

Abstract
Background
Cerebral cavernous malformations (CCM) are vascular lesions of the central nervous system that can be found in sporadic or autosomal dominantly inherited forms and manifest with headaches, seizures, and hemorrhagic stroke. The precise proportion of de novo mutations in the CCM1,CCM2, and CCM3 genes remains unknown.
Methods
We here present a series of six trios with de novo mutations that have been analyzed by amplicon deep sequencing to differentiate between constitutional and postzygotic mutations.
Results
In one case, allelic ratios clearly indicated mosaicism for a CCM3 splice site mutation found in blood and buccal mucosa of a 2‐year‐old boy with multiple CCMs. The remaining five de novo mutations proved to be constitutional. In addition to three CCM3, two CCM1, and one CCM2 de novo point mutations, a deletion of the entire CCM3 gene was identified in an index case that most likely originated from an early postzygotic event. These are the first high‐level mosaic mutations reported in blood samples of isolated CCM cases.
Conclusion
Our data demonstrate that de novo mutations in CCM1‐3 might be more frequent than previously thought. Furthermore, amplicon deep sequencing is useful to discriminate between patients with constitutional and postzygotic mutations, and thereby improves genetic counseling.
Keywords: CCM1, CCM2, CCM3, cerebral cavernous malformation, de novo mutation, deep sequencing, postzygotic mutation
Introduction
Cerebral cavernous malformations (CCM) are low‐flow vascular lesions that are located in the brain or the spinal cord and occur with a prevalence of about 1:650 in the general population (Morris et al. 2009). The familial form (OMIM 116860, 603284, 603285) accounts for up to 20% of all cases and is inherited in an autosomal dominant manner. Heterozygous loss‐of‐function mutations in CCM1 (KRIT1; OMIM *604214), CCM2 (Malcavernin/OSM; *607929), and CCM3 (PDCD10; *609118) have been associated with CCM.
Due to an impaired function of tight and adherens junctions, CCMs are prone to leakiness and rupture. Clinical symptoms mostly arise from recurrent bleedings and may include headaches, seizures, and hemorrhagic stroke. Nevertheless, CCMs are characterized by an incomplete disease penetrance with up to 30–45% of all patients remaining asymptomatic (Denier et al. 2006). For index cases with multiple CCMs or a positive family history, genetic testing is recommended. With the stepwise use of conventional sequencing and multiplex ligation‐dependent probe amplification (MLPA), high mutation detection rates have been achieved. Following the established criteria, pathogenic variants can be found in up to 87% of familial and 57% of isolated cases (Spiegler et al. 2014). De novo mutations have occasionally been reported previously, but their precise prevalence is still unknown (Bergametti et al. 2005; Liquori et al. 2006; Stahl et al. 2008; Riant et al. 2013; Cigoli et al. 2014).
In the study presented here, next‐generation deep sequencing was used to answer the question whether de novo point mutations identified in six out of 60 isolated CCM cases have arisen as postzygotic mutations or may be present as constitutional variants. To the best of our knowledge, we herein describe the first high‐level mosaicism reported in a CCM patient.
Materials and Methods
Probands and DNA samples
Isolated cases of our previously published (Stahl et al. 2008; Spiegler et al. 2014) and consecutively extended cohort were included in this study (see Table 1). Study design was approved by the ethics committee of the University Medicine Greifswald (registration number: BB 047/14). In addition, all probands participated with written informed consent according to the German Gene Diagnostics Act. Genomic DNA was isolated from blood lymphocytes using NucleoSpin® Blood Kit (Macherey‐Nagel, Düren, Germany) or buccal mucosa using Gentra Puregene Buccal Cell Kit (Qiagen, Hilden, Germany). Paternity was confirmed for all de novo cases using PowerPlex 16 System (Promega, Mannheim, Germany).
Table 1.
De novo mutations found in isolated cases harboring multiple CCMs
| Proband | Mutation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Sex | Age at onset | Clinical symptoms and MRI findings | Reference of previously described cases | Gene | Nucleotide changea | Alternate allele read frequency in ADSb | Z‐score | P‐value after BHC | First description of the mutation |
| P1 | M | 2y | Acute left‐sided hemiparesis due to hemorrhage of a brainstem CCM, multiple supra‐ and infratentorial CCMs on MRI scans | Previously unpublished | CCM3 | c.474+5G>A | 35% (1865×) | −2.86 | 0.026 | Liquori et al. (2006) |
| P2 | F | 1y | Recurrent bleedings of a brainstem CCM with deficits of multiple cranial nerves, two additional supratentorial CCMs | Previously unpublished | CCM2 | c.563_564dupGG | 41% (1700×) | −1.52 | 0.260 | Novel |
| P3 | M | 2y | Multiple CCMs identified as incidental finding on MRI scans of a boy with a mental retardation | Previously unpublished | CCM3 | c.395+1G>A | 46% (287×) | −0.40 | 0.829 | D'Angelo et al. (2011) |
| P4 | M | 14y | Multiple CCMs with epileptic seizures | Spiegler et al. (2014) | CCM1 | c.1660_1678del | 44% (1040×) | −0.84 | 0.597 | Spiegler et al. (2014) |
| P5 | F | 15y | Proximal paresis of the left arm and right‐sided dysesthesia, epileptic seizures, multiple CCMs on MRI scans | Stahl et al. (2008) | CCM1 | c.2143‐2A>G | 46% (418×) | −0.40 | 0.691 | Stahl et al. (2008) |
| P6c | F | 19y | Epileptic seizures | Spiegler et al. (2014) | CCM3 | c.391delA | 55% (93×) | 1.61 | 0.320 | Spiegler et al. (2014) |
| P7 | M | 32y | Obstructive hydrocephalus with headaches and an acute brainstem herniation due to a large CCM of the left cerebellar hemisphere | Previously unpublished | CCM3 | Deletion of the entire gene | n.d. | n.d. | n.d. | Bergametti et al. (2005) |
CCM1 (LRG_650t1; ENST00000394507.5), CCM2 (LRG_664t2; ENST00000258781.10), CCM3 (NM_007217.3; ENST00000392750.6).
Calculated as the percentage of variant reads from the total number of reads (total coverage in brackets); y, year; n.d., not done; BHC, Benjamini–Hochberg correction.
The daughter of P6 also carries the familial CCM3 mutation and presented with multiple, symptomatic CCMs (epileptic seizures and acute CCM hemorrhage) at the age of 12 months.
Mutation analyses
Coding exons and adjacent splice sites of CCM1 (LRG_650t1; ENST00000394507.5), CCM2 (LRG_664t2; ENST00000258781.10), and CCM3 (NM_007217.3; ENST00000392750.6) were sequenced as described elsewhere (Spiegler et al. 2014). DNA mutation numbering is based on cDNA sequence with +1 corresponding to the A of the ATG translation initiation codon (http://varnomen.hgvs.org/). SALSA MLPA Kits P130 & P131 were used for the detection of copy number variations (MRC Holland, Amsterdam, Netherlands).
Digital PCR
Absolute quantification of genomic DNA was assessed using the QuantStudio® 3D Digital PCR System (Thermo Fisher Scientific, Waltham, MA, USA) with an Universal Probe Library assay for CCM3 (Roche, UPL#30, Primer left: 5′‐AGCAGAAGAGGTCTAGGGTCAC‐3′, Primer right: 5′‐CTCCTCTTCCGCCCGTAG‐3′) and the TBP gene as reference (Roche, UPL#3, Primer left: 5′‐CAGCCGTTCAGCAGTCAAC‐3′, Primer right: 5′‐TGTGAGTGGAAGAGCTGTGG‐3′).
Deep sequencing and data analysis
Genomic regions covering de novo mutations were PCR‐amplified with the high‐fidelity PrimeSTAR GXL (Takara Clontech, Saint‐Germain‐en‐Laye, France) or KAPA HiFi polymerase (Kapa Biosystems, London, UK). Primer sequences are available upon request. Following purification of the amplicons with Agencourt® AMPure® XP beads (Beckman Coulter, Pasadena, CA, USA), Nextera XT Kit was used for library preparation (Illumina®, San Diego, CA, USA).
Target enrichment of all exons of CCM1‐3 using a custom‐made HaloPlex panel (Agilent, Santa Clara, CA, USA) was performed as previously described (Kohda et al. 2016). All libraries were sequenced on a MiSeq sequencer with 2 × 250 cycles using Reagent Kit v3 (Illumina®). The SeqNext software (JSI Medical Systems, Ettenheim, Germany) was used for read mapping to the reference genome hg19 and variant calling. Z‐scores for allelic ratios of variants were calculated as previously described (Acuna‐Hidalgo et al. 2015). Statistical significance was assumed with a critical value of P < 0.05 after Benjamini–Hochberg correction.
Results
Characteristics of isolated CCM cases
Since 2005, a total of 60 isolated cases with multiple CCMs were analyzed (Stahl et al. 2008; Spiegler et al. 2014). Conventional Sanger sequencing and MLPA analysis identified pathogenic variants in 34 individuals (56%). Despite our best efforts, DNA samples of both parents were available for only eight of all mutation‐positive probands as many of the asymptomatic parents were unavailable or had decided against predictive testing. De novo mutations could be verified in six cases based on conventional sequencing of parental lymphocyte DNA (P1–P6, Table 1). Three CCM3, two CCM1, and one CCM2 de novo point mutations were identified. Nonpaternity was excluded for these cases. Either one parent of further two probands proved to be an asymptomatic carrier of a familial mutation (data not shown).
The clinical symptoms of de novo CCM cases varied from multiple asymptomatic CCMs identified as incidental finding in a 2‐year‐old boy with global mental retardation to acute neurological deficits caused by recurrent bleedings (age of onset: 1–32 years, Table 1). The mean age of the parents at birth of the index probands was 31.8 years (maternal range: 24–38 years, paternal: 20–39).
Amplicon deep sequencing (ADS) identified a postzygotic CCM3 mutation
The DNA samples isolated from blood lymphocytes of each de novo case were reanalyzed by ADS to distinguish between constitutional and postzygotic mutations after cut‐off values for allelic ratios of constitutional variants had been established (lower cut‐off ADS: 0.39; Fig. S1). The mean target sequencing depth was 900× (min: 93×; max: 1865×). Allelic read ratios (range: 41–55%) indicated constitutional status in five of the six de novo cases (P2–P6, Table 1). The respective mutation could not be found in any of the parental blood samples by ADS (Table S1).
However, the variant allelic read ratios in ADS (coverage: 1865×) and HaloPlex enrichment (778×) for the CCM3 splice mutation found in the lymphocyte DNA of case P1 (c.474+5G>A) showed a significant deviation from the expected value of a constitutional variant (ADS: 35%, Z‐score = −2.86; HaloPlex: 33%, Z‐score = −3.18; Fig. 1B,D and Table 1). ADS of DNA isolated from buccal mucosa also indicated mosaicism (Fig. 1D). These results were reproducible in independent library preparations and sequencing runs. Different PCR primers were used to exclude a possible amplification bias. The identified sequence variant had previously been described in the literature as pathogenic (Liquori et al. 2006; Shenkar et al. 2015) and the suspected splice defect was verified by Liquori et al. using transcript analysis (Liquori et al. 2006). The mutation was excluded in both parental blood samples by deep sequencing using HaloPlex enrichment (Fig. 1A,C).
Figure 1.
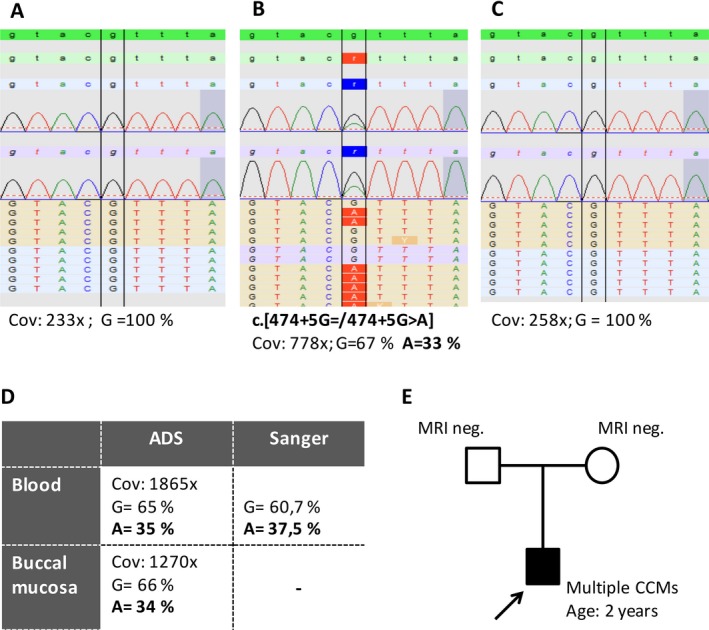
Mosaic CCM3 splice site mutation (NM_007217.3; ENST00000392750.6) identified in P1. Lymphocyte DNA of the father (A), index (B), and mother (C) were used for HaloPlex target enrichment. Pseudo‐electropherograms representing alignments in SeqNext software are depicted. ADS and Sanger sequencing of lymphocyte DNA and/or DNA from buccal mucosa of the index case showed comparable results (D). The pedigree of the family is shown in (E). Cov = coverage; MRI = magnetic resonance imaging; neg. = negative.
Postzygotic deletion of the entire CCM3 gene
If parents are not available for genetic testing, results of molecular analyses have to be interpreted with even greater care. Using lymphocyte DNA, slightly decreased allele dosages (mean ratio after normalization: 0.69) below the lower cut‐off value (<0.75) were observed for all CCM3‐specific probes in MLPA analyses of case P7 (Fig. 2). These results were reproducible using independent blood as well as buccal mucosa samples and could be verified in different laboratories (data not shown). As parental blood samples were not available to confirm the suspected de novo event, a digital PCR assay was applied to determine the CCM3 allele dosage with a second method (Fig. 2C). Equal DNA amounts were used for the TBP gene (TATA box‐binding protein) as reference and for positive and negative controls. Taken together, these results led to the assumption of mosaicism in case P7 with a fraction of about 30% of all CCM3 alleles carrying the deletion.
Figure 2.
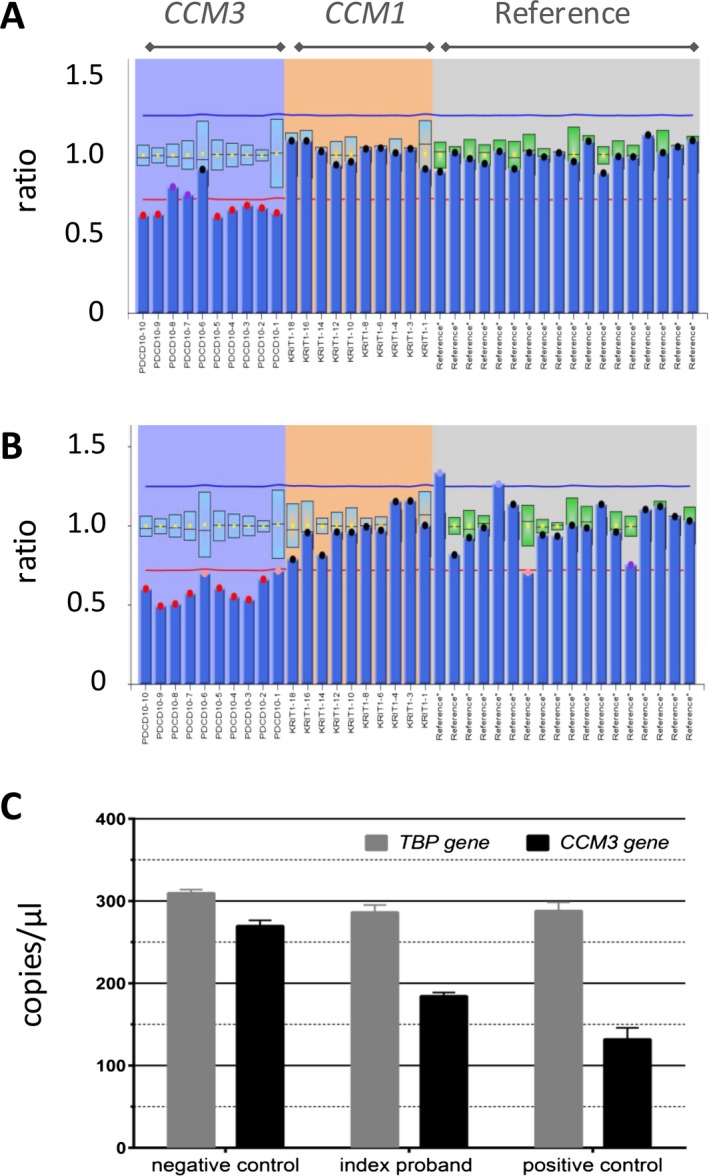
CCM3 deletion (NM_007217.3; ENST00000392750.6) identified by MLPA analysis in lymphocyte DNA (A) and buccal mucosa DNA (B) of case P7. The height of the columns represents the dosage of the segments. The allele dosages of the deleted probes were hardly below the cut‐off value (<0.75) in the blood. Absolute quantification using digital PCR also implicated mosaicism (C). A carrier of a familial CCM3 deletion and a mutation‐negative proband served as controls.
Discussion
Twenty CCM3 (Bergametti et al. 2005; Liquori et al. 2006; Riant et al. 2013; Cigoli et al. 2014; Shenkar et al. 2015) and two CCM1 de novo mutations (Lucas et al. 2001; Stahl et al. 2008) have previously been described in isolated CCM cases. Notably, one of these led to the identification of CCM3 as disease‐associated gene (Bergametti et al. 2005). However, the prevalence of de novo mutations may still be underestimated since parents are not always available for genetic testing. The relatively high number of three CCM3, two CCM1, and one CCM2 de novo point mutations as well as one additional postzygotic CCM3 deletion in our series of isolated CCM cases (7/60; 11.6%) indicates that the prevalence of de novo events may be higher than previously thought. The predominance of CCM3 de novo mutations that have previously been reported or newly identified in our cohort may also imply that CCM3 is more prone to de novo mutational events. Interestingly, CCM3 mutations are known to be associated with an earlier age of manifestation and a higher risk of hemorrhages (Riant et al. 2013; Spiegler et al. 2014; Shenkar et al. 2015). Aside from constitutional variants, somatic mutations in cavernous lesions of isolated CCM probands have been reported (McDonald et al. 2014). These are hardly ever detectable by conventional sequencing of lymphocyte DNA.
The interpretation of molecular diagnostics for isolated CCM cases can be challenging. In the context of genetic counseling, the identification of low‐level mutations as well as the discrimination between high‐level mosaicism and constitutional mutations is important to give a reliable risk assessment for CCM patients and their families. In accordance with previous reports (Jamuar et al. 2014), our data demonstrate that for this discrimination, the specificity of conventional sequencing is insufficient in most cases. Ultradeep sequencing might be useful for this purpose. Nevertheless, the cut‐off values of the expected allelic ratios for true heterozygous variants have to be carefully established with a known set of germline mutations for the respective technique (Acuna‐Hidalgo et al. 2015) (Fig. S1). Using NGS of DNA from blood lymphocytes and buccal mucosa, we have identified a likely mosaic mutation (P1). As comparable ratios of the alternate allele were found in the derivatives from different germ layers (mesoderm and ectoderm), an early postzygotic event rather than a selection against mutant cells in the blood (Huisman et al. 2013) was assumed. Interestingly, the CCM3 mutation had previously been reported as a de novo mutation in an independent CCM case (Liquori et al. 2006).
No significant difference in the phenotype (age of manifestation, bleeding history, number of CCMs) was observed upon comparison of de novo cases carrying constitutional mutations (P2–P6) and postzygotic mutations (P1, P7). Although the mutations were not identified in any of the parental blood samples of constitutional mutation carriers, gonadal mosaicism cannot be excluded and has to be considered.
In conclusion, we recommend a stepwise approach for obvious de novo mutations. In the first step, amplicon deep sequencing should be applied for the index case to search for postzygotic mutations, because the recurrence risk for the parents and the possibility of siblings to be a mutation carrier as well are negligible in these cases. As it is well known that the majority of constitutional de novo mutations originate in the parental germline, high‐sensitivity ultradeep sequencing of parental blood and maybe even of paternal sperm samples might be warranted for the future as a second step to distinguish between low level and gonadal mosaicism.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Distribution of alternate allele read ratios of inherited heterozygous variants identified by amplicon deep sequencing (A) or HaloPlex enrichment (B).
Table S1. Deep sequencing performed for parents of index cases with de novo mutations.
Acknowledgments
The authors thank all patients and their families for their cooperation. C. Sperling, S. Göbel, and K. Hartwig are thanked for their excellent technical assistance. We thank Dr. Marcus Vollmer (Institute of Bioinformatics, University Medicine Greifswald) for fruitful discussion on statistical analysis. S. S. and U. F. are funded by a grant from the German Research Foundation (DFG, Grant No FE432/9‐1). This work was also supported by the Research Network Molecular Medicine of the University Medicine Greifswald (Grant No FOCM‐2014‐04).
References
- Acuna‐Hidalgo, R. , Bo T., Kwint M. P., van de Vorst M., M. Pinelli , Veltman J. A., et al. 2015. Post‐zygotic point mutations are an underrecognized source of de novo genomic variation. Am. J. Hum. Genet. 97:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergametti, F. , Denier C., Labauge P., Arnoult M., Boetto S., Clanet M., et al. 2005. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigoli, M. S. , Avemaria F., De Benedetti S., Gesu G. P., Accorsi L. G., Parmigiani S., et al. 2014. PDCD10 gene mutations in multiple cerebral cavernous malformations. PLoS ONE 9:e110438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo, R. , Marini V., Rinaldi C., Origone P., Dorcaratto A., Avolio M., et al. 2011. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol. 21:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier, C. , Labauge P., Bergametti F., Marchelli F., Riant F., Arnoult M., et al. 2006. Genotype‐phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 60:550–556. [DOI] [PubMed] [Google Scholar]
- Huisman, S. A. , Redeker E. J., Maas S. M., Mannens M. M., and Hennekam R. C.. 2013. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J. Med. Genet. 50:339–344. [DOI] [PubMed] [Google Scholar]
- Jamuar, S. S. , Lam A. T., Kircher M., D'Gama A. M., Wang J., Barry B. J., et al. 2014. Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 371:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda, M. , Kumamoto K., Eguchi H., Hirata T., Tada Y., K. Tanakaya , et al. 2016. Rapid detection of germline mutations for hereditary gastrointestinal polyposis/cancers using HaloPlex target enrichment and high‐throughput sequencing technologies. Fam. Cancer 15:553–562. [DOI] [PubMed] [Google Scholar]
- Liquori, C. L. , Berg M. J., Squitieri F., Ottenbacher M., Sorlie M., Leedom T. P., et al. 2006. Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus. Hum. Mutat. 27:118. [DOI] [PubMed] [Google Scholar]
- Lucas, M. , Costa A. F., Montori M., Solano F., Zayas M. D., and Izquierdo G.. 2001. Germline mutations in the CCM1 gene, encoding Krit1, cause cerebral cavernous malformations. Ann. Neurol. 49:529–532. [PubMed] [Google Scholar]
- McDonald, D. A. , Shi C., Shenkar R., Gallione C. J., Akers A. L., Li S., et al. 2014. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 23:4357–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, Z. , Whiteley W. N., Longstreth W. T. Jr., Weber F., Lee Y. C., Tsushima Y., et al. 2009. Incidental findings on brain magnetic resonance imaging: systematic review and meta‐analysis. BMJ 339:b3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riant, F. , Bergametti F., Fournier H. D., Chapon F., Michalak‐Provost S., Cecillon M., et al. 2013. CCM3 mutations are associated with early‐onset cerebral hemorrhage and multiple meningiomas. Mol. Syndromol. 4:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkar, R. , Shi C., Rebeiz T., Stockton R. A., McDonald D. A., Mikati A. G., et al. 2015. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet. Med. 17:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegler, S. , Najm J., Liu J., Gkalympoudis S., Schroder W., Borck G., et al. 2014. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one‐third of probands are minors. Mol. Genet. Genomic Med. 2:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, S. , Gaetzner S., Voss K., Brackertz B., Schleider E., O. Surucu , et al. 2008. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in‐frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum. Mutat. 29:709–717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of alternate allele read ratios of inherited heterozygous variants identified by amplicon deep sequencing (A) or HaloPlex enrichment (B).
Table S1. Deep sequencing performed for parents of index cases with de novo mutations.