka M, Ishikawa H, Niki T, Sohara Y, Kodama T, Mishimura M, Fukayama M, Doska-Akita H, Auratani H. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers non-small cell lung carcinoma. Clin. Cancer Res. 2005;11:1776–1785. doi: 10.1158/1078-0432.CCR-04-1238. [DOI] [PubMed] [Google Scholar]
- (63).Nagaraj N, Beckers S, Mensah JK, Waigel S, Vigneswaran N, Zacharias W. Cigarette smoke condensate induces cytochrome P450 and aldo keto reductases in oral cancer cells. Toxicol. Lett. 2006;165:182–194. doi: 10.1016/j.toxlet.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Woenckhaus M, Klein-Hitpass L, Grepmeier U, Merk J, Pfeifer M, Wild PJ, Bettstetter M, Wuensch P, Blaszyk H, Hartmann A, Hofstaedter F, Dietmaier W. Smoking and cancer-related gene expression in bronchial epithelium and non-small cell lung cancer. J. Pathol. 2006;210:192–204. doi: 10.1002/path.2039. [DOI] [PubMed] [Google Scholar]
- (65).Gumus ZH, Du B, Kacker A, Boyle JO, Bocker JM, Mukherjee P, Subbaramaiah K, Dannenberg AJ, Weinstein H. Effects of tobacco smoke on gene expression and cellular pathways in a cellular model of oral leukoplakia. Cancer Prev. Res. 2008;1:100–111. doi: 10.1158/1940-6207.CAPR-08-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Zhang X, Sebastiani P, Liu G, Schembri F, Dumas YM, Langer EM, Alekseyev Y, O'Connor GT, Brooks DR, Lenburg ME, Spira A. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol. Genomics. 2010;41:1–8. doi: 10.1152/physiolgenomics.00167.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zhang L, Lee JJ, Tang H, Fan Y-H, Xiao L, Ren H, Kurie J, Morice RC, Hong WK, Mao L. Impact of smoking cessation on global gene expression in the bronchial epithelium of chronic smokers. Cancer Prev. Res. 2008;1:112. doi: 10.1158/1940-6207.CAPR-07-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS One. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Muscarella LA, Parrella P, D'Alessandro V, la Torre A, Barbano R, Fontana A, Tancredi A, Guarnieri V, Balsamo T, Coco M, Copetti M, Pellegrini F, De Bonis P, Bisceglia M, Scaramuzzi G, Maiello E, Valori VM, Merla G, Vendemiale G, Fazio VM. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics. 2011;6:710–719. doi: 10.4161/epi.6.6.15773. [DOI] [PubMed] [Google Scholar]
- (70).Gallego O, Belyaeva OV, Porté S, Ruiz FX, Stetsenko AV, Shabrova EV, Kostereva NV, Farrés J, Parés X, Kedishvili NY. Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem. J. 2006;399:101–109. doi: 10.1042/BJ20051988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wang R, Wang G, Ricard MJ, Ferris B, Strulovici-Barel Y, Salit J, Hackett NR, Gudas LJ, Crystal RG. Smoking-induced upregulation of AKR1B10 expression in the airway epithelium of healthy individuals. Chest. 2010;138:1402–1410. doi: 10.1378/chest.09-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Breyer-Pfaff U, Martin HJ, Ernst M, Maser E. Enantioselectivity of carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab. Dispos. 2004;32:915–922. [PubMed] [Google Scholar]
- (73).Esterbauer H, Schauer RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malodialdehyde and related aldehydes. Free Radical Biol. Med. 1991;11:81–82. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- (74).Pollack M, Yang IY, Kim HY, Blair IA, Moriya M. Translesion DNA Synthesis across the heptanone-etheno-2′-deoxycytidine adduct in cells. Chem. Res. Toxicol. 2006;19:1074–1079. doi: 10.1021/tx0600503. [DOI] [PubMed] [Google Scholar]
- (75).Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J. Biol. Chem. 2009;284:26742–26748. doi: 10.1074/jbc.M109.022897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zhong L, Liu Z, Yan R, Johnson S, Zhao Y, Fang X, Cao D. Aldo-keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem. Biophys. Res. Commun. 2009;387:245–250. doi: 10.1016/j.bbrc.2009.06.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lyon RC, Li D, McGarvie G, Ellis EM. Aldo-keto reductases mediate constitutive and inducible protection against aldehyde toxicity in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 2013;62:113–121. doi: 10.1016/j.neuint.2012.10.007. [DOI] [PubMed] [Google Scholar]
- (78).Matsunaga T, Yamane Y, Iida K, Endo S, Banno Y, El-Kabbani O, Hara A. Involvement of the aldo-keto reductase, AKR1B10, in mitomycin-c resistance through reactive oxygen species-dependent mechanisms. Anti-Cancer Drugs. 2011;22:402–408. doi: 10.1097/CAD.0b013e3283448df0. [DOI] [PubMed] [Google Scholar]
- (79).Le Calvé B, Rynkowski M, Le Mercier M, Bruyère C, Lonez C, Gras T, Haibe-Kains B, Bontempi G, Decaestecker C, Ruysschaert JM, Kiss R, Lefranc F. Long-term in vitro treatment of human glioblastoma cells with Temozolomide increases resistance in vivo through upregulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia. 2010;12:727–739. doi: 10.1593/neo.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Bacolod MD, Lin SM, Johnson SP, Bullock NS, Colvin M, Bigner DD, Friedman HS. The gene expression profiles of medulloblastoma cell lines resistant to preactivated cyclophosphamide. Curr. Cancer Drug Targets. 2008;8:172–179. doi: 10.2174/156800908784293631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kolb NS, Hunsaker LA, Vander Jagt DL. Aldose reductase-catalyzed reduction of acrolein: implications in cyclophosphamide toxicity. Mol. Pharmacol. 1994;45:797–801. [PubMed] [Google Scholar]
- (82).Matsunaga T, Wada Y, Endo S, Soda M, El-Kabbani O, Hara A. Aldo-keto reductase 1B10 and Its role in proliferation capacity of drug-resistant cancers. Front. Pharmacol. 2012;31:3–5. doi: 10.3389/fphar.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Felsted RL, Gee M, Bachur NR. Rat liver daunorubicin reductase. An aldo-keto reductase. J. Biol. Chem. 1974;249:3672–3679. [PubMed] [Google Scholar]
- (84).Takahashi RH, Bains OS, Pfeifer TA, Grigliatti TA, Reid RE, Riggs KW. Aldo-keto reductase 1C2 fails to metabolize doxorubicin and daunorubicin in vitro. Drug Metab. Disp. 2008;36:991–994. doi: 10.1124/dmd.108.020388. [DOI] [PubMed] [Google Scholar]
- (85).Veitch ZW, Guo B, Hembruff SL, Bewick AJ, Heibein AD, Eng J, Cull S, Maclean DA, Parissenti AM. Induction of 1C aldo-ketoreductases and other drug dose-dependent genes upon acquisition of anthracycline resistance. Pharmacogenet. Genomics. 2009;19:477–488. doi: 10.1097/FPC.0b013e32832c484b. [DOI] [PubMed] [Google Scholar]
- (86).Wsol V, Szotakova B, Martin H, Maser E. Aldo-keto reductases (AKR) from the AKR1C subfamily catalyze the carbonyl reduction of the novel anticancer drug oracin in man. Toxicology. 2007;238:111–118. doi: 10.1016/j.tox.2007.05.021. [DOI] [PubMed] [Google Scholar]
- (87).Novotna R, Wsol V, Xiong G, Maser E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo–keto reductase (AKR) 1C3. Toxicol. Lett. 2008;181:1–6. doi: 10.1016/j.toxlet.2008.06.858. [DOI] [PubMed] [Google Scholar]
- (88).Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Roebuck BD, Johnson DN, Sutter CH, Egner PA, Scholl PF, Friesen MD, Baumgartner KJ, Ware NM, Bodreddigari S, Groopman JD, Kensler TW, Sutter TR. Transgenic expression of aflatoxin aldehyde reductase (AKR7A1) modulates aflatoxin B1 metabolism but not hepatic carcinogenesis in the rat. Toxicol. Sci. 2009;109:41–49. doi: 10.1093/toxsci/kfp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Quinn AM, Harvey RG, Penning TM. Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem. Res. Toxicol. 2008;21:2207–2215. doi: 10.1021/tx8002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Penning TM, Ohnishi ST, Ohnishi T, Harvey RG. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase. Chem. Res. Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- (92).Smithgall TE, Harvey RG, Penning TM. Regio- and stereospecificity of homogeneous 3α-hydroxysteroid-dihydrodiol dehydrogenase for trans-dihydrodiol metabolites of polycyclic aromatic hydrocarbons. J. Biol. Chem. 1986;261:6184–6191. [PubMed] [Google Scholar]
- (93).Smithgall TE, Harvey RG, Penning TM. Spectroscopic identification of ortho-quinones as the products of polycyclic aromatic trans-dihydrodiol oxidation catalyzed by dihydrodiol dehydrogenase. A potential route of proximate carcinogen metabolism. J. Biol. Chem. 1988;263:1814–1820. [PubMed] [Google Scholar]
- (94).Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, Penning TM. Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6846–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Zhang L, Huang M, Blair IA, Penning TM. Detoxication of benzo[a]pyrene-7,8-dione by sulfotransferases (SULTs) in human lung cells. J. Biol. Chem. 2012;287:29909–29920. doi: 10.1074/jbc.M112.386052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Zhang L, Huang M, Blair IA, Penning TM. Interception of benzo[a]pyrene-7,8-dione by UDP glucuronosyltransferases (UGTs) in human lung cells. Chem. Res. Toxicol. 2013;26:1570–1578. doi: 10.1021/tx400268q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Zhang L, Jin Y, Chen M, Huang M, Harvey RG, Blair IA, Penning TM. Detoxication of structurally diverse polycyclic aromatic hydrocarbon (PAH) o-quinones by human recombinant catechol-O-methyltransferase (COMT) via O-methylation of PAH catechols. J. Biol. Chem. 2011;286:25644–25654. doi: 10.1074/jbc.M111.240739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Shultz CA, Quinn AM, Park JH, Harvey RG, Bolton JL, Maser E, Penning TM. Specificity of human aldo-keto reductases, NAD(P)H:quinone oxidoreductase, and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 2011;24:2153–2166. doi: 10.1021/tx200294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Zhang X, D'Agostino J, Wu H, Zhang QY, von Weymarn L, Murphy SE, Ding X. CYP2A13: variable expression and role in human lung microsomal metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. J. Pharmacol. Exp. Ther. 2007;323:570–578. doi: 10.1124/jpet.107.127068. [DOI] [PubMed] [Google Scholar]
- (100).Peterson LA, Mathew RB, Murphy SE, Trushin N, Hecht SS. In vivo and in vitro persistence of pyridyloxobutyl DNA adducts from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1991;12:2069–2072. doi: 10.1093/carcin/12.11.2069. [DOI] [PubMed] [Google Scholar]
- (101).Wang M, Cheng G, Villalta PW, Hecht SS. Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2007;20:1141–1148. doi: 10.1021/tx700189c. [DOI] [PubMed] [Google Scholar]
- (102).Peterson LA, Hecht SS. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- (103).Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2007;20:235–245. doi: 10.1021/tx060207r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Zimmerman CL, Wu Z, Upadhyaya P, Hecht SS. Stereoselective metabolism and tissue retention in rats of the individual enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), metabolites of the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 2004;25:1237–1242. doi: 10.1093/carcin/bgh120. [DOI] [PubMed] [Google Scholar]
- (105).Malik V, Black GW. Structural, functional, and mutagenesis studies of UDP-glycosyltransferases. Adv. Protein Chem. Struct. Biol. 2012;87:87–115. doi: 10.1016/B978-0-12-398312-1.00004-4. [DOI] [PubMed] [Google Scholar]
- (106).IARC Working Group on the Evalutaion of Carcinogenic Risks to Humans . IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 105. IARC Monographs; 2014. Diesel and gasoline engine exhausts and some nitroarenes; pp. 9–699. [PMC free article] [PubMed] [Google Scholar]
- (107).Arlt VM, Sorg BL, Osborne M, Hewer A, Seidel A, Schmeiser HH, Phillips DH. DNA adduct formation by the ubiquitous environmental pollutant 3-nitrobenzanthrone and its metabolites in rats. Biochem. Biophys. Res. Commun. 2003;300:107–114. doi: 10.1016/s0006-291x(02)02789-4. [DOI] [PubMed] [Google Scholar]
- (108).Arlt VM, Stiborova M, Hewer A, Schmeiser HH, Phillips DH. Human enzymes involved in the metabolic activation of the environmental contaminant 3-nitrobenzanthrone: Evidence for reductive activation by NADPH: cyctochrome P450 reductase. Cancer Res. 2003;63:2752–2761. [PubMed] [Google Scholar]
- (109).Arlt VM, Stiborova M, Henderson CJ, Osborne MR, Bieler CA, Frei E, Martinek V, Sopko B, Wolf R, Schmeiser HH, Phillips DH. Environmental pollutant and potent mutagen 3-nitrobenzanthrone forms DNA adducts after reduction by NAD(P)H:Quinone oxidoreductase and conjugation by acetyltransferases and sulfotransferases in human hepatic cytosols. Cancer Res. 2005;65:2644–2652. doi: 10.1158/0008-5472.CAN-04-3544. [DOI] [PubMed] [Google Scholar]
- (110).Delclos KB, El-Bayoumy K, Casciano DA, Walker RP, Kadlubar FF, Hecht SS, Shivapurkar N, Mandal S, Stoner GD. Metabolic activation of 6-nitrochrysene in explants of human bronchus and in isolated rat hepatocytes. Cancer Res. 1989;49:2009–2913. [PubMed] [Google Scholar]
- (111).El-Bayoumy K, Shiue GH, Hecht SS. Comparative tumorigenicity of 6-nitrochrysene and its metabolites in newborn mice. Carcinogenesis. 1989;10:369–372. doi: 10.1093/carcin/10.2.369. [DOI] [PubMed] [Google Scholar]
- (112).Osborne MR, Arlt VM, Kliem C, Hull WE, Mirza A, Bieler CA, Schmeiser HH, Phillips DH. Synthesis, characterization and 32P-post-labeling analysis of DNA adducts derived from the environmental contaminant 3-nitrobenzanthrone. Chem. Res. Toxicol. 2005;18:1056–1070. doi: 10.1021/tx0500474. [DOI] [PubMed] [Google Scholar]
- (113).Murata M, Tezuka T, Ohnishi S, Takamura-Enya T, Hisamatsu Y, Kawanishi S. Carcinogenic 3-nitrobenzanthrone induces oxidative damage to isolated and cellular DNA. Free Radical Biol. Med. 2006;40:1242–1249. doi: 10.1016/j.freeradbiomed.2005.11.015. [DOI] [PubMed] [Google Scholar]
- (114).Stiborova M, Dracinska H, Mizerovska J, Frei E, Schmeiser HH, Hudecek J, Hodek P, Phillips DH, Arlt VM. The environmental pollutant and carcinogen 3-nitrobenzanthrone induces cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase in rat lung and kidney, thereby enhancing its own genotoxicity. Toxicology. 2008;247:11–22. doi: 10.1016/j.tox.2008.01.018. [DOI] [PubMed] [Google Scholar]
- (115).Penning TM, Lerman C. Genomics of smoking gene battery. Cancer Prev. Res. 2008;1:80–83. doi: 10.1158/1940-6207.CAPR-08-0047. [DOI] [PubMed] [Google Scholar]
- (116).Gallego O, Ruiz FX, Ardèvol A, Domínguez M, Alvarez R, de Lera AR, Rovira C, Farrès J, Fita I, Parès X. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20764–20769. doi: 10.1073/pnas.0705659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Reginato MJ, Krakow SL, Bailey ST, Lazar MA. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 1998;273:1855–1858. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- (118).Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63:505–512. [PubMed] [Google Scholar]
- (119).Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ, Ride JP, Viant MR, Gunther UL, Mountford JC, Schrewe H, Green RM, Murray JA, Drayson MT, Bunce CM. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One. 2009;4:e8147. doi: 10.1371/journal.pone.0008147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol. Cell. Endocrinol. 2006;248:126–135. doi: 10.1016/j.mce.2005.10.009. [DOI] [PubMed] [Google Scholar]
- (121).Wang Y, Wang Y, Zhang Z, Park JY, Guo D, Liao H, Yi X, Zheng Y, Zhang D, Chambers SK, Zheng W. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-AKR1C1 pathway. Oncotarget. 2016;7:10363–10372. doi: 10.18632/oncotarget.7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013;137:136–149. doi: 10.1016/j.jsbmb.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Adeniji A, Uddin MJ, Zang T, Tamae D, Wangtrakuldee P, Marnett LJ, Penning TM. Discovery of (R)-2-(6-methoxynaphthalen-2-yl)butanoic acid as a potent and selective aldoketo reductase 1C3 inhibitor. J. Med. Chem. 2016;59:7431–7444. doi: 10.1021/acs.jmedchem.6b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J. Med. Chem. 2012;55:2311–2323. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg. Med. Chem. Lett. 2012;22:3492–3497. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Kikuchi A, Furutani T, Azami H, Watanabe K, Niimi T, Kamiyama Y, Kuromitsu S, Baskin-Bey E, Heeringa M, Ouatas T, Enjo K. In vitro and in vivo characterization of ASP9521: a novel selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydrogenase type 5 (17β-HSD5; AKR1C3) Invest. New Drugs. 2014;32:860–870. doi: 10.1007/s10637-014-0130-5. [DOI] [PubMed] [Google Scholar]
- (127).Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J. Med. Chem. 2013;56:2429–2446. doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Loriot Y, Fizazi K, Jones RJ, Van den Brande J, Molife RL, Omlin A, James ND, Baskin-Bey E, Heeringa M, Baron B, Holtkamp GM, Ouatas T, de Bono JS. Safety, tolerability and anti-tumor activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest. New Drugs. 2014;32:995–1004. doi: 10.1007/s10637-014-0101-x. [DOI] [PubMed] [Google Scholar]
- (129).Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, Narayanan R. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin. Cancer Res. 2013;19:5613–5625. doi: 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- (130).Yin YD, Fu M, Brooke DG, Heinrich DM, Denny WA, Jamieson SMF. The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17β-estradiol production and function in castration resistant prostate cancer and ER-positive breast cancer. Front. Oncol. 2014;4:1–12. doi: 10.3389/fonc.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Chung F-L, Conaway CC, Rao CV, Reddy BS. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis. 2000;21:2287–2291. doi: 10.1093/carcin/21.12.2287. [DOI] [PubMed] [Google Scholar]
- (132).Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Wattenberg LW, Bueding E. Inhibitory effects of 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (Oltipraz) on carcinogenesis induced by benzo[a]pyrene, diethylnitrosamine and uracil mustard. Carcinogenesis. 1986;7:1379–1381. doi: 10.1093/carcin/7.8.1379. [DOI] [PubMed] [Google Scholar]
- (134).Glintborg B, Weimann A, Kensler TW, Poulsen HE. Oltipraz chemoprevention trial in Qidong, People's Republic of China: unaltered oxidative biomarkers. Free Radical Biol. Med. 2006;41:1010–1014. doi: 10.1016/j.freeradbiomed.2006.06.015. [DOI] [PubMed] [Google Scholar]
- (135).Kensler TW, Wakabayashi N. Nrf2 friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Primiano T, Egner PA, Sutter TR, Kelloff GJ, Roebuck BD, Kensler TW. Intermittent dosing with Oltipraz: relationship between chemoprevention of aflatoxin-induced tumorigenesis and induction of glutathione S-transferases. Cancer Res. 1995;55:4319–4324. [PubMed] [Google Scholar]
- (138).Anderson L, Steele VK, Kelloff GJ, Sharma S. Effects of oltipraz and related chemoprevention compounds on gene expression in rat liver. J. Cell. Biochem. 1995;22:108–116. doi: 10.1002/jcb.240590814. [DOI] [PubMed] [Google Scholar]
- (139).Guo Y, Yu S, Zhang C, Kong AN. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radical Biol. Med. 2015;88:337–349. doi: 10.1016/j.freeradbiomed.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Hayes DP. Nutritional hormesis. Eur. J. Clin. Nutr. 2007;61:147–157. doi: 10.1038/sj.ejcn.1602507. [DOI] [PubMed] [Google Scholar]
- (141).Eaton DL, Klassen CD. Principles in Toxicology. In: Klassen CD, editor. Casarett and Doull's Toxicology. McGraw-Hill; New York: 2001. [Google Scholar]