

Abstract
An enzyme-mediated approach for the assembly of singly modified RNA constructs in which specific G residues are replaced with thG, an emissive isomorphic G surrogate, is reported. Transcription in the presence of thG and native nucleoside triphosphates enforces initiation with the unnatural analogue, yielding 5′-end modified transcripts that can be mono-phosphorylated and ligated to provide longer site-specifically modified RNA constructs. The scope of this unprecedented enzymatic approach to non-canonical purine-containing RNAs is explored via the assembly of several altered hammerhead (HH) ribozymes and a singly modified HH substrate. By strategically modifying key positions, a mechanistic insight into the ribozyme-mediated cleavage is gained. Additionally, the emissive features of the modified nucleoside and its responsiveness to environmental changes can be used to monitor cleavage in real time by steady state fluorescence spectroscopy.
Keywords: fluorescence, hammerhead ribozyme, nucleosides, RNA, transcription initiation
Graphical Abstract
An enzyme-mediated approach is used for the assembly of singly modified RNA constructs in which specific G residues are replaced with thG, an emissive isomorphic G surrogate. By strategic modifications, a mechanistic insight into the ribozyme-mediated cleavage is gained. Emissive features of the modified nucleoside and responsiveness to environmental changes are used to monitor cleavage in real time by steady-state fluorescence spectroscopy.
Transcription initiation is a critical step in gene expression. In eukaryotes and prokaryotes it requires a specific promoter sequence and a hierarchical assembly of specific polymerases and accessory proteins and has been thought to have a limited tolerance for the initiating nucleotides.[1] Recent observations have suggested, however, that non-canonical nucleotides can serve as initiators in bacterial transcription, yielding unusual 5′-capped structures.[2] While significant, academic and industrial researchers still rely on phage-based polymerases (e.g., T7 RNA pol) for producing and studying RNA. The ability of such enzymes to initiate transcription with non-canonical nucleosides and nucleotides, and in particular with unnatural analogues, remains rather unexplored. Herein we investigate transcription initiation with thG, an isomorphic G surrogate. Along with fundamental observations showing the tolerance of the enzyme to such non-native purine mimics, the unique structural and photophysical features of this nucleoside can be exploited for mechanistic and spectroscopic studies, respectively.
We have previously introduced thG, an isomorphic guanosine analogue, displaying favorable biophysical, biochemical, and photophysical features (Figure 1).[3] We demonstrated that thG triphosphate (thGTP), as a GTP surrogate, successfully facilitated initiation of in vitro transcription reactions and elongation of the growing transcripts catalyzed by T7 RNA polymerase.[4] In the resulting transcripts all guanosine residues were replaced with thG. When applied to the minimal hammerhead (HH) ribozyme HH16, a small functional RNA, the impact of individual residues on catalysis was obscured.[4]
Figure 1.

An enzyme-mediated approach for the assembly of singly modified RNA constructs, replacing a G residue with thG.
To facilitate refined mechanistic studies of functional RNAs and the role of individual G residues, we disclose an enzyme-mediated approach for the assembly of singly modified RNA constructs in which specific G residues are judiciously replaced with thG (Figure 1). It relies on transcription in the presence of excess thG and native nucleoside triphosphates, which enforces initiation with the unnatural analogue. The resulting 5′-end modified transcripts are then mono-phosphorylated and ligated to provide longer site-specifically modified RNA constructs (Figure 1).[5] We further exploit the emissive features of thG and its responsiveness to environmental changes and demonstrate that cleavage can be monitored in real time by steady-state fluorescence spectroscopy. The observations reported herein illustrate the accommodation of thG by three distinct enzymes, namely a polymerase, a kinase, and a ligase, and provide a method that can be exploited to address questions in mechanistic RNA biochemistry and for the assembly of fluorescence-based assays for RNA structure/function.
Transcription initiation with thG was first explored with a model template 1, thG, all native NTPs, and T7 RNA polymerase (Figure 2a).[4,6] thG-initiated RNA constructs and the unmodified native products were successfully separated using PAGE (Figure 2b, lane 2, indicated by the white arrow). UV illumination (302 nm) visualizes the product and truncated transcripts, which are all highly fluorescent (Figure 2b). The purified full-length thG initiated transcript (1b) and native transcript (1a) were quantified by measuring their UV absorption (λ = 260 nm), as described in Section S2.1 in the Supporting Information, and analyzed by mass spectrometry (Figure 2c). The relative transcript yield (1b/1a) was 0.55 ± 0.02.
Figure 2.

Transcription reactions with template 1. a) T7 promoter and template 1 depicting the enzymatic incorporation reaction using natural NTPs with or without the presence of thG resulting in different transcripts. b) Transcription reaction using template 1 with 1 mM of all natural NTPs (lane 1 and 1′), 1 mM of all natural NTPs and 5 mM of thG (lanes 2 and 2′). The white arrow indicates transcript 1b (in lane 2). UV shadowing was observed at 254 nm, photoluminescence (PL) was observed at 302 nm. c) MALDI analysis of transcript 1 b.
To identify the optimal thG/GTP ratio for the production of thG-initiated full-length transcripts, the concentration of thG was varied (1–13 mM), while keeping the concentration of Mg2+ and all native NTPs, including GTP, constant (1 mM). The relative yield of transcript 1b increased from 0.29 to 1.09 as thG concentrations were elevated, while the total yield of RNA remained comparable (Supporting Information, Figure S1). Scaled-up transcription reactions of template 1 were therefore carried out with 5 mM of thG. We note, however, that the optimal conditions for thG-initiated transcription reactions were template-dependent.
Our previous studies showed that the activity of the fully modified HH16 ribozyme, containing 13 thG residues, was severely diminished, suggesting that the substitution of G for thG interferes with either folding and/or catalysis.[4] The impact of specific residues could not be assessed. The method described herein, facilitating single-site modification, allows this very fundamental question to be probed. We therefore focused on G residues within the catalytic core, including G8, G10.1, G12, which have been demonstrated to be important for the cleavage reaction (Figure 3).[7] We also replaced G11.4, which is part of helix II, and thus is not directly involved in the catalytic process, with thG. To facilitate the preparation of internally, singly modified HH16 enzymes, thG-terminated oligonucleotides have been synthesizes as described above and then phosphorylated and ligated to the corresponding unmodified oligonucleotides (Supporting Information, Figures S2–S5). The ligation reactions were found to be effective, providing the desired HH enzymes in 20–40% yield (Supporting Information, Figure S6). All the thG-terminated donor strands and ligated oligonucleotides were characterized by mass spectrometry (Supporting Information, Figures S7–S16), and E5 was also digested using S1 nuclease and dephosphorylated before being subjected to HPLC analysis (Supporting Information, Figure S17a), confirming the presence and stoichiometry of the modified intact nucleoside. To validate the position of modifications, enzymatic digestion with T1 nuclease, which is an N-7 dependent RNase that cleaves single-stranded RNA 3′ to G residues,[8] was also applied to all the modified HH16 enzyme strands (Supporting Information, Figures S17b, S18). A comparison of the T1 RNase cleavage pattern obtained for the native RNA E1 and the singly modified E5 shows a footprint at position 12, where thG replaces G (compare lanes 3 and 6 in the Supporting Information, Figure S17b), thus further substantiating the presence of thG at this position.
Figure 3.

Hammerhead ribozymes and representation of cleavage reactions of a natural substrate (S1) with native (E1), thG8 (E2), thG10.1 (E3), thG11.4 (E4), and thG12 (E5) enzymes. The enzyme—substrate association step (typically characterized as k1) is not shown.
Hammerhead ribozymes with thG-modified enzymes (E2–E5) were assembled with a native 32P-labeled substrate S1 (Figure 3), and tested for strand cleavage (Supporting Information, Figure S19), using conditions similar to those previously published.[4,9] The initial rate constants obtained were 0.13 ± 0.02, 0.13 ± 0.01, and 0.47 ± 0.02 min−1, for E5-S1, E1-S1, and E4-S1, respectively (Figure 4a). However, the cleavage of S1 by E2 and E3 was significantly prohibited (Figure 4b), with only 4.9 ± 0.6% and 18.4 ± 0.8%, respectively, observed after 40 minutes (Table 1).
Figure 4.

Cleavage of 32P-labeled S1 by of HH enzyme strands with replacement of G for thG at different positions. a) Initial kinetics of S1-E1 (black), S1-E5 (blue) and S1-E4 (green). The pseudo first-order rate constants (k2) of the cleavage reactions are determined as the slope of semi-logarithmic plot of the fraction cleaved (S/S0) as function of time. b) Ribozyme-mediated cleavage curves as determined by 32P data for S1-E1 (black), S1-E2 (orange), S1-E3 (purple) S1-E4 (green), and S1-E5 (blue). Fraction cleaved (S/S0) was determined by dividing the amount of cleaved substrate by the sum of the full length and cleaved substrate.
Table 1.
Cleavage data for HH ribozymes.
| E1-S1 | E2-S1 | E3-S1 | E4-S1 | E5-S1 | |
|---|---|---|---|---|---|
| k2[a] | 0.13 ± 0.01 | n.d. | n.d. | 0.47 ± 0.02 | 0.13 ± 0.02 |
| S/S0 (20 min)[b] | 0.90 ± 0.01 | 0.03 ± 0.01 | 0.10 ± 0.01 | 0.89 ± 0.02 | 0.74 ± 0.06 |
| S/S0 (40 min)[b] | – | 0.05 ± 0.01 | 0.18 ± 0.01 | – | – |
k2 is the pseudo-first-order rate constant [min−1] and it is equal to the slope of the semi-logarithmic plot in Figure 4a.
Fraction cleaved (S/S0) was determined after 20 and/or 40 minutes by dividing the amount of cleaved substrate (S) by the sum of the full length and cleaved substrate (S0).
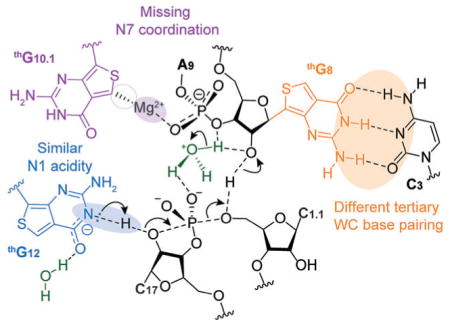
As articulated below, some observations can be straightforwardly rationalized, while others appear more challenging to explain, suggesting a rather high susceptibility of certain HH16 positions to nucleobase alterations. In the proposed ribozyme-mediated cleavage mechanism,[7] a divalent metal ion, which plays a key role in stabilizing the tertiary structure of the folded HH ribozyme, coordinates G10.1 through its N7, which is missing in thG. This could therefore explain the low activity of E3 (Figure 5).
Figure 5.

Representation of the HH cleavage reaction. The putative role and interactions of each thG nucleotide are highlighted.
The HH ribozyme has been proposed to undergo a conformational rearrangement, which requires an optimal combination of sufficient stability and conformational flexibility of the stem-loop II.[10,11] Previous studies have shown that improved cleavage rate could be obtained by specific mutations at stem-loop II of HH ribozymes.[7,12] It is plausible that differences in aromaticity and hydrophobic characters of thG in comparison to G affect the dynamics of E4, favoring its catalytically active conformations, leading to its high cleavage potency.
Interestingly, E5 cleaves the native substrate at a comparable rate to that of E1, the native enzyme (Figure 4a, b). The nucleobase of G12 is involved in the cleavage reaction where the putatively deprotonated N1 acts as a base to deprotonate the 2′-OH of C17, which then attacks the adjacent 3′ phosphate, leading to strand cleavage (Figure 5). Previously reported theoretical calculations and our experimental determination (Supporting Information, Figure S20) suggest high similarity between the acidity of N1H in thG (Supporting Information, Table S1) compared to the native G (pKa 10.1 and 9.2–9.6 respectively).[13,14] Thus, the HH ribozyme cleavage process is not significantly impacted by the replacement of G12 with thG, a synthetic guanosine surrogate.
Perhaps surprisingly, the substitution of G8 with thG in E2 severely reduces the cleavage rate of the native substrate strand, although only the ribose of this nucleotide has been proposed to be directly involved in catalysis.[7] We speculate that substituting the invariant G8 for thG might subtly impact the tertiary WC base-pairing with the invariant C3. This tertiary pair appears to be the Achilles heel of HH16, as any modification significantly diminishes cleavage, and even the “compensatory” G·C to C·G double mutation has been shown to only partially restore activity.[15,16] Hammann and coworkers have indicated that tolerance to the exchange of WC base pair between position 3 and 8 depends on the respective sequence context.[17] While thG has been shown to form highly stable WC pairs,[3a] tertiary pairing has not yet been explored. Inferring from the elevated stability of thG·C vs. G·C in duplexes, which is likely due to the higher stackability of the former, such a replacement may again impact the dynamics of the HH16 fold, thus impacting its active conformer accessibility.[16] It is plausible that minor structural (N vs. C nucleoside) and stability differences between the modified and native base pairs lowers the population of the active conformation.[16,17]
To explore the impact of incorporating thG into the substrate at the cleavage site (position 1.1 in Figure 3), modified substrates with either thG [thG1.1-S (S3)] or guano-sine [G1.1-S (S2)] residues replacing A1.1 in S1, were prepared, 32P labeled and hybridized to E6, a modified enzyme containing a complementary C at position 2.1 (Supporting Information, Figure S21). No major differences between the cleavage rates of E6-S2 and E6-S3 were seen (0.11 ± 0.01 and 0.08 ± 0.01 min−1, respectively; Supporting Information, Figure S22a, b), indicating that the cleavage reaction was not significantly impacted by replacing a native G1.1 with thG, even at the cleavage site of the ribozyme.
The cleavage reaction of E6-S3 (k2 = 0.08 ± 0.01 min−1) was also followed with a nonradiolabeled substrate by monitoring emission changes under the same experimental conditions as for the radiolabeled constructs but in a slightly larger scale (Figure 6b; Supporting Information, Figure S22b). The initial increased fluorescence intensity (Figure 6a; Supporting Information, Figure S23) was likely due to the cleavage of S3, which releases product 3 from the duplex with thG1.1 at its 5′-terminus. Good agreement between the radioactively monitored HH reaction and the fluorescence-monitored one was seen (Figure 6b).[9b]
Figure 6.

a) Ribozyme-mediated cleavage curves as determined by 32P data for E6 with S2 (black) and with S3 (red). Fraction cleaved (S/S0) was determined by dividing the amount of cleaved substrate by the sum of the full length and cleaved substrate. b) Ribozyme-mediated cleavage curves as determined by 32P data (red) as well as fluorescence spectroscopy (cyan). See the Supporting Information for additional details.
The results described above reflect the intricate molecular interactions involved in a ribozyme-mediated cleavage (Supporting Information, Figure S23). We are cognizant of the fact that any substitution of a native residue for a synthetic one, regardless how isomorphic the modification might be, impacts multiple molecular and supramolecular features (as a result of different H bonding strengths, pKa values, stackability, and so on). Yet, while one might question the actual insight gained by such isomorphic replacement, we stress that a HH16 substrate, where thG replaces G at the cleavage site (S3), undergoes the expected cleavage reaction at essentially the same rate as native RNA. While this thG-containing substrate can be used to monitor the reaction by fluorescence, avoiding 32P labeling, it serves as a critical control illustrating the true functionality of this G surrogate. This, in turn, suggests, that the differences seen in the various thG-modified enzymes, while not always fully decipherable, indeed reflect subtle molecular features that impact the dynamics and conformation of the ribozyme.
Site-specific modification has served as a powerful tool for probing structure, function and mechanisms of biologically-relevant RNA.[18] Owing to the somewhat relaxed restriction of the 5′-triphosphate of the initiation nucleotide during transcription, some A or G analogues that are functionalized through 5′-monophosphate have been developed to initiate transcription (Supporting Information, Table S2).[19,20] All other currently known initiator molecules generate transcripts with blocked 5′-end and cannot be further ligated.[21] Some dinucleotides, usually composed of GMP residue linked to a modified nucleotide, have also been introduced to favor the initiation step,[22] though the additional nucleotide limits their utility. To the best of our knowledge, enzymatic transcription reactions initiated with a nucleoside possessing a modified nucleobase with a free 5′-hydroxy have not yet been reported. Our results show that thG, as a free nucleoside, can initiate in vitro transcription reactions, thus generating 5′-end modified RNA constructs. These emissive strands can be easily phosphorylated and ligated to provide longer internally modified RNAs, illustrating the accommodation of thG by three commonly used and distinct enzymes. The modified RNAs containing single thG substitution, in addition to potentially serving as fluorescence probes, can be exploited to provide mechanistic insight into RNA folding and dynamics as well as into the impact of specific G residues on RNA function and recognition.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for generous support (GM 069773) and the Chemistry and Biochemistry MS Facility. We are greatly indebted to Professor Venkat Gopalan (Ohio State University) for his insight, suggestions, and encouragement.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201609327.
Contributor Information
Yao Li, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358 (USA).
Dr. Andrea Fin, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358 (USA)
Dr. Lisa McCoy, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358 (USA)
Prof. Yitzhak Tor, Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358 (USA)
References
- 1.a) Reoder RG. Trends Biochem Sci. 1996;21:327–335. [PubMed] [Google Scholar]; b) Kadonaga JT, Jones KA, Tijian R. Trends Biochem Sci. 1986;11:20– 23. [Google Scholar]
- 2.Bird JG, Zhang Y, Tian Y, Panova N, Barvik I, Greene L, Liu M, Buckley B, Krasny L, Lee KK, Kaplan CD, Ebright RH, Nickels BE. Nature. 2016;535:444– 447. doi: 10.1038/nature18622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Shin D, Sinkeldam RW, Tor Y. J Am Chem Soc. 2011;133:14912–14915. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu W, Shin D, Tor Y, Cooperman BS. ACS Chem Biol. 2013;8:2017–2023. doi: 10.1021/cb400256h. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sinkeldam RW, McCoy LS, Shin D, Tor Y. Angew Chem Int Ed. 2013;52:14026–14030. doi: 10.1002/anie.201307064. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:14276–14280. [Google Scholar]; d) Mizrahi RA, Shin D, Sinkeldam RW, Phelps KJ, Fin A, Tantillo DJ, Tor Y, Beal PA. Angew Chem Int Ed. 2015;54:8713– 8716. doi: 10.1002/anie.201502070. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2015;127:8837–8840. [Google Scholar]; e) Sholokh M, Sharma R, Shin D, Das R, Zaporozhets OA, Tor Y, Mély Y. J Am Chem Soc. 2015;137:3185–3188. doi: 10.1021/ja513107r. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sholokh M, Improta R, Mori M, Sharma R, Kenfack C, Shin D, Voltz K, Stote RH, Zaporozhets OA, Botta M, Tor Y, Mély Y. Angew Chem Int Ed. 2016;55:7974– 7978. doi: 10.1002/anie.201601688. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2016;128:8106– 8110. [Google Scholar]
- 4.McCoy LS, Shin D, Tor Y. J Am Chem Soc. 2014;136:15176– 15184. doi: 10.1021/ja5039227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang K, Micura R. Nat Protoc. 2008;3:1457– 1466. doi: 10.1038/nprot.2008.135. [DOI] [PubMed] [Google Scholar]
- 6.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Nucleic Acids Res. 1987;15:8783– 8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martick M, Lee TS, York DM, Scott GW. Chem Biol. 2008;15:332– 342. doi: 10.1016/j.chembiol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pace CN, Heinemann U, Hahn U, Saenger W. Angew Chem Int Ed Engl. 1991;30:343– 360. [Google Scholar]; Angew Chem. 1991;103:351–369. [Google Scholar]
- 9.a) Hertel KJ, Herschiag D, Uhlenbeck OC. Biochemistry. 1994;33:3374–3385. doi: 10.1021/bi00177a031. [DOI] [PubMed] [Google Scholar]; b) Kirk SR, Luedtke NW, Tor Y. Bioorg Med Chem. 2001;9:2295– 2301. doi: 10.1016/s0968-0896(01)00123-7. [DOI] [PubMed] [Google Scholar]
- 10.Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Biochemistry. 1999;43:14363– 14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 11.a) Long DM, Uhlenbeck OC. Proc Natl Acad Sci USA. 1994;91:6977–6981. doi: 10.1073/pnas.91.15.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Persson T, Hartmann RK, Eckstein F. ChemBioChem. 2002;3:1066– 1071. doi: 10.1002/1439-7633(20021104)3:11<1066::AID-CBIC1066>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 12.a) Burgin AB, Jr, Gonzalez C, Matulic-Adamic J, Karpeisky AM, Usman N, McSwiggen JA, Beigelman L. Biochemistry. 1996;35:14090–14097. doi: 10.1021/bi961264u. [DOI] [PubMed] [Google Scholar]; b) Clouet-d’Orva B, Uhlenbeck OC. Biochemistry. 1997;36:9087– 9092. doi: 10.1021/bi9710941. [DOI] [PubMed] [Google Scholar]
- 13.a) Bundari S. The Merck Index. 12. Merck and Co., Inc; Whitehouse Station, NJ: 1996. [Google Scholar]; b) Sigel H, Massoud SS, Corfù NA. J Am Chem Soc. 1994;116:2958–2971. [Google Scholar]; c) Kampf G, Kapinos LE, Griesser R, Lippert B, Sigel H. J Chem Soc Perkin Trans 2. 2002:1320–1327. [Google Scholar]; d) Thapa B, Schlegel HB. J Phys Chem A. 2015;119:5134– 5144. doi: 10.1021/jp5088866. [DOI] [PubMed] [Google Scholar]
- 14.a) Samanta PK, Manna AK, Pati SK. J Phys Chem B. 2012;116:7618–7626. doi: 10.1021/jp301752k. [DOI] [PubMed] [Google Scholar]; b) Lee YJ, Jang YH, Kim Y, Hwang S. Bull Korean Chem Soc. 2012;33:4255–4257. [Google Scholar]; c) Samanta PK, Pati SK. New J Chem. 2013;37:3640–3646. [Google Scholar]; d) Gedik M, Brown A. J Photochem Photobiol A. 2013;259:25–32. [Google Scholar]; e) Samanta PK, Pati SK. Phys Chem Chem Phys. 2015;17:10053– 10058. doi: 10.1039/c5cp00134j. [DOI] [PubMed] [Google Scholar]
- 15.Martick M, Scott GW. Cell. 2006;126:309– 320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson JA, Uhlenbeck OC. RNA. 2008;14:43– 54. doi: 10.1261/rna.717908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Przybilski R, Hammann C. RNA. 2007;13:1625– 1630. doi: 10.1261/rna.631207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Allerson CR, Chen SL, Verdine GL. J Am Chem Soc. 1997;119:7423–7433. [Google Scholar]; b) Earnshaw DJ, Gait MJ. Biopolymers. 1998;48:39–55. [Google Scholar]; c) Silverman SK, Cech TR. Biochemistry. 1999;38:14224–14237. doi: 10.1021/bi991333f. [DOI] [PubMed] [Google Scholar]; d) Strobel SA. Curr Opin Struct Biol. 1999;9:346–352. doi: 10.1016/S0959-440X(99)80046-3. [DOI] [PubMed] [Google Scholar]; e) Vrma S, Vaish NK, Eckstein F. In: RNA. Söll D, Nishimura S, Moore P, editors. Elsevier; Amsterdam: 2001. pp. 259–275. [Google Scholar]; f) Chow CS, Mahto SK, Lamichhane TN. ACS Chem Biol. 2008;3:30–37. doi: 10.1021/cb7002225. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Onizuka K, Taniguchi Y, Sasaki S. Bioconjugate Chem. 2009;20:799–803. doi: 10.1021/bc900009p. [DOI] [PubMed] [Google Scholar]; h) Onizuka K, Taniguchi Y, Nishioka T, Sasaki S. Nucleic Acids Symp Ser. 2009;53:67–68. doi: 10.1093/nass/nrp034. [DOI] [PubMed] [Google Scholar]; i) Solomatin S, Herschlag D. Methods Enzymol. 2009;469:47–68. doi: 10.1016/S0076-6879(09)69003-0. [DOI] [PubMed] [Google Scholar]; j) Bramsen JB, Malgorzata M, Pakula MM, Hansen TB, Bus C, Langkjær N, Odadzic D, Smicius R, Wengel SL, Chattopadhyaya J, Engels JW, Herdewijn P, Wengel J, Kjems J. Nucleic Acids Res. 2010;38:5761–5773. doi: 10.1093/nar/gkq341. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Sasaki S, Onizuka K, Taniguchi Y. Chem Soc Rev. 2011;40:5698–5706. doi: 10.1039/c1cs15066a. [DOI] [PubMed] [Google Scholar]; l) Phelps K, Beal PA. ACS Chem Biol. 2012;7:100–109. doi: 10.1021/cb200422t. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Schulz D, Holstein JM, Rentmeister A. Angew Chem Int Ed. 2013;52:7874– 7878. doi: 10.1002/anie.201302874. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:8028–8032. [Google Scholar]; n) Edwards TE, Sigurdsson ST. In: Handbook of RNA Biochemistry. Hartmann RK, Bindereif A, Schön A, Westhof E, editors. Wiley-VCH; Weinheim: 2014. pp. 151–171. [Google Scholar]; o) Oshiro I, Jitsuzaki D, Onizuka K, Nishimoto A, Taniguchi Y, Sasaki S. ChemBioChem. 2015;16:1199– 1204. doi: 10.1002/cbic.201500084. [DOI] [PubMed] [Google Scholar]
- 19.Srivatsan SG, Tor Y. J Am Chem Soc. 2007;129:2044– 2053. doi: 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Sampson JR, Uhlenbeck OC. Proc Natl Acad Sci USA. 1988;85:1033–10337. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Williamson D, Cann MJ, Hodgson DRW. Chem Commun. 2007:5096–5098. doi: 10.1039/b712066d. [DOI] [PubMed] [Google Scholar]; c) Huang FQ, He J, Zhang YL, Guo YL. Nat Protoc. 2008;3:1848–1861. doi: 10.1038/nprot.2008.185. [DOI] [PubMed] [Google Scholar]; d) Paredes E, Das SR. ChemBioChem. 2011;12:125–131. doi: 10.1002/cbic.201000466. [DOI] [PubMed] [Google Scholar]; e) Lee GH, Lim HK, Jung W, Hah SS. Bull Korean Chem Soc. 2012;33:3861–3863. [Google Scholar]
- 21.a) Fusz S, Srivatsan SG, Ackermann D, Famulok M. J Org Chem. 2008;73:5069–5077. doi: 10.1021/jo800639p. [DOI] [PubMed] [Google Scholar]; b) Seelig B, Jäschke A. Tetrahedron Lett. 1997;38:7729–7732. [Google Scholar]; c) Fiammengo R, Musilek K, Jaschke A. J Am Chem Soc. 2005;127:9271–9276. doi: 10.1021/ja051179m. [DOI] [PubMed] [Google Scholar]; d) Kim I, Shin S, Jeong Y, Hah SS. Tetrahedron Lett. 2010;51:3446–3448. [Google Scholar]
- 22.a) Samanta A, Krause A, Jäschke A. Chem Commun. 2014;50:1313–1316. doi: 10.1039/c3cc46132g. [DOI] [PubMed] [Google Scholar]; b) Wolf J, Dombos V, Appel B, Muller S. Org Biomol Chem. 2008;6:899–907. doi: 10.1039/b716151d. [DOI] [PubMed] [Google Scholar]; c) Pitulle C, Kleineidam RG, Sproat B, Krupp G. Gene. 1992;112:101– 105. doi: 10.1016/0378-1119(92)90309-d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.