

Abstract
Pediatric brain and extracranial solid tumors are a diverse group of malignancies that represent almost half of pediatric cancers. Standard therapy includes various combinations of surgery, cytotoxic chemotherapy and radiation, which can be very harmful to a developing child, and survivors carry a substantial burden of long term morbidities. While these therapies have improved survival rates for children with solid tumors, outcomes still remain extremely poor for subsets of patients. Recently, immunosuppressive checkpoint molecules that negatively regulate immune cell function have been described. When found on malignant cells or in the tumor microenvironment, they contribute to immune evasion and tumor escape. Agents designed to inhibit these proteins have demonstrated significant efficacy in human adult solid tumor studies. However, there is limited research focusing on immune checkpoint molecules and inhibitors in pediatric solid tumors. In this review, we examine the current knowledge on immune checkpoint proteins with an emphasis on Cytotoxic T Lymphocyte Antigen-4 (CTLA-4); Programmed cell Death protein-1 (PD-1) and Programmed Death Ligand 1 (PD-L1); OX-2 membrane glycoprotein (CD200); and Indoleamine 2,3-dioxygenase (IDO). We review T cell signaling, the mechanisms of action of these checkpoint molecules, pediatric preclinical studies on checkpoint proteins and checkpoint blockade, pediatric checkpoint inhibitor clinical trials conducted to date, and future immunotherapy opportunities for childhood cancers.
Keywords: immunotherapy, checkpoint proteins, pediatric solid tumors, pediatric brain tumors, PD-1
Introduction
Pediatric solid tumors are a heterogeneous group of malignancies that represent approximately 50% of pediatric cancers (1). They can be divided into extracranial solid tumors and central nervous system (CNS) tumors, which are the most common solid tumors in children accounting for 20-25% of childhood malignancies. Neuroblastoma, Wilms tumor, and sarcomas, including rhabdomyosarcoma, osteosarcoma and Ewing sarcoma, represent the most common extracranial solid tumors in childhood. The mainstay of therapy includes various combinations of surgery, cytotoxic chemotherapy and radiation, which can be toxic, impair normal development, and result in long-term morbidities. While these therapies have improved survival rates for children with solid tumors, outcomes are extremely poor for subsets of patients such as those with high-grade, refractory, or metastatic disease. Novel, targeted therapies are being developed to improve outcomes and lessen toxicities from conventional therapies.
Recently, immunosuppressive checkpoint molecules that negatively regulate immune cell function and enable local tumor escape have been described in adult malignancies (2, 3). These molecules exert their immunosuppressive effects by down-regulating the normal T cell response and increasing FoxP3+ T regulatory cell numbers and activation. These checkpoint molecules are normally expressed on a variety of cells in the body and likely play a crucial role in peripheral immune tolerance and regulation. When found on malignant cells or in the tumor microenvironment, they can contribute to immune evasion and tumor escape. Agents designed to inhibit these proteins have been developed and have shown significant efficacy in human adult solid tumor studies with several approved by the FDA (See Table 1 for summary of checkpoint inhibitors advanced to clinical trials and approved for human use) (4-10). There is limited research focusing on immune checkpoint molecules and the potential benefit of checkpoint blockade in children with solid tumors. In this review, we examine immune checkpoint proteins with an emphasis on cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed cell death protein-1 (PD-1)/programmed death-ligand 1 (PD-L1), OX-2 membrane glycoprotein (CD200) and Indoleamine 2,3-dioxygenase (IDO). We review their mechanism of action, pediatric preclinical studies related to these checkpoint proteins, pediatric checkpoint inhibitor clinical trials, and future immunotherapy opportunities.
Table 1.
Summary of checkpoint inhibitors advanced to clinical trials
| Protein | Drug (Trade Name) | Manufacturer | FDA Approval Diseases |
|---|---|---|---|
| CTLA-4 | Ipilimumab (Yervoy) | Bristol-Myers Squibb | Stage III and metastatic melanoma |
| Tremelimumab | MedImmune (AstraZeneca) | ||
| AGEN-1884 | Agenus | ||
| AGEN-2041 | Agenus | ||
| PD-1 | Nivolumab (Opdivo) | Bristol-Myers Squibb | Melanoma, metastatic squamous NSCLC, RCC, classical Hodgkin lymphoma |
| Pembrolizumab (Keytruda) | Merck | Advanced melanoma, metastatic NSCLC | |
| AMP-224 | MedImmune (AstraZeneca) | ||
| BGB-A317 | BeiGene | ||
| JS001 | Shanghai Junshi Biosciences | ||
| MEDI0680 | MedImmune (AstraZeneca) | ||
| PDR001 | Novartis | ||
| REGN2810 | Regeneron | ||
| SHR-1210 | Incyte | ||
| TSR-042 | TESARO | ||
| PD-L1 | Atezolizumab (TECENTRIQ) | GeneTech/Roche | Urothelial carcinoma |
| Avelumab | Pfizer | ||
| Durvalumab | MedImmune (AstraZeneca) | ||
| CA-170 | Curis | ||
| CD200 | Samalizumab | Alexion Pharmaceuticals | |
| IDO | Epacadostat | Incyte | |
| Indoximod | NewLink Genetics | ||
| GDC-0919 | NewLink Genetics | ||
| PF-06840003 | Pfizer |
NSCLC, non-small cell lung carcinoma; RCC, renal cell carcinoma
T cell signaling
T cell signaling plays a critical role in the adaptive immune system. T cells first recognize foreign antigens associated with the major histocompatibility complex (MHC) on antigen presenting cells (APCs) through the CD3 T-cell receptor (TCR). Second, CD28 on T cells interacts with CD80 (B7-1)/CD86 (B7-2) on APCs resulting in amplification of TCR signaling, production of interleukin (IL)-2, and growth and expansion of T cells while preventing the death of activated T cells (11). Checkpoint molecules can result in downregulation of this T cell response (Figure 1). Myeloid derived suppressor cells (MDSCs) and FoxP3+ T regulatory cells (Tregs) normally work to provide signals for the physiologic termination of the immune response but can also be upregulated in various malignancies (12). Tumor-infiltrating Tregs are often associated with a poor clinical outcome likely by limiting antitumor immune responses and promoting immunologic tolerance to cancer cells (13). Tregs mediate immune regulation through direct cytolytic activity, metabolic disruption, suppression of dendritic cells, and secretion of soluble or membrane-bound immunosuppressive molecules (12). Increased Treg cell numbers and activation is associated with expression of CTLA-4, PD-1/PD-L1, CD200 and IDO (14-17).
Figure 1. Interaction of checkpoint proteins and receptors on tumor cells, antigen presenting cells (APCs), T cells and T regulatory cells.
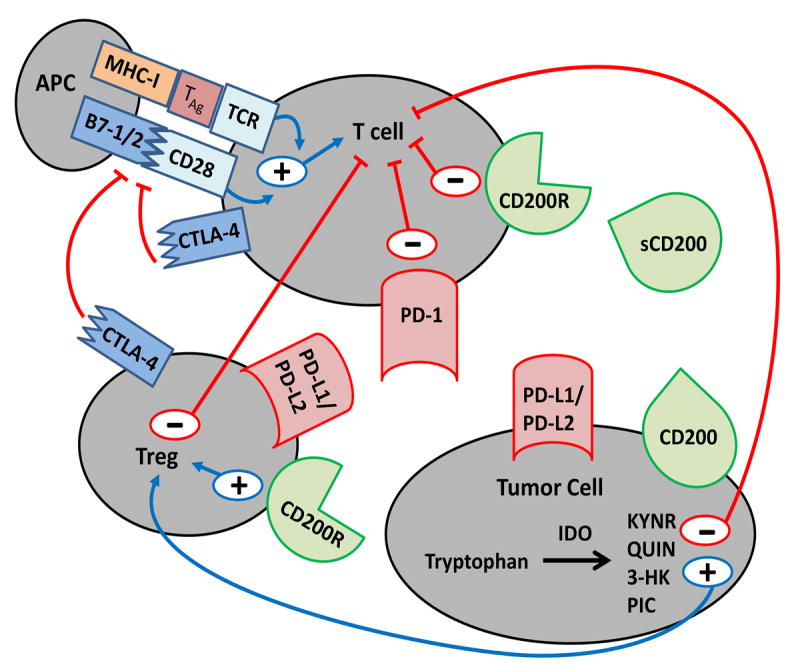
Programmed Death Ligand 1/2 (PD-L1/2), and CD200 are expressed on the tumor cell surface. Soluble CD200 (sCD200) exists in the serum of patients with solid tumors. Binding their ligands on T cells (PD-1, and CD200R, respectively) results in downregulation of the activated T cell immune response. Cytotoxic T lymphocyte Antigen-4 (CTLA-4) on the surface of Tregs and T cells can bind to B7-1/2 on APCs and interfere with T cell activation and proliferation via disruption of the B7-1/2-CD28 costimulatory complex. Indoleamine 2,3-dioxygenase (IDO), an enzyme found in the cytoplasm of tumor cells, catabolizes tryptophan into active metabolites, Kynurenic acid (KYNR), Quinolinic acid (QUIN), 3-Hydroxykynurenine (3-HK) and Picolinic acid (PIC), which contributes to T cell inhibition directly and indirectly through T regulatory cell (Treg) activation. Treg activation results in T cell inhibition through Treg cell surface CTLA-4 and PD-L1/2. The interaction between sCD200, tumor CD200 and Treg CD200R results in Treg activation and further T cell inhibition.
CTLA-4
CTLA-4 is a cell surface homodimeric glycoprotein belonging to the human immunoglobulin gene superfamily found on CD4+ and CD8+ T cells as well as Tregs. It shares 30% homology with CD28 and binds the B7 family of proteins with very high affinity. The molecule is predominantly expressed within the intracellular compartment of T cells and can be exported to the cell surface in response to constitutive T cell activation and high levels of IL-2 (18, 19). Once on the cell surface, the extracellular domain of CTLA-4 competes with CD28 by interacting with CD80 (B7-1)/CD86 (B7-2), resulting in inhibition of IL-2, interferon (IFN)-γ, IL-4 production, IL-2 receptor expression and cell cycle progression, thereby decreasing activation and expansion of T cells and accelerating death of activated T cells (11).
Human germline mutations in the CTLA-4 gene are associated with severe immune dysregulation and lymphocytic infiltration of target organs (20). CTLA-4 knockout mice developed profound lymphoproliferation and succumbed to early death (21). CTLA-4 may be critical for circulating Tregs to maintain immunological self-tolerance and homeostasis. Spontaneous development of systemic lymphoproliferation occurred when Tregs were deficient in CTLA-4 (22). In colon adenocarcinoma models, anti-CTLA-4 antibodies mediated a rapid and dramatic reduction of Tregs and expansion of CD8+ T cells at the tumor site (23). The combination of direct enhancement of T effector cell function and inhibition of Treg cell activity is essential for mediating the full therapeutic effects of anti-CTLA-4 antibodies (14).
While preclinical studies in adult malignancies have demonstrated increased CTLA-4 expression and significant responses to anti-CTLA-4 antibodies, few pediatric studies exist (24-26). Patients with newly diagnosed or relapsed osteosarcoma and Ewing sarcoma had increased expression of peripheral blood CTLA-4+ T cells compared to healthy controls (27). While the significance of CTLA-4 expression on tumor cells has not been fully elucidated, cytoplasmic and surface expression of CTLA-4 in pediatric neuroblastoma, rhabdomyosarcoma and osteosarcoma cell lines have been reported (see Table 2 for summary of checkpoint protein expression by pediatric tumor type) (28). In vitro treatment of human CTLA-4-expressing osteosarcoma cells lines with recombinant forms of the CTLA-4-ligands B7-1 and B7-2 induced caspase-dependent tumor cell apoptosis suggesting that tumor surface CTLA-4 may be targetable. Genetic polymorphisms in the CTLA-4 gene have been shown to influence T-cell activation and susceptibility to malignancies; the presence of CTLA-4 +49G>A (rs231775) polymorphism, which results in greater affinity of CTLA-4 to bind B7-1 molecule resulting in increased inhibition of T cell activation, is associated with increased risk of malignant bone tumors, including osteosarcoma and Ewing sarcoma (29, 30).
Table 2.
Summary of checkpoint protein expression by pediatric tumor type
| Tumor Type | CTLA-4 | PD-1/PD-L1 | CD200 | IDO | References |
|---|---|---|---|---|---|
| Extracranial Solid Tumors | |||||
| Ewing Sarcoma | ND | -/++/+++ | ND | ND | (38-40) |
| Hodgkin’s Lymphoma | ND | ND | ND | +++ | (71) |
| Non-Hodgkin’s Lymphoma | ND | +++ | ND | ND | (40) |
| Neuroblastoma | +++ | -/+/+++ | +++ | +++ | (28), (38, 40, 42, 45), (58), (63) |
| Osteosarcoma | +++ | ++/+++ | ND | +++ | (28), (38, 47) , (69) |
| Retinoblastoma | ND | +++ | ND | ND | (44) |
| Alveolar RMS | ND | +++ | ND | ND | (38) |
| Embryonal RMS | +++ | ++ | ND | ND | (28), (38) |
| Wilms, favorable | ND | + | ND | ND | (41) |
| Wilms, anaplastic | ND | ++ | ND | ND | (41) |
| Intracranial Solid Tumors | |||||
| Ependymoma | ND | ND | +++ | ND | (59) |
| Germinoma | ND | +++ | ND | ND | (42) |
| Glioblastoma | ND | ++ | ND | ND | (40) |
| Medulloblastoma | ND | -/+++ | ND | ND | (40) |
Checkpoint protein expression was graded as follows:
, no expression;
, 1-19% of tumors tested;
, 20-59% of tumors tested;
, 60-100% of tumors tested.
ND; no data. RMS; rhabdomyosarcoma.
Two CTLA-4 antibodies, ipilimumab and tremelimumab (Table 1), have been tested in adult solid tumor clinical trials alone or in combination with other targeted drugs or standard chemotherapy agents (4-7, 31, 32). These studies demonstrated improved survival in advanced melanoma (4-7). The most frequent immune related adverse events (irAEs) were grade 1-2 skin or gastrointestinal. Grade 3-4 irAEs, most commonly gastrointestinal, hepatic, or endocrine, occurred in approximately one-third of patients, and treatment-related deaths were seen in 1-2% of patients. Most immune toxicities responded to early, aggressive high-dose corticosteroids, which did not appear to adversely affect treatment outcomes (31). Post-treatment examination of tumor tissue showed decreased Treg infiltration and an increased CD4+/CD8+ to Treg ratio (32). Interestingly, ipilimumab used to treat metastatic renal cell carcinoma showed a direct association between autoimmune events and tumor regression suggesting that breaking normal immune tolerance may be important (31).
The first pediatric checkpoint inhibitor study was a phase 1 trial of ipilimumab in patients <21years old with recurrent or refractory solid tumors (See Table 3 for summary of pediatric trials) (33). Thirty-three patients (28 months to 21 years) with melanoma (n=12), sarcoma (n=17), renal or bladder carcinoma (n=3), or neuroblastoma (n=1) received 1, 3, 5, or 10mg/kg intravenously every 3 weeks with response assessed as a secondary objective at 6 and 12 weeks, and then every 3 months. Overall, 55% developed any grade irAE and 27% developed grade 3 or 4 irAE with a similar spectrum of immune-related events as described in adults including pancreatitis, pneumonitis, colitis, endocrinopathies and transaminitis. Nine subjects developed irAE after a single dose, and dose limiting toxicities occurred at 5mg/kg and 10mg/kg dose levels. While no fatal events occurred, one patient developed hypophysitis and subsequent panhypopituitarism, and another patient developed colitis that responded to steroids but resulted in colonic perforation. Although there were no partial or complete responses, stable disease was seen in six subjects for 4-10 cycles, and overall survival was increased in patients with immune-related toxicities.
Table 3.
Summary of pediatric checkpoint inhibitor trials
| Protein | Drug | Status | NCT Identifier | References |
|---|---|---|---|---|
| CTLA-4 | Ipilimumab | Completed Phase I trial for patients <21 years old with recurrent or refractory solid tumors | NCT01445379 | (33) |
| Ipilimumab | Terminated Phase II trial for children 12-17 years old with previously treated or untreated, unresectable stage III or stage IV melanoma | NCT01696045 | ||
| PD-1 | Nivolumab | Active Phase II trial for patients >1 year old with glioblastoma | NCT02550249 | |
| Nivolumab ± Ipilimumab | Active Phase I/II trial for patients 1-30 years old with recurrent or refractory solid tumors | NCT02304458 | ||
| Nivolumab + Cyclophosphamide ± radiation | Active European proof-of- Concept therapeutic stratification trial of molecular anomalies in children ≤18 years old with relapsed or refractory tumors | NCT02813135 | ||
| Pembrolizumab | Active safety/efficacy trial for patients 1-21 years old with progressive or recurrent high-grade glioma | NCT02359565 | ||
| CTLA-4 + PD-L1 | Tremelimumab + Durvalumab | Pending Phase II study in patients 16 years and older with advanced rare tumors | NCT02879162 | |
| IDO | Indoximod | Active Phase I trial for patients 3-21 years old with primary malignant brain tumors | NCT02502708 |
An ongoing phase I/II pediatric trial through the Children’s Oncology Group (COG) is investigating the safety of PD-1 inhibitor nivolumab with and without ipilimumab in patients with recurrent or refractory solid tumors (NCT02304458). The primary aims are to define toxicities, characterize pharmacokinetics, and assess immunogenicity by measuring anti-drug levels. Lastly, a phase II study of ipilimumab in children 12-17 years old with previously treated or untreated, unresectable stage III/IV melanoma (NCT01696045) recently was terminated due to slow accrual.
PD-1/PD-L1
PD-L1 (B7-H1) is a member of the B7 family of costimulatory molecules involved in the regulation of cellular and humoral immune responses through the PD-1 receptor on activated T and B cells (2). PD-L1 exists as a 40kDa type 1 transmembrane cell surface glycoprotein on hematopoietic and parenchymal cells (34). Interaction of PD-1 with PD-L1 dramatically inhibits T cell receptor mediated proliferation and production of IL-2 and IFN-γ. PD-1 regulates peripheral tolerance and autoimmunity; mice deficient for PD-1 develop features of lupus-like disease and dilated cardiomyopathy (35).
PD-L1 expressed on the surface of malignant cells can suppress the antitumor immune response leading to tumor growth and immune escape (2). Studies in adult malignancies indicate that increased PD-L1 expression is associated with increased disease stage, presence of metastases, and refractory or relapsed disease (36, 37). Several studies have examined PD-L1 expression in pediatric solid tumors by immunohistochemistry (IHC) (Table 2) (38-42). Results have been variable likely due to a lack of standardized methods for scoring and reporting stains and because there are over a dozen PD-L1 antibodies that differ in their targeted epitope, isotype, source, and binding affinity (43). Despite these limitations, moderate to high PD-L1 expression was seen in pediatric sarcomas, whereas expression in Wilms tumor was low; however expression was more likely to occur in anaplastic Wilms and was associated with an increased risk of recurrence in favorable histology tumors (38, 39, 41, 42). Importantly, pediatric tumors with the highest proportion of PD-L1 positivity showed the poorest survival (38).
In pediatric preclinical studies, PD-L1 expression was upregulated in response to immunogenic stimuli in retinoblastoma and neuroblastoma cells. (44, 45). PD-L1 antibody treatment enhanced T cell activation and proliferation implicating PD-L1 in the negative regulation of the antitumor immune response. In a murine neuroblastoma model, targeting PD-1/PD-L1 with blocking antibodies was insufficient to control tumor growth alone; however combining PD-1/PD-L1 blockade with a selective colony stimulating factor-1 receptor inhibitor (BLZ945) that blocks induction of suppressive MDSCs resulted in significant tumor responses suggesting that combined immunotherapy approaches may be necessary (46).
Differences in PD-L1 expression and response to PD-L1 blockade were seen in primary versus metastatic tumors and within different subtypes of the same tumor (47, 48). Human metastatic osteosarcomas but not primary tumors expressed PD-L1, and CD8+ tumor-infiltrating lymphocytes in metastatic osteosarcoma expressed PD-1 suggesting that PD-L1+ tumors are immunogenic but able to tolerize infiltrating T cells within the tumor microenvironment through this pathway (47). An anti-PD-L1 antibody significantly increased survival and improved function of infiltrating lymphocytes in a metastatic murine osteosarcoma model. Group 3 murine medulloblastoma had a higher percentage of CD8+/PD-1+ T cells and were more sensitive to PD-1 blockade than Shh murine tumors (48). These studies indicate that immunologic differences within the tumor microenvironment may exist and can be leveraged for therapeutic benefit.
There are several FDA approved anti-PD-1/anti-PD-L1 monoclonal antibodies that have improved survival in multiple adult malignancies (Table 1) (6-10). Importantly, PD-L1 expression correlated with response to treatment but PD-L1 negativity did not preclude a treatment response, and PD-L1 tumor expression was neither prognostic nor predictive in efficacy endpoints in trials using anti-PD-L1 antibodies (10, 49). The irAEs seen with PD-1/PD-L1 inhibition were similar to those seen with CTLA-4 blockade with skin, gastrointestinal, hepatic and endocrine toxicities occurring most frequently (6-10). Grade 3-4 toxicities occurred in 16% of adult melanoma patients treated with nivolumab (PD-1 inhibitor) alone; however this rate increased to 55% when nivolumab was combined with ipilimumab (7).
While there are no completed pediatric studies, several studies are ongoing and a recent report of significant responses to nivolumab in two young siblings with recurrent multifocal glioblastoma and biallelic mismatch repair deficiency suggests the potential of PD-1/PD-L1 blockade in children (50). A safety/efficacy study to assess the dose limiting toxicities and define a recommended Phase II dose of pembrolizumab (PD-1 inhibitor) in children with recurrent or refractory high-grade glioma and diffuse intrinsic pontine glioma (DIPG) is active through the Pediatric Brain Tumor Consortium (NCT02359565). Initial starting dose is at the recommended dose in adults (2mg/kg every 3 weeks). Secondary objectives include correlating potential biomarkers (PD-L1 and PD-1 tumor expression, patient immunophenotype, cytokine expression profiles, RNA signature profile, and tumor gene expression profile) with outcomes, and exploring whether Quantitative magnetic resonance spectroscopy and diffusion-weighted imaging can predict early tumor response and differentiate between progressive disease and pseudoprogression.
As discussed above, a pediatric phase I/II trial investigating the safety of nivolumab alone and with ipilimumab is ongoing. This study will explore whether correlations exist between PD-L1 tumor expression levels and antitumor effects of nivolumab alone and in combination with ipilimumab. A phase II study of neoadjuvant nivolumab in patients >1 year old with primary or recurrent glioblastoma is currently recruiting patients in Spain (NCT02550249). Patients that require surgery will receive neoadjuvant nivolumab 3mg/kg IV every 2 weeks. Changes in PD-L1 expression on tumor cells and lymphocytes will be assessed at baseline and response rate will be measured. Lastly, two other trials are exploring combination approaches. A European basket trial that is stratifying patients ≤ 18 years old with relapsed or refractory tumors based on tumor molecular anomalies includes an arm combining nivolumab and cyclophosphamide with and without radiation (NCT02813135), and a Canadian Phase II clinical trial testing the safety of durvalumab and tremelimumab in patients 16 years and older with advanced rare tumors is planned (NCT02879162).
CD200
CD200 is a type 1a transmembrane protein related to the B7 family of costimulatory receptors involved in T cell signaling. It is normally expressed on lymphoid and neuronal tissue. (51). Its receptor, CD200R, is expressed on APCs and T cells. Interaction of CD200 with CD200R inhibits monocyte/macrophage production of IL-2 and IFN-γ (52, 53) and downregulates the T cell mediated immune response through augmented production of Tregs (54, 55). CD200 deficient mice demonstrate myeloid dysregulation and autoimmune inflammation (52, 53). CD200 can be expressed on the surface of malignant cells and result in tumor immune escape (54, 56). In adult AML, expression of CD200 correlated with lower natural killer (NK) cell numbers, increased frequency of Tregs, and a worse prognosis (16, 57).
Few preclinical studies in pediatric cancers exist. Two neuroblastoma samples expressed surface CD200, and Th1 cytokines, that are necessary for efficient cytotoxic T-cell function, IL2 and IFN-γ, were downregulated when CD200-expressing, but not CD200-negative tumor cell lines, were added to mixed lymphocyte reactions (58). Inclusion of an anti-CD200 antibody restored Th1 cytokine responses suggesting CD200 suppresses antitumor immune responses. In a murine glioma model, mice treated with a CD200 antagonist with OVA+poly:ICLC, which induces an antigen specific cellular immune response, had prolonged survival compared to untreated mice. Treated mice had increased numbers of antigen-specific T cells and production of tumor necrosis factor-α and IFN-γ implicating CD200 in suppressing the immune response (59). Several pediatric brain tumor types including ependymoma, medulloblastoma and DIPG had higher CD200 expression by western blot compared to normal brain tissue (59). Increased CD200 mRNA expression was seen in supratentorial compared to posterior fossa ependymoma and in group 4 compared to Shh or group 3 medulloblastoma. Further research is needed to elucidate the cause of immunologic differences seen between tumor types and within tumor subtypes.
There is one reported clinical trial of monoclonal anti-CD200 antibody samalizumab in 26 adults with advanced B-cell chronic lymphocytic leukemia (B-CLL) or multiple myeloma (60). Samalizumab was well tolerated; the most common adverse events were fatigue, fever and rash, and grade ≥ 3 events included neutropenia and infections. The drug exhibited a dose dependent effect on CD200+ B-CLL, CD4+ T cells and Tregs. Some patients showed disease stabilization and two patients who received >9 cycles had a reduced tumor burden. There are currently no active adult or pediatric clinical trials targeting CD200.
IDO
Indoleamine 2,3-dioxygenase (IDO) is an intracellular enzyme that catalyzes the initial and rate limiting step in the kynurenine pathway which is the major pathway of L-tryptophan catabolism in mammals (61). The kynurenine pathway produces many metabolites, including L-kynurenine, kynurenic acid (KYNA), quinolinic acid (QUIN), 3-hydroxykynurenine (3-HK), and picolinic acid (PIC). QUIN is a potent NMDA receptor agonist, and QUIN, L-kynurenine, and PIC inhibit T and NK cell proliferation (62). 3-HK has immunomodulatory properties indirectly by the production of free radicals. T and NK cells are also inhibited by the depletion of available tryptophan, which occurs in the presence of high levels of IDO. IFN-γ induces IDO in monocytes and tumor cells which suggests that IDO is upregulated in the setting of inflammation, likely to quell the inflammatory cascade (63). IDO is expressed in a wide variety of normal cells, and likely plays a fundamental role in immunosuppression and peripheral tolerance (64, 65). IDO is expressed in the placenta early in pregnancy and has been implicated in the prevention of allogenic fetus rejection (66). Mucosal biopsies of patients with inflammatory bowel disease revealed IDO overexpression suggesting that IDO has an anti-inflammatory mechanism to counterbalance the tissue-damaging effects of activated T cells (67).
Many adult tumors constitutively express IDO, and increased IDO expression correlated with aggressive tumor growth and resistance to T cell targeting therapies due to the recruitment and activation of MDSCs through a Treg-dependent mechanism (17, 68). Expression of IDO in immunogenic murine tumor cells prevented rejection by mice preimmunized with tumor antigen and resulted in decreased T cell accumulation at the tumor site (68). Furthermore, IDO inhibition with 1-methyl-tryptophan (1MT) significantly slowed tumor progression.
Similar to other checkpoint molecules, there are few studies examining IDO in pediatric tumors. Urakawa et al. scored IDO expression from 0-5 by IHC in 30 pediatric osteosarcoma patient samples. Patients with high (≥4+) IDO expression had significantly lower metastasis-free survival (53% vs. 81%) and 5-year overall survival (60% vs. 92%) compared to patients with lower (<4+) expression suggesting that the immune tolerance mediated by IDO may have an important role in the metastatic potential of osteosarcoma and may impact clinical outcome (69). IDO expression was also seen in 17/20 (85%) pediatric Hodgkin lymphoma patient samples and in human Ewing sarcoma, grown in NOD/scid mice (70, 71).
Indoximod (1-methyl-tryptophan), an orally available IDO inhibitor, is currently being studied in several adult phase I and II trials alone, in combination with conventional chemotherapy and/or radiation, and with ipilimumab, pembrolizumab or nivolumab. In a phase I study of indoximod in 48 adults with advanced extracranial solid tumors, a maximum tolerated dose was not reached and the only irAE was grade 2 hypophysitis in three patients (6%) previously treated with other checkpoint inhibitors (72). A phase I trial using indoximod in combination with temozolomide for children with primary malignant brain tumors is currently recruiting patients 3-21 years old (NCT02502708). Patients will receive indoximod orally in escalating doses, beginning at 12.8 mg/kg/dose twice daily and increasing to 22.4 mg/kg/dose twice daily along with temozolomide 200mg/m2 daily for five days. Primary outcome measures are incidence of regimen limiting toxicities and objective response rate, and secondary outcomes are indoximod pharmacokinetics, progression free and overall survival.
Future Directions
Based on dramatic, durable responses seen in a variety of adult cancers and the initial preclinical data indicating that many pediatric solid tumors express checkpoint molecules, immune checkpoint inhibition is a promising therapy for pediatric malignancies. Adult checkpoint inhibitor studies resulted in some significant, potentially fatal autoimmune side effects with increased toxicity seen when combining PD-1 and CTLA-4 inhibitors. Increased vigilance and aggressive treatment of these adverse events was essential to decrease morbidity and mortality. Initial pediatric checkpoint inhibitor trials have shown similar results with some autoimmune side effects. Further studies and larger clinical trials are needed to evaluate the full range of side effects in children. The short and long term consequences of using checkpoint inhibitors in children with naïve immune systems and developing organ systems are unknown.
Checkpoint blockade may be beneficial for children with relapsed or refractory solid tumors as monotherapy, in combination with other checkpoint inhibitors, or with standard chemotherapies or radiotherapy. The role of checkpoint molecules in immune suppression and tumor escape suggests that patients need an intact immune system to achieve maximum therapeutic benefit from these agents. Thus, checkpoint inhibitors may have less therapeutic benefit when combined with myelosuppressive chemotherapy. An ideal response from checkpoint inhibitors is likely to be achieved with the combination of a potent immunogen to activate the immune system, a cytokine to further amplify the antitumor immune response, and a checkpoint inhibitor to further amplify and sustain the immune response (Figure 2).
Figure 2. Maximizing the Antitumor Immune Response with Checkpoint Blockade.
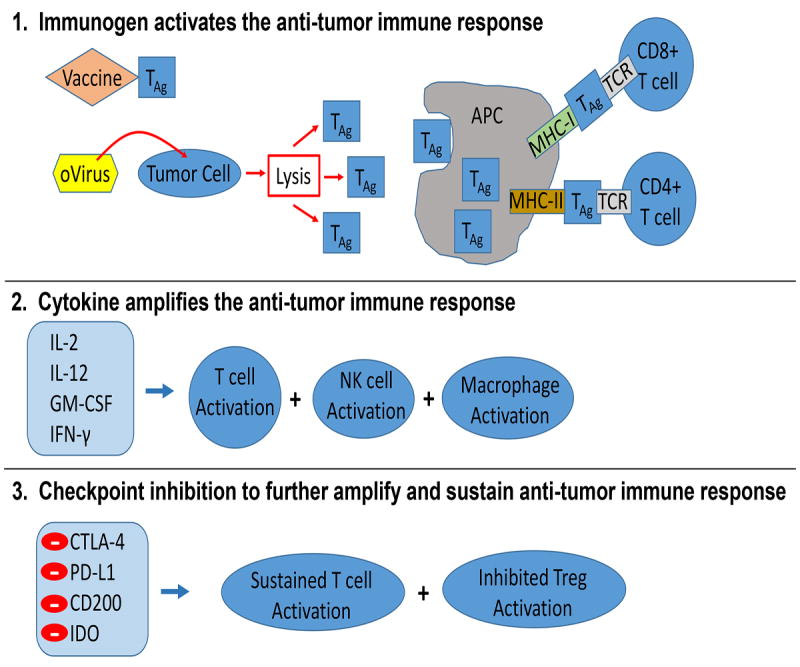
1) Activation of the immune response can occur through various approaches such as oncolytic viruses (oVirus) or vaccinations with tumor associated antigens (TAg) or other antigens such as tetanus toxoid. Antigen presenting cells (APC) process and present TAg to the T cell receptor (TCR) on T cells through major histocompatibility complex (MHC) I and II. 2) Various cytokines can be used to augment the antitumor immune response through stimulation of T cells, natural killer (NK) cells and/or macrophages. 3) Checkpoint protein inhibition enables continued activation of T cells to sustain the antitumor immune response and suppresses the development of immune tolerance to TAg mediated by T regulatory cells (Tregs).
Agents designed to stimulate the tumor immune response such as tumor vaccines or oncolytic viruses may be attractive therapies to combine with immune checkpoint inhibition. A phase Ib/III study using pembrolizumab (anti PD-1) with or without talimogene laherparepvec (T-VEC; Imlygic (Amgen)), an oncolytic herpes simplex virus-1 which produces granulocyte macrophage colony stimulating factor (GM-CSF) and is the first FDA approved oncolytic virus, is currently recruiting patients for treatment of stage IIIB-IV melanoma (NCT02263508). Other emerging immune checkpoint molecules, such as Fibrinogen-like protein 2 (FGL2), lymphocyte activation gene-3 (LAG-3), and colony stimulating-1 receptor (CSF-1R) have shown immunomodulatory roles in human cancer and require further studies in pediatric cancers (73-75). Additional pediatric clinical trials and a more complete understanding of the tumor microenvironment and tumor-host interaction will aid in realizing the full potential of immune checkpoint blockade in pediatric cancers.
Acknowledgments
Grant Support:
St. Baldrick’s Foundation, the Rally Foundation for Childhood Cancer Research, The Truth 365, Department of Defense (W81XWH-15-1-0108), Kaul Pediatric Research Institute to GKF, and the National Institutes of Health (P20CA151129 to GYG and P01CA071933 to JMM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense.
Footnotes
Conflict of Interest: authors have no conflicts of interest
References
- 1.Ries L, Smith M, Gurney J, Linet M, Tamra T, Young J, et al. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program. NIH Pub No 99-4649; Bethesda, MD: 1999. [Google Scholar]
- 2.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–30. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 7.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. The New England journal of medicine. 2015;373:1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157:636–42. [PubMed] [Google Scholar]
- 12.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162–71. doi: 10.1158/0008-5472.CAN-11-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–25. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, et al. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia. 2012;26:2146–8. doi: 10.1038/leu.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015;13:412–24. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–70. [PubMed] [Google Scholar]
- 19.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–43. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- 20.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (New York, NY) 2014;345:1623–7. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (New York, NY) 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 22.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (New York, NY) 2008;322:271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 23.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 24.Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10067–71. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8099–103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (New York, NY) 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani P, Maas ML, Gustafson MP, Dickman P, Adams RH, Watanabe M, et al. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) HLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J Immunother Cancer. 2015;3:35. doi: 10.1186/s40425-015-0082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer. 2005;117:538–50. doi: 10.1002/ijc.21155. [DOI] [PubMed] [Google Scholar]
- 29.Yu F, Miao J. Significant association between cytotoxic T lymphocyte antigen 4 +49G>A polymorphism and risk of malignant bone tumors. Tumour Biol. 2013;34:3371–5. doi: 10.1007/s13277-013-0908-7. [DOI] [PubMed] [Google Scholar]
- 30.Sun T, Zhou Y, Yang M, Hu Z, Tan W, Han X, et al. Functional genetic variations in cytotoxic T-lymphocyte antigen 4 and susceptibility to multiple types of cancer. Cancer Research. 2008;68:7025–34. doi: 10.1158/0008-5472.CAN-08-0806. [DOI] [PubMed] [Google Scholar]
- 31.Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30:825–30. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010;16:3485–94. doi: 10.1158/1078-0432.CCR-10-0505. [DOI] [PubMed] [Google Scholar]
- 33.Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22:1364–70. doi: 10.1158/1078-0432.CCR-15-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001;22:265–8. doi: 10.1016/s1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 36.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 37.Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947–53. doi: 10.1158/1078-0432.CCR-04-1469. [DOI] [PubMed] [Google Scholar]
- 38.Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton-Key M, Gray JC. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. Oncoimmunology. 2015;4:e1029701. [Google Scholar]
- 39.Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin KH, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC cancer. 2016;16:434. doi: 10.1186/s12885-016-2451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel B, Santi-Vincini M, et al. Assessment of PD-L1 expression and tumor-associated lymphocytes in pediatric cancer tissues. Cancer Research. 2015;75:249. [Google Scholar]
- 41.Routh JC, Ashley RA, Sebo TJ, Lohse CM, Husmann DA, Kramer SA, et al. B7-H1 expression in Wilms tumor: correlation with tumor biology and disease recurrence. J Urol. 2008;179:1954–9. doi: 10.1016/j.juro.2008.01.056. discussion 9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aoki T, Hino M, Koh K, Kyushiki M, Kishimoto H, Arakawa Y, et al. Low Frequency of Programmed Death Ligand 1 Expression in Pediatric Cancers. Pediatric blood & cancer. 2016;63:1461–4. doi: 10.1002/pbc.26018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhaijee F, Anders RA. PD-L1 Expression as a Predictive Biomarker: Is Absence of Proof the Same as Proof of Absence? JAMA Oncol. 2016;2:54–5. doi: 10.1001/jamaoncol.2015.3782. [DOI] [PubMed] [Google Scholar]
- 44.Usui Y, Okunuki Y, Hattori T, Takeuchi M, Kezuka T, Goto H, et al. Expression of costimulatory molecules on human retinoblastoma cells Y-79: functional expression of CD40 and B7H1. Invest Ophthalmol Vis Sci. 2006;47:4607–13. doi: 10.1167/iovs.06-0181. [DOI] [PubMed] [Google Scholar]
- 45.Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett. 2015;361:49–56. doi: 10.1016/j.canlet.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 46.Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-1912. [DOI] [PubMed] [Google Scholar]
- 47.Lussier DM, O’Neill L, Nieves LM, McAfee MS, Holechek SA, Collins AW, et al. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J Immunother. 2015;38:96–106. doi: 10.1097/CJI.0000000000000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pham CD, Flores C, Yang C, Pinheiro EM, Yearley JH, Sayour EJ, et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin Cancer Res. 2016;22:582–95. doi: 10.1158/1078-0432.CCR-15-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Calio A, et al. Differential Activity of Nivolumab, Pembrolizumab and MPDL3280A according to the Tumor Expression of Programmed Death-Ligand-1 (PD-L1): Sensitivity Analysis of Trials in Melanoma, Lung and Genitourinary Cancers. PLoS One. 2015;10:e0130142. doi: 10.1371/journal.pone.0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016;34:2206–11. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 51.Wright GJ, Jones M, Puklavec MJ, Brown MH, Barclay AN. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology. 2001;102:173–9. doi: 10.1046/j.1365-2567.2001.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science (New York, NY) 2000;290:1768–71. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 53.Jenmalm MC, Cherwinski H, Bowman EP, Phillips JH, Sedgwick JD. Regulation of myeloid cell function through the CD200 receptor. J Immunol. 2006;176:191–9. doi: 10.4049/jimmunol.176.1.191. [DOI] [PubMed] [Google Scholar]
- 54.McWhirter JR, Kretz-Rommel A, Saven A, Maruyama T, Potter KN, Mockridge CI, et al. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1041–6. doi: 10.1073/pnas.0510081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gorczynski RM, Lee L, Boudakov I. Augmented Induction of CD4+CD25+ Treg using monoclonal antibodies to CD200R. Transplantation. 2005;79:1180–3. [PubMed] [Google Scholar]
- 56.Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol. 2007;178:5595–605. doi: 10.4049/jimmunol.178.9.5595. [DOI] [PubMed] [Google Scholar]
- 57.Damiani D, Tiribelli M, Raspadori D, Sirianni S, Meneghel A, Cavalllin M, et al. Clinical impact of CD200 expression in patients with acute myeloid leukemia and correlation with other molecular prognostic factors. Oncotarget. 2015;6:30212–21. doi: 10.18632/oncotarget.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siva A, Xin H, Qin F, Oltean D, Bowdish KS, Kretz-Rommel A. Immune modulation by melanoma and ovarian tumor cells through expression of the immunosuppressive molecule CD200. Cancer Immunol Immunother. 2008;57:987–96. doi: 10.1007/s00262-007-0429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moertel CL, Xia J, LaRue R, Waldron NN, Andersen BM, Prins RM, et al. CD200 in CNS tumor-induced immunosuppression: the role for CD200 pathway blockade in targeted immunotherapy. J Immunother Cancer. 2014;2:46. doi: 10.1186/s40425-014-0046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahadevan D LM, Wheldon M, Faas SJ, Ulery TL, Kukreja A, LiL MS, Bedrosian CL. In: Heffner L, editor. First-inhuman phase I dose escalation study of a humanized anti-CD200 antibody (Samalizumab) in patients with advanced stage B cell chronic lymphocytic leukemia (B-CLL) or multiple myeloma (MM); ASH annual meeting; 2010; Orlando, FL. 2010. p. 2465. [Google Scholar]
- 61.Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem Biophys Res Commun. 2005;338:12–9. doi: 10.1016/j.bbrc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 62.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–68. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G, Wachter H. Characteristics of interferon induced tryptophan metabolism in human cells in vitro. Biochimica et biophysica acta. 1989;1012:140–7. doi: 10.1016/0167-4889(89)90087-6. [DOI] [PubMed] [Google Scholar]
- 64.Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005;49:15–23. doi: 10.1002/glia.20090. [DOI] [PubMed] [Google Scholar]
- 65.Dai X, Zhu BT. Indoleamine 2,3-dioxygenase tissue distribution and cellular localization in mice: implications for its biological functions. J Histochem Cytochem. 2010;58:17–28. doi: 10.1369/jhc.2009.953604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (New York, NY) 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 67.Wolf AM, Wolf D, Rumpold H, Moschen AR, Kaser A, Obrist P, et al. Overexpression of indoleamine 2,3-dioxygenase in human inflammatory bowel disease. Clin Immunol. 2004;113:47–55. doi: 10.1016/j.clim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 68.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 69.Urakawa H, Nishida Y, Nakashima H, Shimoyama Y, Nakamura S, Ishiguro N. Prognostic value of indoleamine 2,3-dioxygenase expression in high grade osteosarcoma. Clin Exp Metastasis. 2009;26:1005–12. doi: 10.1007/s10585-009-9290-7. [DOI] [PubMed] [Google Scholar]
- 70.Max D, Kuhnol CD, Burdach S, Niu L, Staege MS, Foll JL. Indoleamine-2,3-dioxygenase in an immunotherapy model for Ewing sarcoma. Anticancer Res. 2014;34:6431–41. [PubMed] [Google Scholar]
- 71.Shahlaee A, Al-Quran S, Hou W, Schwartz C, Munn D. Aberrant indoleamine 2, 3 dioxygenase (IDO) expression is present in pediatric patients with Hodgkin’s lymphoma. ASCO Annual Meeting Proceedings; 2007; 2007. p. 9531. [Google Scholar]
- 72.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7:22928–38. doi: 10.18632/oncotarget.8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan J, Kong LY, Hu J, Gabrusiewicz K, Dibra D, Xia X, et al. FGL2 as a Multimodality Regulator of Tumor-Mediated Immune Suppression and Therapeutic Target in Gliomas. J Natl Cancer Inst. 2015:107. doi: 10.1093/jnci/djv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li B, VanRoey M, Triebel F, Jooss K. Lymphocyte activation gene-3 fusion protein increases the potency of a granulocyte macrophage colony-stimulating factor-secreting tumor cell immunotherapy. Clin Cancer Res. 2008;14:3545–54. doi: 10.1158/1078-0432.CCR-07-5200. [DOI] [PubMed] [Google Scholar]
- 75.Chen Q, Lu XJ, Li MY, Chen J. Molecular cloning, pathologically-correlated expression and functional characterization of the colonystimulating factor 1 receptor (CSF-1R) gene from a teleost, Plecoglossus altivelis. Dongwuxue Yanjiu. 2016;37:96–102. doi: 10.13918/j.issn.2095-8137.2016.2.96. [DOI] [PMC free article] [PubMed] [Google Scholar]