

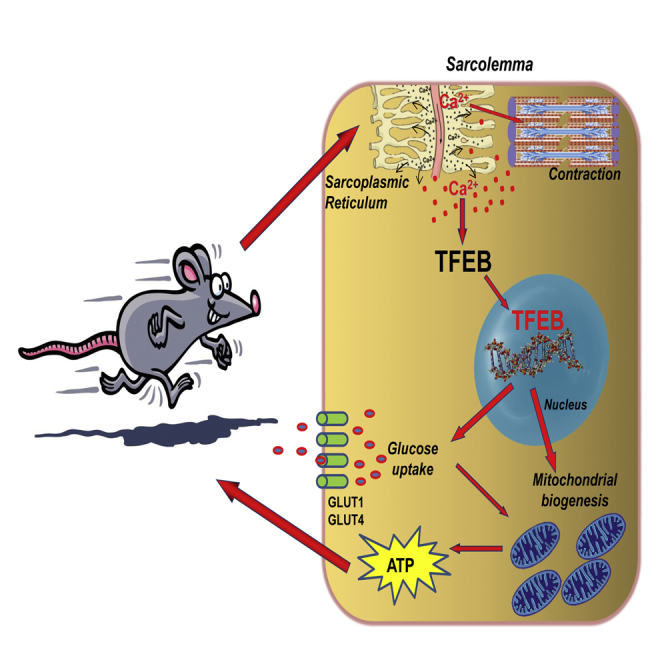
Summary
The transcription factor EB (TFEB) is an essential component of lysosomal biogenesis and autophagy for the adaptive response to food deprivation. To address the physiological function of TFEB in skeletal muscle, we have used muscle-specific gain- and loss-of-function approaches. Here, we show that TFEB controls metabolic flexibility in muscle during exercise and that this action is independent of peroxisome proliferator-activated receptor-γ coactivator1α (PGC1α). Indeed, TFEB translocates into the myonuclei during physical activity and regulates glucose uptake and glycogen content by controlling expression of glucose transporters, glycolytic enzymes, and pathways related to glucose homeostasis. In addition, TFEB induces the expression of genes involved in mitochondrial biogenesis, fatty acid oxidation, and oxidative phosphorylation. This coordinated action optimizes mitochondrial substrate utilization, thus enhancing ATP production and exercise capacity. These findings identify TFEB as a critical mediator of the beneficial effects of exercise on metabolism.
Keywords: TFEB, autophagy, exercise, glucose, mitochondria, diabetes, mitochondrial fusion, PGC1alpha, metabolic flexibility, insulin
Graphical Abstract
Highlights
-
•
TFEB regulates mitochondrial biogenesis and function in muscle
-
•
Glucose homeostasis in skeletal muscle requires TFEB
-
•
The effects of TFEB on muscle metabolism are independent from PGC1α
-
•
TFEB coordinates metabolic flexibility during physical exercise
Mansueto et al. show that TFEB acts as a central coordinator of skeletal muscle insulin sensitivity, glucose homeostasis, lipid oxidation, and mitochondrial function in the adaptive metabolic response to physical exercise in a PGC1α-independent manner.
Introduction
Exercise elicits several beneficial effects by acting on mitochondrial content/function, fatty acid oxidation, and glucose homeostasis (Hawley, 2002, Holloszy and Coyle, 1984, Holloszy et al., 1998). Indeed, muscle activity is important to counteract disease progression in diabetes, obesity, and metabolic syndrome. The signaling pathways that control the contraction-mediated beneficial effects on mitochondria and glucose/lipid homeostasis are distinct from insulin signaling and mainly rely on AMPK and PGC1α. We have recently found that exercise leads to nuclear translocation of the helix-loop-helix leucine zipper transcription factor EB (TFEB) (Medina et al., 2015), an important regulator of lysosomal biogenesis and autophagy (Sardiello et al., 2009, Settembre et al., 2011). Upregulation of TFEB has been found in several tissues after food deprivation, including liver and skeletal muscle. We have previously shown that in liver, TFEB regulates genes involved in lipid catabolism, fatty acid oxidation, and ketogenesis (Settembre et al., 2013). Some of these effects are elicited by TFEB-mediated induction of PGC1α (Settembre et al., 2013), a transcriptional coactivator, which interacts with and enhances the activity of transcription factors involved in mitochondrial biogenesis, glucose homeostasis, and lipid oxidation (Kelly and Scarpulla, 2004, Puigserver et al., 1998).
In the presence of nutrients, TFEB is sequestered in the cytoplasm by mTORC1-mediated phosphorylation, whereas in nutrient-depleted conditions, mTORC1 is inactive and dephosphorylated TFEB translocates to the nucleus, where it induces the transcription of target genes (Martina et al., 2012, Roczniak-Ferguson et al., 2012, Settembre et al., 2012). The dephosphorylation of TFEB is mediated by the calcium-dependent phosphatase, calcineurin, which is necessary for TFEB activation (Medina et al., 2015). Importantly, exercise-dependent calcium influx activates calcineurin, which dephosphorylates TFEB, leading to nuclear localization. The calcineurin-mediated induction of TFEB is independent from mTORC1 activity, indicating that calcium-dependent signaling is a rate-limiting step of TFEB activation (Medina et al., 2015).
Previous studies implicated calcineurin in a variety of physiological processes, in particular in skeletal muscle adaptation to exercise (Gehlert et al., 2015). Muscle-specific transgenic mice that overexpress an activated form of calcineurin show increased glucose uptake, glycogen accumulation, and lipid oxidation (Long et al., 2007). Interestingly, calcineurin promotes the nuclear translocation of another family of transcription factors, NFAT, which, depending on the type of physical activity, modulate the expression of the different myosin isoforms (Calabria et al., 2009, Long et al., 2007, McCullagh et al., 2004).
Here we show that the calcineurin-TFEB axis plays a major role in the metabolic adaptations that occur during physical exercise. By using gain- and loss-of-function approaches, we show that TFEB regulates mitochondrial biogenesis and glucose uptake independently of PGC1α. Indeed, TFEB controls genes involved in glucose metabolism such as GLUT1 and GLUT4, hexokinase I and II, TBC1 domain family member 1 (TBC1D1), and glycogen synthase (GYS), leading to glycogen accumulation to sustain energy production during exercise.
Results
Genome-wide Analyses Identified Glucose-Related and Mitochondrial Genes as Downstream Targets of TFEB
We previously demonstrated that TFEB promotes lipid catabolism in the liver and protects against diet-induced weight gain and insulin resistance (Settembre et al., 2013). Here we studied the physiological relevance of TFEB in skeletal muscle, an important insulin- and autophagy-dependent tissue (Grumati and Bonaldo, 2012, Mammucari et al., 2007, Masiero et al., 2009). Transcriptome analysis was performed by whole-genome gene expression profiling experiments (SuperSeries-GSE62980) in skeletal muscle from both TFEB-overexpressing and TFEB knockout (KO) mice. Overexpression of Tcfeb, the murine homolog of human TFEB, in muscle was achieved by means of intramuscular viral-mediated gene transfer using the adeno-associated virus (AAV) system. Adult mice were injected intramuscularly with either AAV2.1-CMV-TFEB or AAV2.1-CMV-GFP control vector and animals were sacrificed after 21 days, a time that allows efficient TFEB expression (Figure S1A, available online). Muscle-specific conditional TFEB KO mice were generated by crossing Tcfeb floxed (Settembre et al., 2013) with MLC1f-Cre transgenic mice (Bothe et al., 2000). Efficiency and specificity of the gene deletion were confirmed by quantitative real-time PCR analysis on multiple tissues (Figure S1B).
Overexpression of TFEB in muscle resulted in the upregulation of 1,514 genes and the downregulation of 1,109 genes (GSE62975), while genetic ablation of TFEB increased 496 genes and suppressed 458 genes (GSE62976). The up- or downregulated genes are highlighted in red and green, respectively, in Tables S1 and S2. To identify the main cellular compartments (CCs) and the principal biological process (BPs) for which the TFEB-dependent genes were enriched, we performed a gene ontology enrichment analysis (GOEA). The GOEA was performed on the lists of genes whose expression was either increased or decreased in transfected muscle or in the TFEB KO mice. Interestingly, several gene categories related to cellular metabolism, including lipid and glucose homeostasis, were found upregulated in TFEB-overexpressing muscle and downregulated in TFEB KO (Figure 1A; Table S3). Strikingly, genes involved in mitochondrial biogenesis were oppositely regulated by gain- and loss-of-function approaches. Indeed, 38 genes involved in mitochondrial function were induced in AAV2.1-TFEB-infected muscles (Table S4), while 73 genes were inhibited in TFEB KO muscles (Table S5).
Figure 1.

TFEB Induces Mitochondrial Biogenesis
(A) Genes involved in lipid and glucose metabolism that were affected by TFEB expression are shown in colored circles. The upregulated (red circles) and downregulated genes (green circles) are shown in TFEB-overexpressing and TFEB KO, respectively. The genes were divided into functional sub-categories.
(B–D) EM analysis of skeletal muscles from WT mice transfected with AAV2.1-GFP (B) and AAV2.1-TFEB (C), and from TFEB KO mice (D). Normal mitochondria are indicated by black arrows (B and C), while abnormal mitochondria in TFEB KO muscle (D) are indicated by empty arrows. Accumulation of glycogen is shown by white arrows in (C). The scale bars represent 500 nm.
(E and F) Morphometrical analyses of mitochondrial number (E) and size (F) in TFEB-overexpressing and TFEB KO muscles compared to controls. Error bars represent mean ± SE, n = 3; ∗∗p < 0.01.
(G) mtDNA analysis of muscles infected with AAV2.1-TFEB and from TFEB KO compared to WT mice. Quantitative real-time PCR of mtDNA copy numbers. Data are shown as mean ± SE, n = 3; ∗∗p < 0.01.
(H) Quantification of abnormal mitochondria in TFEB KO gastrocnemius (GCN) muscles compared to WT muscles. Data are expressed as percentage of abnormal mitochondria on total mitochondria of 16 pictures (for detailed description of the criteria, see Experimental Procedures). Error bars represent mean ± SE; ∗p < 0.05.
To better identify the network of genes regulated by TFEB in muscle, we performed sequence analysis to identify putative TFEB target sites, previously referred to as CLEAR sites (coordinated lysosomal expression and regulation) (Palmieri et al., 2011), in the promoter regions of the downregulated genes in TFEB KO mice. Interestingly, we found that 79% of these genes contain a CLEAR sequence and are, therefore, potential direct targets of TFEB (Table S6).
TFEB Regulates Mitochondrial Biogenesis in Muscle
To examine potential effects of TFEB in mitochondrial function, we analyzed mitochondrial morphology in muscles overexpressing or lacking TFEB. Electron microscopy (EM) analyses showed a striking increase of mitochondrial density and volume in TFEB-overexpressing muscles (Figures 1B, 1C, 1E, and 1F). Interestingly, mitochondrial density and size were normal in the TFEB KO muscles (Figures 1E and 1F). Consistent with the EM data, increase of mitochondrial DNA (mtDNA) was found in TFEB transgenic muscles, while no differences were observed in TFEB KO muscles (Figure 1G). However, while the cristae shape, matrix density, and outer membrane morphology were normal in TFEB-overexpressing muscles (Figure 1C), abnormalities were found in approximately 10% of the mitochondria from TFEB KO muscles (Figures 1D and 1H). An increase in the number of mitochondria was also observed in C2C12 muscle cells transfected with TFEB-GFP, as detected by immunofluorescence confocal and confirmed by EM analyses (Figures S2A and S2B). Quantitative real-time PCR also revealed an increase of mtDNA content in TFEB-overexpressing cells (Figure S2C).
Importantly, quantitative real-time PCR analysis revealed that TFEB overexpression in muscle and in C2C12 cells induces the expression of many genes involved in mitochondrial biogenesis and function, including the master gene of mitochondrial biogenesis, PGC1α, a known direct target of TFEB (Settembre et al., 2013) (Figures 2A and S2D). Moreover, another PGC-1 family member, PGC1β, was also upregulated by TFEB overexpression. Consistently, we found a significant induction of peroxisome proliferator-activated receptor α (PPARα), PPARβ/δ, and PPARγ in TFEB-overexpressing muscles. However, TFEB deletion did not affect the expression of PGC1α/β and PPAR genes, with the exception of PPARα, which was downregulated. In order to elucidate the possible mechanisms underlying the induction of mitochondrial biogenesis observed in TFEB-transfected muscles, we examined the expression of nuclear respiratory factors 1 and 2 (NRF1and NRF2). The mRNA levels of NRF2 were increased, as well as NRF downstream genes, including mitochondrial transcription factor A (TFAM). Chromatin immunoprecipitation (ChIP) experiments confirmed the direct recruitment of TFEB on NRF1 and NRF2 promoters (Figure 2B), but not on TFAM promoter (data not shown). Finally, overexpression of TFEB in skeletal muscle increased the expression of mitochondrial enzymes. Subunits of the four respiratory chain complexes and the ATP synthase, as well as genes encoding electron transport and tricarboxylic acid cycle proteins, were induced by TFEB overexpression and were reduced by TFEB deletion (Figure 2A). Importantly, immunoblotting analyses confirmed the increase of complex I (NDUFA9), complex II (SDHA), and complex IV (COX5a) proteins in TFEB-transfected muscles (Figures 2C and 2D). TFEB deletion did not alter NDUFA9 and COX5a expression but significantly reduced the level of SDHA protein (Figures 2C and 2D). To better characterize the involvement of TFEB in mitochondrial respiration, we analyzed the specific activities of enzymes involved in oxidative phosphorylation. Biochemical analysis of muscle samples infected with AAV2.1-TFEB compared to wild-type (WT) muscles showed increase of citrate synthase, mitochondrial respiratory chain complex I (CI), CII, CIII, and CIV activities (Figure 2E). Consistent with the western blot analyses, TFEB deletion led to decrease of CII activity, the complex that contains the SDHA flavoprotein, while the other respiratory complexes had normal activities (Figure 2E). The changes in respiratory chain activities were corroborated by histochemical analyses for COX and SDH activity in AAV2.1-TFEB-transfected and TFEB KO muscles. Indeed, only SDH activity was greatly reduced in the absence of TFEB, while both COX and SDH were increased in TFEB-overexpressing muscles (Figure 2F). To further investigate the role of TFEB in mitochondrial function, we generated an inducible muscle-specific transgenic mouse line. Acute activation of TFEB by tamoxifen treatment in adult mice recapitulated the phenotype of AAV2.1-TFEB overexpression on mitochondria biogenesis (Figure S3). Consistent with the increase of SDH and respiratory chain complex activity, mitochondrial respiration was significantly enhanced by TFEB expression in adult transgenic muscles (Figures S3A and S3B).
Figure 2.

TFEB Regulates Mitochondrial Function
(A) Quantitative real-time PCR of genes related to mitochondrial biogenesis and function in muscles of WT (blue bar), TFEB-overexpressing (red bar), and TFEB KO (green bar) mice. Data were normalized for GAPDH and expressed as fold induction relative to the WT. Data are expressed as mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01.
(B) ChIP analysis in muscles from TFEB-overexpressing and WT mice. The CLEAR elements of NRF1 and NRF2 promoters are depicted as red boxes. TSS (transcriptional start site) indicates the first codon. The histograms show the amount of immunoprecipitated DNA as detected by quantitative real-time PCR assay. Data represent mean ± SE of three independent experiments; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) Western blots of mitochondrial respiratory chain proteins. Blot is representative of four independent experiments.
(D) Densitometric quantification of blots shown in (B). Data are shown as means ± SE (n = 4); ∗p < 0.05.
(E) Mitochondrial respiratory chain activity in muscles of WT, AAV2.1-TFEB transfected, and TFEB KO mice. Data are expressed as a percentage relative to the WT. Data are expressed as mean ± SE, n = 4; ∗p < 0.05, ∗∗p < 0.01.
(F) H&E, succinate dehydrogenase (SDH), and cytochrome oxidase (COX) staining of AAV2.1-GFP, AAV2.1-TFEB transfected, and muscles from TFEB KO mice. The scale bars represent 100 μm. Magnification, 20×.
(G) ATP production rate per gram of tissue from TFEB-overexpressing and TFEB KO mice compared to controls. Data are shown as means ± SE (n = 5); ∗∗p < 0.01, ∗∗∗p < 0.001.
(H) Mitochondrial membrane potential of myofibers isolated from FDB muscles of WT and TFEB KO mice. Where indicated, 6 μM oligomycin or 4 μM FCCP were added. Each trace represents the tetramethylrhodamine methyl ester (TMRM) fluorescence of a single fiber. The fraction of myofibers with depolarizing mitochondria is indicated for each condition.
(I) Densitometric quantification of the carbonylated proteins extracted from WT and TFEB KO mice. Values are mean ± SEM (n = 4; ∗p < 0.05). A representative immunoblot for carbonylated proteins is shown in Figure S3C.
We next assessed whether these TFEB-mediated changes of mitochondrial morphology and function had any impact on energy production. ATP levels were higher in TFEB-transfected muscles and lower in TFEB-deficient muscles compared to controls (Figure 2G). To understand the mechanisms underlying the significant decrease of ATP in TFEB KO muscles, we checked mitochondrial function in these animals. Fluorescent dyes, like tetramethylrhodamine methyl ester (TMRM), monitor the mitochondrial membrane potential (Δψm), the critical parameter that drives ATP production. Therefore, we checked the status of Δψm in isolated adult fibers of WT and TFEB KO muscles. As expected, in control mice oligomycin-dependent inhibition of ATP synthase did not alter Δψm (Figure 2H), and mitochondrial depolarization was achieved after membrane permeabilization by the protonophore carbonylcyanide-p-trifluoromethoxy phenylhydrazone (FCCP). Conversely, mitochondria of TFEB null fibers underwent a significant depolarization after oligomycin treatment (Figure 2G), suggesting that these fibers were at least in part relying on reverse activity of ATP synthase to preserve their membrane potential as a consequence of proton membrane leak. In addition, oxidative stress, revealed by protein carbonylation, was significantly higher in TFEB KO mice than controls (Figures 2I and S3C).
TFEB Regulates Mitochondrial Biogenesis in Skeletal Muscle through a PGC1α- and PGC1β-Independent Mechanism
Both PGC1α and PGC1β are master regulators of mitochondrial biogenesis and oxidative metabolism. However, recent findings suggest the presence of an independent pathway that regulates mitochondrial biogenesis during exercise (Rowe et al., 2012). Therefore, we examined whether TFEB is the missing sensor of physical activity that coordinates the metabolic responses independently of PGC1α. First, we checked expression and localization of endogenous TFEB in PGC1α KO mice before and after exercise. TFEB was expressed at lower levels and more cytosolic in PGC1α KO mice when compared to controls (Figures 3A and 3B). Importantly, exercise restored a normal TFEB expression, induced TFEB nuclear translocation, and triggered upregulation of genes related to mitochondrial biogenesis (Figures 3C and S4A).
Figure 3.

Exercise Induces TFEB Expression and Nuclear Localization in PGC1α−/− Muscle
(A) TFEB mRNA levels of PGC1α−/− muscles infected with AAV2.9 control virus (light gray) or AAV2.9-TFEB (dark gray). Data were compared with TFEB mRNA level of WT mice (white). The levels of TFEB were measured in sedentary condition and post-exercise as indicated. Error bars represent mean ± SE for n = 3; ∗p < 0.05, ∗∗∗p < 0.001.
(B) TFEB immunohistochemical analysis of PGC1α−/− muscles from sedentary and exercised mice. GCN muscle cryosections were immunostained by using anti-TFEB antibody. Control means endogenous TFEB in muscles infected with AAV2.9 control virus. TFEB means the transgene TFEB in muscles infected with AAV2.9-TFEB. Arrows indicate exercise-induced TFEB nuclear localization. The scale bars represent 50 μm.
(C) Analysis of genes related to mitochondrial biogenesis in PGC1α−/− skeletal muscle infected with AAV2.9 control or with AAV2.9-TFEB virus. The mRNA levels were measured before and after acute exercise, as indicated. Data are shown as mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D and E) Muscle fatigue and physical performance are improved by TFEB expression in PGC1α-deficient muscle.
(D) The mice were systemically injected with AAV2.9-TFEB or AAV2.9 control virus. After 3 weeks from the virus injection, mice were subjected to run until exhaustion, as described in the Experimental Procedures. Data are shown as mean ± SE, n = 6; ∗p < 0.05.
(E) Soleus muscles were transected by intramuscular injection of AAV2.1-TFEB or AAV2.1 control virus, which resulted in 100% transfection efficacy. Index for fatigue means tetanic muscle force generated after 100 s of stimulation divided by the maximal tetanic tension produced during the fatigue protocol. Data are shown as mean ± SE, n = 4; ∗p < 0.05, ∗∗∗p < 0.001.
EM analysis of overexpressed TFEB in PGC1α KO mice revealed increased mitochondrial volume and density (Figure S4B). TFEB overexpression in PGC1α KO muscle also resulted in increased COX and SDH activity along with increased activities of CI, II, III, and IV (Figures S4C and S4D). The levels of PGC1α targets such as TFAM, NRF1, and NRF2 were also significantly upregulated by TFEB overexpression, even in the absence of PGC1α (Figure S5A). Importantly, systemic delivery of TFEB in muscle-specific PGC1α KO mice improved their exercise tolerance (Figure 3D). Indeed, TFEB expression was able to restore normal fatigue index when expressed in PGC1α KO muscle (Figure 3E). Altogether, these findings suggest that the induction of mitochondrial biogenesis in TFEB-overexpressing muscles does not depend on the presence of PGC1α.
To determine whether PGC1β may compensate for the lack of PGC1α, we measured the effect of TFEB overexpression on mitochondrial biogenesis in cells that were silenced for PGC1α and PGC1β. Importantly, inhibition of both PGC1 factors did not prevent or reduce TFEB-mediated induction of genes related to mitochondrial biogenesis and mitochondrial respiratory chain activity (Figure S5B).
TFEB Controls Energy Balance in Skeletal Muscle during Exercise
Physical activity has a major impact on glucose homeostasis and mitochondrial biogenesis and function. Therefore, we checked whether acute exhausting versus mild and chronic exercise regiments are equally able to activate TFEB. As a readout of TFEB activation, we monitored its nuclear localization. While acute exhausting contraction led to nuclear translocation of TFEB (Figure 4A), mild exercise did not (Figure 4B). However, 7 weeks of training with progressive increase of intensity without reaching exhaustion induced a massive TFEB nuclear translocation with concomitant cytosolic depletion (Figure 4B). Therefore, intensity and duration of training are critical factors that affect TFEB nuclear translocation and transcriptional regulation of genes related to mitochondrial biogenesis and function (Figure 4C).
Figure 4.

Exercise Induces TFEB Nuclear Translocation
(A) GCN tissue sections of TFEB KO and WT mice were immunostained with anti-TFEB antibody and counter-stained with hematotoxylin as indicated. Arrows indicate nuclear localization of endogenous TFEB in WT mice after an acute bout of exercise. The scale bars represent 50 μm.
(B) Endogenous TFEB was immunostained in muscle cryosections of sedentary, 4 days mild exercised, and 7 weeks intense trained WT mice. Arrows indicate TFEB nuclear localization.
(C) Expression analysis of genes related to mitochondrial biogenesis and mitochondrial respiratory chain in WT skeletal muscle before and after acute exercise. Data are shown as mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01.
To determine the physiological consequences of TFEB translocation during physical activity, we examined exercise performance in both TFEB KO and inducible muscle-specific TFEB transgenic mice. High-intensity exercise revealed significant training intolerance of TFEB KO mice compared to controls (Figure 5A). Conversely, acute muscle-specific activation of TFEB enhanced physical performance (Figure 5A). To better understand the exercise intolerance of the TFEB KO mice, we examined energy expenditure during treadmill running. While WT mice maintained constant levels of energy expenditure during physical activity, the TFEB KO mice displayed a drop after 15 min of physical exercise (Figure 5B). Metabolic analyses revealed that in basal condition, TFEB KO mice have a higher respiratory exchange rate (RER) than controls (Figure 5C). These data suggest that TFEB KO mice depend on glucose oxidation more than controls. In addition, while WT mice maintained a relatively constant RER during running period, TFEB KO mice showed a drop in RER after 20 min (Figure 5C). This decrease indicates a shift in substrate usage from glucose to fat metabolism. Finally, we measured glucose and fatty acid levels in muscle and blood from TFEB KO and TFEB transgenic mice before and after exercise. TFEB KO mice showed lower blood glucose levels compared to WT mice in basal condition (Figure 5D). Exercise caused a 50% reduction of blood glucose in TFEB KO, TFEB transgenic, and control mice (Figure 5D). Insulin levels mirrored the changes of blood glucose, as they were reduced in basal condition in TFEB KO mice and dropped after exercise in the different genotypes (Figure 5E).
Figure 5.

TFEB Controls Energy Balance in Skeletal Muscle
(A) High-intensity exhaustive exercise. To determine exercise capacity, mice were run on a treadmill. During high-intensity exercise, transgenic mice (red) ran more, while TFEB KO mice (gray) ran half as much as WT mice (white). Data are shown as mean ± SE, n = 10; ∗∗p < 0.01.
(B and C) Energy expenditure (B) and RER (C) were determined during exercise in TFEB KO and WT mice. The mice ran at a fixed speed of 10 m/min and an incline of 20°. The figure shows the mean RER measured at peak oxygen consumption. Data were transformed by Blom’s method to obtain both normally distributed data and normally distributed residual. Two-way ANOVA was used for comparison of RERs in the two groups during the entire resting period, whereas t test was used for comparison of individual time points, n = 8.
(D–F) Blood and muscle metabolites before and after high-intensity exercise in WT (white), TFEB transgenic (red), and TFEB KO mice (gray). Data are shown as mean ± SE, n = 8; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(G) Enzymatic quantification of muscle glycogen before and after high-intensity exercise in WT (white), TFEB transgenic (red), and TFEB KO mice (gray). Data are shown as mean ± SE, n = 8; ∗∗p < 0.01.
(H) Periodic acid-Schiff (PAS) staining of cryosections from AAV2.1-GFP, AAV2.1-TFEB transfected, and TFEB KO mice. Inserts show PAS staining after glycogen breakdown. The scale bars represent 100 μm.
(I and J) Quantitative analysis of serum (I) and muscle (J) NEFA before and after high-intensity exercise in TFEB KO (gray) and WT mice (white). Data are shown as mean ± SE, n = 8; ∗p < 0.05, ∗∗p < 0.01.
(K) Enzymatic quantification of B-hydroxybutyrate before and after high-intensity exercise in TFEB KO (gray box) and WT mice (white box). Data are shown as mean ± SE, n = 8; ∗p < 0.05.
(L) TFEB ablation results in a decreased rate of insulin-stimulated GIR. EU clamps were used to assess whole-body insulin sensitivity by determining the GIR required to maintain euglycemia in WT (white) and TFEB KO (gray) mice. Data represent the means ± SE from five to six individual mice per group; ∗p < 0.05.
(M) TFEB deletion results in a reduction of insulin-stimulated glucose uptake in muscle. Insulin-stimulated glucose uptake into muscle tissues was determined by 2-deoxy-d-[1-14C]glucose injection during the last 35 min of insulin infusion during the EU clamp. These data represent the means ± SE of five to six mice per group; ∗p < 0.05. WT, white; TFEB KO, gray.
(N) The amount of glucose conversion to glycogen in GCN muscles from WT (white) and TFEB KO (gray) mice was determined during the EU clamp by infusion of [3-3H]glucose. Data represent the means ± SE from five to six mice per group; ∗p < 0.05.
(O and P) TFEB KO mice do not show any significant difference in glucose homeostasis of liver or adipose tissue. (O) Glucose uptake into adipose tissue (EPI) and (P) insulin suppression of hepatic glucose output (HGP, liver) were determined during the EU clamp by infusion of 2-deoxy-d-[1-14C]glucose or [3-3H]glucose infusion into WT mice (white) and TFEB KO (gray) mice. These data represent the means ± SE from five to six mice per group.
TFEB KO mice are hypoglycemic, contain dysfunctional mitochondria, and produce less ATP. Thus, we reasoned that they use anaerobic glycolysis to produce energy. Consistent with this hypothesis, we found higher levels of lactate in the blood of TFEB KO mice before and after exercise compared to controls (Figure 5F). Conversely, lactate of transgenic mice was already lower than controls in resting condition and did not increase after exercise (Figure 5F). Therefore, TFEB transgenic mice better utilize glucose for energy production. To further confirm this finding, we measured glycogen levels in muscle. Glycogen levels were remarkably lower in TFEB KO mice and higher in TFEB transgenic in basal condition compared to WT. Enzymatic quantification showed that glycogen content was 3-fold less after TFEB ablation (Figure 5G), while it was 10-fold higher after TFEB overexpression compared to controls (Figure 5G). This was confirmed by periodic acid-Schiff (PAS) staining (Figure 5H). Exercise led to glycogen consumption in both TFEB KO muscles and controls (Figure 5G). The lower glycogen content detected in sedentary TFEB KO muscles explains the decrease of RER observed after a 15 min exercise (Figure 5C). Since glycogen is rapidly depleted in TFEB KO mice, the additional need for energy during exhausting exercise requires a switch from glycolysis to fatty acid oxidation. Consistently, while blood free fatty acid concentrations were reduced after exercise in both TFEB KO mice and controls (Figure 5I) and blood levels of non-esterified fatty acids (NEFAs) did not differ between genotypes (Figure 5I), their muscle content dramatically decreased after exercise only in TFEB KO mice (Figure 5J). Importantly, ketones did not differ between TFEB KO and controls (Figure 5K). These findings confirm a change in metabolic flexibility in the absence of TFEB that forced muscle cells to use lipids for ATP production. The exhaustion of the lipid fuel in KO mice results in inability to maintain the same exercise intensity of controls.
TFEB Controls Metabolic Flexibility and Energy Balance Independently of Autophagy
We and others have found that autophagy is important for mitochondrial quality control and is activated by exercise to clear dysfunctional mitochondria (Lo Verso et al., 2014). Thus, we checked whether TFEB controls autophagy in adult skeletal muscles. Surprisingly, TFEB activation was not sufficient to enhance autophagy flux and TFEB deletion did not impair autophagy flux in the presence or absence of nutrients (Figures S6A and S6B). Moreover, activation of TFEB did not induce protein breakdown and muscle loss. In fact, most of the atrophy-related genes belonging to the ubiquitin proteasome and autophagy-lysosome systems were not induced by TFEB expression (Figure S6C). Similarly, mitophagy genes were not upregulated. Since protein degradation is not affected by TFEB activation, we checked whether genes related to protein synthesis were modulated by TFEB. However, when we checked a cross-sectional area, we found a shift toward smaller size in transgenic mice, suggesting that protein synthesis was not induced. This decrease in fiber size is due to a metabolic shift because oxidative fibers are smaller than glycolytic skeletal muscle fibers (Figure S7A). Furthermore, we did not find any significant difference in myosin distribution between transgenic and control muscles (Figure S7B). Therefore, TFEB controls myofiber metabolism, but not myosin content/type, independently of autophagy or proteostasis.
TFEB Controls Glucose Homeostasis and Insulin Sensitivity Independently of PGC1α
Because muscles from TFEB KO mice contain lower glycogen levels than controls, we reasoned that they may have abnormal regulation of glucose homeostasis. Thus, we performed euglycemic-hyperinsulinemic (EU) clamps and observed that the glucose infusion rate (GIR) that is required to maintain a constant glycemia during insulin treatment was significantly reduced in TFEB KO compared to control mice (Figure 5L). The reduction of GIR was consequent to a decrease in skeletal muscle glucose uptake (Figure 5M), which caused a decrease of glycogen synthesis (Figure 5N). Although there was an apparent small decrease in adipose tissue glucose uptake, this was not statistically significant (Figure 5O). Moreover, insulin was equally effective in suppressing hepatic glucose output during the clamp experiment (Figure 5P). Together, these data demonstrate that TFEB deficiency in skeletal muscle results in peripheral insulin resistance-reduced glucose uptake and decreased glycogen content.
These findings are consistent with the transcriptomic signature of TFEB-overexpressing and TFEB KO muscles. To further confirm these findings, we monitored the expression levels of glucose homeostasis-related genes in muscles injected with AAV-TFEB and in muscles of TFEB transgenic mice and found a significant increase in the expression of GLUT1 and GLUT4, the GTPase involved in GLUT4 translocation (TBC1D1), and the rate-limiting enzymes of glycolysis (hexokinase 1 and 2) (Figure 6A). Immunoblotting analyses confirmed increased protein levels of GLUT1 in TFEB-overexpressing muscles (Figure 6B). The induction of genes involved in glucose uptake was also coupled with an increase of transcript and protein of GYS (Figures 6C and 6D).
Figure 6.

TFEB Controls Genes Related to Glucose Metabolism
(A and C) Quantitative real-time PCR of glucose uptake and glycogen biosynthesis-related genes in GCN muscles infected with AAV2.1-TFEB (black bar) and in TFEB transgenic muscles (red bar) compared with WT (white bar) muscles. Data were normalized for GAPDH and expressed as fold induction relative to the WT. Data are shown as mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01.
(B) Western blot of GLUT1 and densitometric quantification. Values are normalized for GAPDH; ∗p < 0.05.
(D) Western blot analysis of total and phosphorylated GYS from extracts of GCN. Representative blot images are shown (left panel). Densitometric quantification is depicted on the right panel. Data are shown as mean ± SE, n = 3; ∗p < 0.05.
(E–G) TFEB regulates glycogen synthesis independently of PGC1α.
(E) Quantitative real-time PCR of genes related to glucose uptake and glycogen biosynthesis in PGC1α−/− and WT mice that were infected with AAV2.1-GFP (white) or AAV2.1-TFEB (black). Data are shown as mean ± SE, n = 3; ∗p < 0.05, ∗∗p < 0.01.
(F) PAS staining of cryosections from PGC1α−/− muscles that were transfected by AAV2.1-GFP or AAV2.1-TFEB. The scale bars represent 100 μm.
(G) Enzymatic quantification of basal glycogen levels in PGC1α−/− and WT muscle, infected with AAV2.1-GFP (white) or AAV2.1-TFEB (black).
GYS activity is negatively regulated by phosphorylation of the C-terminal region by GYS kinases (GSK3s) (Jensen and Lai, 2009). However, the phospho-GYS levels were unchanged in AAV-TFEB muscles, and therefore, the pGYS/GYS ratio was dramatically decreased in TFEB-overexpressing muscles when compared to controls (Figure 6D). Furthermore, expression analysis of genes related to glucose metabolism did not reveal any significant difference between PGC1α KO and controls after TFEB overexpression (Figure 6E). Finally, EM showed an accumulation of glycogen in PGC1α KO mice that were infected by AAV2.1-TFEB (Figure S4B). Quantitative and qualitative analyses of glycogen showed that TFEB induced a higher glycogen accumulation in PGC1α KO muscle than in WT (Figures 6F and 6G). These data indicate that TFEB is able to control muscle glycogen content in a PGC1α-independent manner.
Glucose-Related Signaling Pathways Are Affected by TFEB
The activity of GLUT transporters is tightly controlled by several pathways. Thus, we checked whether TFEB impinges not only on GLUT1/4 expression but also on glucose-related signaling. Previous studies have shown that nitric oxide (NO) controls several metabolic aspects of skeletal muscle, including mitochondrial biogenesis and glucose uptake. NO is formed by nitric oxide synthase (NOS) via the conversion of L-arginine to L-citrulline. Skeletal muscles express neuronal (nNOS), endothelial (eNOS), and inducibile (iNOS) isoforms (Andrew and Mayer, 1999). Moreover, nNOS is the major isoform involved in AMPK-dependent regulation of GLUT4 (Lira et al., 2007). We found that the nNOS transcription and protein expression were increased in TFEB-overexpressing muscle. Conversely, real-time PCR and immunoblotting experiments showed a decrease of nNOS expression in TFEB KO muscle, as compared with controls (Figures 7A and 7B). To test whether nNOS and glucose transporter genes are direct targets of TFEB, we analyzed their promoters and identified CLEAR sites. ChIP experiments showed that TFEB is recruited on nNOS, GLUT1, and GLUT4 promoters (Figure 7C). Since AMPK is a downstream target of nNOS (Lira et al., 2010), we monitored the activation of AMPK. TFEB overexpression in muscle showed a significant induction of AMPK phosphorylation and of its downstream target acetyl-CoA carboxylase (ACC). Interestingly, TFEB overexpression triggered protein kinase B (AKT) activation. However, no changes in pAMPK and pACC were found in TFEB KO muscles (Figure 7D). Consistent with the presence of insulin resistance, we detected decreased AKT phosphorylation in TFEB KO muscles.
Figure 7.

TFEB Also Controls Glucose Uptake via Regulation of AMPK Signaling Pathways
(A) Quantitative real-time PCR of nNOS transcript in WT (white bar), TFEB-overexpressing (black), and TFEB KO muscles (gray). Values are expressed as fold induction compared to controls. Data are shown as mean ± SE, n = 8; ∗p < 0.05, ∗∗p < 0.01.
(B) Western blot of nNOS and densitometric quantification. Value are normalized for GAPDH; ∗p < 0.05; ∗∗∗p < 0.001.
(C) ChIP analyses of muscles from TFEB transgenic and WT mice on nNOS, GLUT1, and GLUT4 promoters. The histograms show the amount of immunoprecipitated DNA as detected by quantitative real-time PCR assay. Data represent mean ± SE of three independent experiments; ∗p < 0.05.
(D) Western blot of AMPK, PAMPK, PACC, and PAKT performed on protein extracts from GCN muscles of AAV2.1-TFEB transfected, TFEB KO, or WT mice. Representative images (left) and densitometric quantification (right) are shown; ∗p < 0.05.
Discussion
The beneficial effects of physical activity on mitochondrial content/function, fatty acid oxidation, and glucose homeostasis are well known (Hawley, 2002, Holloszy and Coyle, 1984, Holloszy et al., 1998). Indeed, muscle activity is important to counteract disease progression in diabetes, obesity, and metabolic syndrome. Here we have found that TFEB is a major regulator of glucose homeostasis and mitochondrial biogenesis to provide the energetic support to maintain muscle contraction. Our in vivo data show that the absence of TFEB causes accumulation of morphologically abnormal and dysfunctional mitochondria that display impairment in respiratory chain complex II activity and proton leakage, leading to a defect in ATP production and exercise intolerance. Conversely, overexpression of TFEB induces mitochondrial biogenesis, improves respiratory chain complex activities, and increases ATP production. These findings are surprising since the documented effects of TFEB are mainly related to lysosomal biogenesis and autophagy regulation.
The positive effects of TFEB on the mitochondrial network in skeletal muscle appear to be independent from PGC1α. We found that TFEB is sufficient to induce the expression of NRF2 and Tfam, two major master regulators of mitochondrial biogenesis in muscle, even in the absence of both PGC1α and PGC1β, and is both sufficient and required for PPARα expression. Therefore, TFEB acts independently of PGC1α to promote oxidation of glucose and lipids, which are critical substrates during the early and late phase of strenuous contraction, respectively. Therefore, TFEB is at least one missing factor that explains why PGC1α is dispensable for exercise and mitochondrial biogenesis (Rowe et al., 2012). Moreover, TFEB directly controls glucose homeostasis via GLUT1/4 expression and insulin sensitivity via nNOS. Indeed, muscle from TFEB KO mice showed decreased glucose uptake during EU clamps, nearly completely absent glycogen stores, and reduced AKT phosphorylation under resting conditions. The insulin resistance of the TFEB-deficient fibers prevents glucose oxidation and therefore drives the exercising muscle to use fatty acid oxidation, which consequently blocked pyruvate dehydrogenase (PDH) enzyme (Figure 2A), resulting in lactate accumulation. Altogether, these findings suggest that TFEB is a critical player of metabolic flexibility during physical activity.
In a previous study, we reported that TFEB regulates lipid metabolism in liver, and this effect appears to be mediated, at least in part, by PGC1α (Settembre et al., 2013). Conversely, our findings in muscle show that TFEB is directly involved in mitochondrial function and glucose homeostasis independently from PGC1α. These observations indicate that the networks of genes regulated by TFEB are context specific, for the gene expression profiles have significant tissue-specific changes, supporting distinct tissue-specific metabolic functions.
Calcium signaling is greatly affected by exercise, and the calcium-dependent phosphatase calcineurin is one of the most important players for muscle adaptation to physical activity. We have recently found that exercise triggers TFEB nuclear localization in a calcineurin-dependent fashion (Medina et al., 2015). Calcineurin dephosphorylates TFEB serine residues that play a critical role in determining TFEB subcellular localization and promotes its nuclear translocation (Medina et al., 2015). Importantly, calcium-dependent signaling also modulates exercise-dependent, glucose-related pathways. In fact, muscle-specific transgenic mice that overexpress an activated form of calcineurin show increased glycogen accumulation and lipid oxidation (Long et al., 2007) and upregulation of PGC1α, several glycolytic enzymes, mitochondrial genes, genes related to lipid metabolism, and GLUT4 (Long and Zierath, 2008). Conversely, muscle-specific calcineurin KO mice show exercise intolerance when subjected to exhausting physical activity (Pfluger et al., 2015). Finally, immunosuppression therapy by high-dose treatment with calcineurin inhibitors cylosporin A or tacrolimus (FK-506) has been associated with a higher risk for developing obesity and diabetes in patients (T D Correia et al., 2003). Altogether, these data suggest that calcineurin plays a major role in glucose and lipid metabolism, although the mechanistic insights of these beneficial effects of calcineurin are unknown. Our data suggest that TFEB is a critical calcineurin downstream target that coordinates metabolic adaptations such as glucose uptake and mitochondria function to optimize energy production to sustain muscle contraction.
TFEB not only regulates expression of glucose transporters and critical glycolytic enzymes but also factors that impinge on AMPK regulation, such as nNOS. It was shown that nNOS controls GLUT4 expression in skeletal muscle cells through AMPK activation. In addition, endogenous nNOS is required for the upregulation of AMPK activity by the AMP mimetic, AICAR (Higaki et al., 2001, Lira et al., 2010, McConell et al., 2010).
In summary, our results position TFEB as a central coordinator of insulin sensitivity, glucose homeostasis, lipid oxidation, and mitochondrial function, further emphasizing the importance of this regulatory pathway in the metabolic response to energy-demanding conditions such as physical exercise.
Experimental Procedures
Cell Culture, Plasmids, and Transfection Reagent
Detailed information in the Supplemental Experimental Procedures.
Real-Time PCR
Quantitative real-time PCR analyses were performed on a LigthCycler 480 II (Roche). For detailed information on preparation and gene expression analysis, see the Supplemental Experimental Procedures.
Generation of Muscle-Specific TFEB KO and Inducible Muscle-Specific Transgenic Mice and In Vivo Experimental Procedures
Detailed information in the Supplemental Experimental Procedures.
Western Blot Analysis and Antibodies
Total homogenates were prepared in RIPA buffer (50 mM Tris HCl [pH 8], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) with the addition of a protease inhibitor cocktail (Roche). Protein concentration was determined by the Lowry method. Aliquots, 50 μg each, were run through an SDS-PAGE and electroblotted onto a PVDF membrane, which was then matched with different antibodies. For the antibodies used in western blot analysis, see the Supplemental Experimental Procedures.
Biochemical Analysis of Mitochondrial Respiratory Chain Complex
Muscle samples stored in liquid nitrogen were homogenized in 10 mM phosphate buffer (pH 7.4), and the spectrophotometric activity of cI, cII, cIII, and cIV, as well as citrate synthase (CS), was measured as described (Bugiani et al., 2004). Detailed information in the Supplemental Experimental Procedures.
Morphological Analysis
Histochemical and ultrastructural analyses were performed as described (Sciacco and Bonilla, 1996). Detailed information in the Supplemental Experimental Procedures.
Isolation of Skeletal Myofibers and Measurements of Mitochondrial Membrane Potential
Muscle fibers were isolated from FDB muscle and mitochondrial membrane potential measured by epifluorescence microscopy on the basis of the accumulation of TMRM fluorescence, as previously described (Lo Verso et al., 2014). Fibers were considered as depolarizing when they lost more than 10% of the initial value of TMRM fluorescence. Imaging was performed with a Zeiss Axiovert 100 TV inverted microscope equipped with a 12-bit digital cooled charge-coupled device camera (Micromax, Princeton Instruments). The results were analyzed with MetaFluor imaging software (Universal Imaging).
Acute Exercise, Training, and Fatigue Experiments
For acute and chronic exercise studies, 16-week-old mice performed concentric exercise on a treadmill (Biological Instruments, LE 8710 Panlab Technology 2B) with a 10° incline, according to the protocol of exercise previously described (Medina et al., 2015). Total running distance was recorded for each mouse. Detailed information in the Supplemental Experimental Procedures.
Immunohistochemistry
Detailed information in the Supplemental Experimental Procedures.
Electron Microscopy
Detailed information in the Supplemental Experimental Procedures.
Plasma Chemistry Analysis
Blood was collected from the orbital plexus under isoflurane (Vedco) anesthesia. Plasma was frozen in aliquots at −20°C or used immediately after collection. Specific enzymatic kits were used for determination of serum NEFAs (Wako) and lactate (Abcam). Plasma glucose was monitored by a glucometer. Insulin was measured by ELISA (Mercodia).
Tissue Metabolite Quantification
Detailed information in the Supplemental Experimental Procedures.
Whole-Body Indirect Calorimetry
Metabolic measurements were performed using an Oxymax indirect calorimetry system (Columbus Instruments) (Zong et al., 2011). Detailed information in the Supplemental Experimental Procedures.
In Vivo Assessment of Insulin Action and Glucose Metabolism
Four days before the experiment, the mice were anesthetized and an indwelling catheter was introduced into the left internal jugular vein. The mice were fully recovered from the surgery before the in vivo experiments, as reflected by their reaching preoperative weight. After an overnight fast, EU clamps were conducted in conscious mice as previously described (Ayala et al., 2006, Zong et al., 2012). Detailed information in the Supplemental Experimental Procedures.
ChIP Assays
We performed ChIP assays on adult skeletal muscle overexpressing TFEB3XFlag, and on WT muscle as control, by using the ChIP assay kit (Upstate) according to Milan et al. (2015). For immunoprecipitation, we used anti-FLAG antibody (F7425, Sigma-Aldrich). Oligonucleotide primers for amplification of a TFEB binding site on the GLUT1, GLUT4, nNOS, NRF1, NRF2, and TFAM promoters are listed in Table S7.
Microarray Data Analysis
Detailed information in the Supplemental Experimental Procedures.
Statistical Analysis
Data are expressed as mean values ± SE. Results were evaluated by repeated-measures ANOVA, multivariate ANOVA (MANOVA), or Student’s two-tailed t test. p < 0.05 was considered statistically significant. In all figures, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
G.M., M.S., and A.B. designed the project and experiments and wrote the manuscript. G.M., A.A., and C.V. performed experiments and analyzed and interpreted data. G.M. and A.A. generated transgenic mice. E.V.P., C.L., I.D.M., V.R., H.Z., B.B., F.S., and C.T. contributed to experiments and data collection; P.G. and P.B. provided reagents and discussed data; R.D.C. performed computational analysis; and L.D., S.M., and P.K.S. provided technical expertise. J.E.P., M.Z., M.S., and A.B. discussed, reviewed, and edited the manuscript. M.S. and A.B. supervised the work.
Acknowledgments
We thank N. Brunetti, D. Medina, and C. Settembre for suggestions. We are grateful to E. Nusco and TIGEM Bioinformatic and HCS Facilities for technical support, in particular D. Carrella and S. Montefusco. We acknowledge support from the Italian Telethon Foundation to A.B. (TCR09003) and M.S. (TCP04009), the European Research Council (ERC) to A.B. (250154-CLEAR) and M.S. (282310-MyoPHAGY), the Italian Ministry of Education (MiUR) (PRIN 2010/2011) to M.S., CARIPARO and Foundation Leducq to M.S., the Pennsylvania University and Beyond Batten Disease Foundation to A.B., the Core Grant MRC-MBU and ERC FP7-322424 to M.Z., GR-2010-2306-756 and the Italian Ministry of Health to C.V., and the National Science Foundation of China (H0315: 81370531) to H.Z.
Published: December 20, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.11.003.
Contributor Information
Marco Sandri, Email: marco.sandri@unipd.it.
Andrea Ballabio, Email: ballabio@tigem.it.
Supplemental Information
Related to Figure 1. Complete list of differentially expressed genes in TFEB overexpression muscle microarray dataset (GSE62975). The genes significantly (FDR<0.05; FoldChange ≥ 1.5) upregulated and downregulated are highlighted in red and in green, respectively.
Related to Figure 1. Complete list of differentially expressed genes in TFEB-KO muscle microarray dataset (GSE62976). The genes significantly (FDR<0.05) upregulated and downregulated are highlighted in red and in green, respectively.
Related to Figure 1. List of Lipid and Glucose Metabolism related terms related to the GOEA induced by TFEB overexpression and inhibited by TFEB-KO. For each term the genes significantly induced by TFEB overexpression (Supplemental Table BPALL_UP_OverTFEB) and inhibited by TFEB-KO (Supplemental Table BPALL_DOWN_KO-TFEB) are reported in table.
Related to Figure 2. Position and sequence of the CLEAR sites (i.e., the consensus TFEB binding-site) in the promoter region (1Kb upstream the TSS in the human and Mouse promoters) of mitochondrial genes downregulated in TFEB-KO muscle.
Related to Figures 2 and 7. Position and sequence of TFEB binding site on the GLUT1, GLUT4, nNOS, TFAM, NRF1 and NRF2 promoter.
References
- Andrew P.J., Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999;43:521–531. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- Ayala J.E., Bracy D.P., McGuinness O.P., Wasserman D.H. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- Bothe G.W., Haspel J.A., Smith C.L., Wiener H.H., Burden S.J. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Calabria E., Ciciliot S., Moretti I., Garcia M., Picard A., Dyar K.A., Pallafacchina G., Tothova J., Schiaffino S., Murgia M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc. Natl. Acad. Sci. USA. 2009;106:13335–13340. doi: 10.1073/pnas.0812911106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert S., Bloch W., Suhr F. Ca2+-dependent regulations and signaling in skeletal muscle: from electro-mechanical coupling to adaptation. Int. J. Mol. Sci. 2015;16:1066–1095. doi: 10.3390/ijms16011066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P., Bonaldo P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells. 2012;1:325–345. doi: 10.3390/cells1030325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley J.A. Adaptations of skeletal muscle to prolonged, intense endurance training. Clin. Exp. Pharmacol. Physiol. 2002;29:218–222. doi: 10.1046/j.1440-1681.2002.03623.x. [DOI] [PubMed] [Google Scholar]
- Higaki Y., Hirshman M.F., Fujii N., Goodyear L.J. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes. 2001;50:241–247. doi: 10.2337/diabetes.50.2.241. [DOI] [PubMed] [Google Scholar]
- Holloszy J.O., Coyle E.F. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Holloszy J.O., Kohrt W.M., Hansen P.A. The regulation of carbohydrate and fat metabolism during and after exercise. Front. Biosci. 1998;3:D1011–D1027. doi: 10.2741/a342. [DOI] [PubMed] [Google Scholar]
- Jensen J., Lai Y.C. Regulation of muscle glycogen synthase phosphorylation and kinetic properties by insulin, exercise, adrenaline and role in insulin resistance. Arch. Physiol. Biochem. 2009;115:13–21. doi: 10.1080/13813450902778171. [DOI] [PubMed] [Google Scholar]
- Kelly D.P., Scarpulla R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Lira V.A., Soltow Q.A., Long J.H., Betters J.L., Sellman J.E., Criswell D.S. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007;293:E1062–E1068. doi: 10.1152/ajpendo.00045.2007. [DOI] [PubMed] [Google Scholar]
- Lira V.A., Brown D.L., Lira A.K., Kavazis A.N., Soltow Q.A., Zeanah E.H., Criswell D.S. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Verso F., Carnio S., Vainshtein A., Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014;10:1883–1894. doi: 10.4161/auto.32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y.C., Zierath J.R. Influence of AMP-activated protein kinase and calcineurin on metabolic networks in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008;295:E545–E552. doi: 10.1152/ajpendo.90259.2008. [DOI] [PubMed] [Google Scholar]
- Long Y.C., Glund S., Garcia-Roves P.M., Zierath J.R. Calcineurin regulates skeletal muscle metabolism via coordinated changes in gene expression. J. Biol. Chem. 2007;282:1607–1614. doi: 10.1074/jbc.M609208200. [DOI] [PubMed] [Google Scholar]
- Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero E., Agatea L., Mammucari C., Blaauw B., Loro E., Komatsu M., Metzger D., Reggiani C., Schiaffino S., Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- McConell G.K., Ng G.P., Phillips M., Ruan Z., Macaulay S.L., Wadley G.D. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J. Appl. Physiol. 2010;108:589–595. doi: 10.1152/japplphysiol.00377.2009. [DOI] [PubMed] [Google Scholar]
- McCullagh K.J., Calabria E., Pallafacchina G., Ciciliot S., Serrano A.L., Argentini C., Kalhovde J.M., Lømo T., Schiaffino S. NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. USA. 2004;101:10590–10595. doi: 10.1073/pnas.0308035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan G., Romanello V., Pescatore F., Armani A., Paik J.H., Frasson L., Seydel A., Zhao J., Abraham R., Goldberg A.L. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- Pfluger P.T., Kabra D.G., Aichler M., Schriever S.C., Pfuhlmann K., García V.C., Lehti M., Weber J., Kutschke M., Rozman J. Calcineurin links mitochondrial elongation with energy metabolism. Cell Metab. 2015;22:838–850. doi: 10.1016/j.cmet.2015.08.022. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A., Petit C.S., Froehlich F., Qian S., Ky J., Angarola B., Walther T.C., Ferguson S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe G.C., El-Khoury R., Patten I.S., Rustin P., Arany Z. PGC-1α is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PLoS ONE. 2012;7:e41817. doi: 10.1371/journal.pone.0041817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., De Cegli R., Mansueto G., Saha P.K., Vetrini F., Visvikis O., Huynh T., Carissimo A., Palmer D., Klisch T.J. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T D Correia M.I., Rego L.O., Lima A.S. Post-liver transplant obesity and diabetes. Curr. Opin. Clin. Nutr. Metab. Care. 2003;6:457–460. doi: 10.1097/01.mco.0000078994.96795.d8. [DOI] [PubMed] [Google Scholar]
- Zong H., Wang C.C., Vaitheesvaran B., Kurland I.J., Hong W., Pessin J.E. Enhanced energy expenditure, glucose utilization, and insulin sensitivity in VAMP8 null mice. Diabetes. 2011;60:30–38. doi: 10.2337/db10-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H., Armoni M., Harel C., Karnieli E., Pessin J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2012;302:E532–E539. doi: 10.1152/ajpendo.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 1. Complete list of differentially expressed genes in TFEB overexpression muscle microarray dataset (GSE62975). The genes significantly (FDR<0.05; FoldChange ≥ 1.5) upregulated and downregulated are highlighted in red and in green, respectively.
Related to Figure 1. Complete list of differentially expressed genes in TFEB-KO muscle microarray dataset (GSE62976). The genes significantly (FDR<0.05) upregulated and downregulated are highlighted in red and in green, respectively.
Related to Figure 1. List of Lipid and Glucose Metabolism related terms related to the GOEA induced by TFEB overexpression and inhibited by TFEB-KO. For each term the genes significantly induced by TFEB overexpression (Supplemental Table BPALL_UP_OverTFEB) and inhibited by TFEB-KO (Supplemental Table BPALL_DOWN_KO-TFEB) are reported in table.
Related to Figure 2. Position and sequence of the CLEAR sites (i.e., the consensus TFEB binding-site) in the promoter region (1Kb upstream the TSS in the human and Mouse promoters) of mitochondrial genes downregulated in TFEB-KO muscle.
Related to Figures 2 and 7. Position and sequence of TFEB binding site on the GLUT1, GLUT4, nNOS, TFAM, NRF1 and NRF2 promoter.