

Abstract
Granulocyte colony stimulating factor (G-CSF) is a hematopoietic cytokine that stimulates neutrophil production and hematopoietic stem cell mobilization by initiating the dimerization of homodimeric granulocyte colony stimulating factor receptor (G-CSFR). Different mutations of CSF3R have been linked to a unique spectrum of myeloid disorders and related malignancies. Myeloid disorders caused by the CSF3R mutations include severe congenital neutropenia (SCN), chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML). In this review, we provide an analysis of G-CSFR, various mutations and their roles in the SCN, CNL and malignant transformation as well as the clinical implications and some perspective on approaches that could expand our knowledge with respect to the normal signaling mechanisms and those associated with mutations in the receptor.
Keywords: CSF3R, G-CSF, G-CSFR, severe congenital neutropenia (SCN), chronic neutrophilic leukemia (CNL), acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), transmembrane proximal mutations, intracellular truncation mutations, JAK/STAT signaling, suppressor of cytokines (SOCSs)

Introduction
Hematopoietic stem cells (HSCs) lead to the formation of all blood cell types. HSCs divide to self-renew as well as to give rise to multipotent and lineage committed hematopoietic progenitor cells (HPCs) (1). Due to their short life span of about 5.4 days, mature granulocytes require constant replenishment from their respective precursor cells (1). Production of 0.5–1.0 × 1011 granulocytes every day in an adult human shows the enormous nature of the overall hematopoietic process (1). Neutrophils are one of the most abundant (50% –70%) blood cell types and play a seminal role in the innate immune system (2). They are motile in nature and function as phagocytes (2). Neutrophils are one of the first innate immune responders to a site of infection or injury caused by bacterial infection, harmful environmental exposure, and various cancers (2). Granulocyte colony stimulating factor (G-CSF) also known as colony stimulating factor 3 (CSF3) is the main growth factor that controls both the proliferation and differentiation of myeloid progenitor cells into neutrophils (3). G-CSF is the primary ligand for granulocyte colony stimulating factor receptor (G-CSFR). G-CSFR is encoded by the colony stimulating factor 3 receptor (CSF3R) gene. Since G-CSF instructs neutrophil production, it is widely used for the treatment of a bone marrow failure syndrome termed severe congenital neutropenia (SCN) with a diagnostic feature of an overall neutrophil counts below 0.5 million per liter. G-CSF is also used to treat cases involving the leukemic progression of SCN (3).
Granulocyte colony stimulating factor (G-CSF) and its receptor
G-CSF and its receptor G-CSFR is a cytokine receptor which has important roles in myeloid progenitor's proliferation and differentiation leading to neutrophil development. Other similar receptors responsible for myeloid developmental processes are macrophage colony stimulating factor receptor (M-CSFR), and granulocyte macrophages colony stimulating factor receptor (GM-CSFR) encoded by CSF1R and CSF2R genes respectively. Macrophage development is orchestrated by M-CSFR whereas GM-CSFR modulates the development of granulocytes and macrophages. While G-CSFR and GM-CSFR are part of the hematopoietin/cytokine receptor superfamily, M-CSFR is a receptor tyrosine kinase. Each receptor is activated by their cognate cytokine, colony stimulating factor (CSF), and the incidence of cross-talk among these cytokine receptors is largely unknown. CSFs are smaller molecular weight secreted glycoproteins which bind to the cell surface receptors of hematopoietic stem cells. The intracellular signaling induced by CSFs binding on their respective receptors in turn activates downstream signaling pathways leading to proliferation and differentiation of stem and progenitor cells into mature granulocyte and macrophages. G-CSF and its receptor G-CSFR play an essential role in the production of neutrophils under basal as well as under the emergency physiological conditions caused by various infections (3, 4).
G-CSFR is a transmembrane protein consisting of 813 amino acids: 604 as an extracellular domain, 26 in the transmembrane domain and 184 in the intracellular cytoplasmic domain (figure 1) (5). The extracellular domain of the receptor is comprised of an immunoglobulin-like (Ig-like) domain, a cytokine receptor homology domain (CRH domains) and three fibronectin type III (FN III) domains (figure 1) (5). Four highly conserved cysteine residues and a WSXWS motif of the CRH domains are required for ligand binding and the receptor activation (5, 6).
Figure 1.

Topography of Granulocyte-Colony Stimulating Factor Receptor (G-CSFR) showing various structural domains and the mutations associated with specific domain leading to neutropenia and leukemia.
G-CSFR has no intrinsic tyrosine kinase activity (7–9); however, upon ligand binding the receptor undergoes a conformational change leading to the activation of several downstream pathways including JAK/STAT, PI3K/AKT and MAPK/ERK (figure 2). The cytoplasmic tail of the G-CSFR receptor is divided into the three distinct box regions (Box 1–3). Box 1 and box 2 are required for proliferation signals and box 3 is essential for inducing the differentiation of myeloid progenitor cells (figure 1) (7). Besides box regions, four tyrosine residues at 704, 729, 744 and 764 play important roles in the signaling of the receptor (7, 8). These tyrosine residues provide docking sites for Src homology 2 (SH2) domain containing signaling proteins after activation mediated by phosphorylation (7, 8). Ubiquitination also plays an important role in G-CSFR signaling which is controlled by E3 ubiquitin ligase activity. E3 ubiquitin ligase mediated modification mainly occurs at five lysine residues 632, 672, 681, 682 and 762 of the receptor (8, 9). Ubiquitination at lysine 632 controls routing of activated G-CSFR from the early endosome vesicles to lysosomes and subsequent degradation (8, 9). An equilibrium between tyrosine phosphorylation/dephosphorylation and ubiquitination of lysine residues by E3 ubiquitin ligase maintains G-CSFR signaling and routing/recycling of the receptor (9). Neutropenia is the major disease phenotype caused by the abnormal G-CSFR signaling (10–13).
Figure 2.

Major downstream signaling pathways of normal and variant G-CSFR. (A) In normal G-CSFR, major pathways activated downstream of the receptor are JAK/STAT, PI3K/AKT and MAPK/ERK, whereas SOCS acts as negative regulator of the overall signaling. (B) G-CSFR with a mutated extracellular receptor leads to disruption of G-CSF binding causing the cessation of downstream signaling. (C) Proximally mutated receptor becomes auto activated and facilitates the constitutive activation of the downstream pathways even in the absence of ligand. (D) Several truncations in the cytoplasmic domains of G-CSFR leads to hyper response to G-CSF and uncontrolled signaling downstream of the receptor upon ligand binding.
JAK-STAT pathway
The major downstream kinases activated by G-CSFR include JAK1, JAK2, TYK2 and SRC family kinases, e.g., LYN, HCK (14–17). Additional kinases activated by the receptor are SYK and TNK (14–17). Phosphorylation of JAK kinases is mediated by the activated G-CSFR. Upon ligand binding, monomeric receptor forms the activated dimeric structure which in turn phosphorylates JAK2. The activation of JAK2 is mediated by tryptophan (W) 650 residue of G-CSFR as demonstrated by site directed mutagenesis studies (18). Activated JAK2 binds to the membrane proximal cytoplasmic region of the receptor. In addition to JAK2, G-CSFR has been shown to activate SRC kinases as well; however, the mechanism of SRC kinases activation is not known (17). Due to lack of intrinsic kinase activity, granulocyte receptors rely on JAK and SRC family kinases for signal transduction. Activated JAKs phosphorylate the tyrosine residues of STAT proteins and the G-CSFR. Phosphorylated STATs form dimers that translocate to the nucleus leading to the activation of target genes (16). The tyrosine residues at 704 and 744 of G-CSFR have been reported to assist in the activation of STAT3 by providing docking sites and thereafter STAT3 gets phosphorylated/activated by JAK2 (14–17). STAT3 has a pro-differentiation role in myeloid lineage development by stimulating the expression of the cyclin dependent kinase (CDK) inhibitor, p27kip1 which assist in cell cycle exit (6). Under normal conditions, STAT3 induces SOCS3, and SOCS3 binds the activated G-CSFR leading to degradation of the receptor and cessation of signaling (9, 10) (figure 2 (A)).
Another important signaling mediator downstream of G-CSFR is STAT5, which induces proliferation and survival (10). STAT5 is directly controlled by activated JAKs through phosphorylation (9). Similar to their biological impact, the overall activation kinetics for STAT3 and STAT5 downstream of the receptor is significantly different. STAT5 activation is transient and returns to the basal level in about 30 minutes (6, 9). In contrast, STAT3 activation is sustained for several hours (6, 9). Thus, G-CSF mediated differential activation of STAT3 and STAT5 might play an important role in modulating myeloid lineage proliferation versus differentiation.
PI3K/AKT pathway
In addition to JAK/STAT signaling, the membrane proximal region of G-CSFR has been reported to stimulate phosphatidylinositol-3 kinase (PI3K) signaling (10–14). PI3K is a lipid kinase that activates pleckstrin homology (PH) domain-containing proteins including AKT, upon G-CSF treatment (15, 16). It is not clear whether the activation of PI3K/AKT pathway is dependent on activated JAK2 downstream of G-CSFR. However, from studies of myeloproliferative neoplasm (MPN) where a combination of JAK2 and PI3K inhibitors showed a synergistic effect, suggest that activation of the PI3K/AKT pathway is, at least in part, independent from JAK2 signaling (19). Nonetheless, this possibility still requires experimental validation with respect to G-CSFR signaling. Another route of PI3K/AKT activation is likely through SRC since SRC kinases have also been shown to stimulate the PI3K/AKT pathway downstream of activated receptor (14, 19, 20). Regardless of the activation mechanism, the PI3K/AKT pathway plays an important role in the regulation of cell cycle by directly controlling proliferation and cellular quiescence (6, 9, 15, 16). Activated PI3K/AKT pathway has also been shown to block apoptosis (13, 14) through inhibition of the interaction between pro-apoptotic protein Bad and anti-apoptotic proteins Bcl2/Bcl-xl (14, 15, 20). Thus, this pathway has a pro-survival role and aids in the proliferation of myeloid progenitor cells; however additional details regarding the mechanism controlling PI3K/AKT activation by G-CSFR (either directly or through JAK2) remains to be elucidated for this receptor. Overall, G-CSF mediates proliferation of myeloid progenitor cells leading to an increase in number of mature granulocytes is likely dependent on the PI3K/AKT pathway given that this pathway also promotes cell proliferation.
MAPK/ ERK pathway
Phosphorylation of tyrosine 764 of the cytoplasmic domain of the G-CSFR activates MAPK/ERK pathway (10-14). The activation is mediated through the intermediate proteins Shc and Grb2 of p21/Ras pathway (14). Loss of 764 phosphorylation leads to a decrease in p21/Ras activation and increase neutrophils differentiation (15). In contrast, adding back tyrosine 764 to a null receptor background increases the proliferation of myeloid progenitors (15, 16). Therefore, p21/Ras pathway facilitates the proliferation of myeloid progenitors and abrogates the differentiation. Several studies have shown that the ERK1/2 MAP kinases are the major effectors downstream of p21/Ras and are directly involved in proliferative signaling in myeloid progenitor cells (17). The other MAP kinases controlled by tyrosine 764 of G-CSFR are p38MAP kinase and Jun N-terminal kinase (JNK) (20). While a direct role of p38MAP and JNK kinases in G-CSFR signaling has not been defined, there are clear indication that G-CSF-induced proliferative signals are mediated through MAPK/ERK. Future studies of the control mechanism of this pathway in mutated G-CSFR involving some of these and other downstream kinases likely will provide a greater understanding of the proliferative response causing leukemia like phenotypes.
CSF3R mutations and myeloid disorders
There are a number of myeloid disorders that have been related to mutations in CSF3R including (SCN, CNL, Myelodysplastic Syndrome (MDS), Acute Myeloid Leukemia (AML), atypical Chronic Myelogenous Leukemia (aCML)) (10-14). The most prevalent of these diseases is SCN. Many SCN patients treated with G-CSF (induction therapy) regain sufficient levels of neutrophils to reduce infection related mortality (21). However, a major concern is the leukemic progression of SCN into MDS or AML (22–27). Specifically, SCN patients on G-CSF treatment can acquire somatic mutations in the CSF3R, some of which have been shown to cause truncations of the cytoplasmic region of the receptor (27). Initially, it was thought that these CSF3R mutations drive clonal expansion by nullifying G-CSF hypo-responsiveness of HSCs and lead to leukemic progression of the SCN patients (27).
Surprisingly though, other CSF3R mutations are frequently observed de novo in CNL and aCML (28, 29) diseases both characterized by hyper-responsiveness to growth factors leading to an excess of neutrophils and/or myeloid cells. However, the specific connection between SCN and CNL/aCML, which are characterized respectively by severe loss or an excess of neutrophils remains unresolved. Some possibilities that could account for these contrasting phenotypes includes, disruption of the negative feedback loop of G-CSFR signaling through the loss of the activity of specific phosphatases (e.g. SHIPs, SHPs) perhaps leading to uncontrolled neutrophil counts in CNL; or conditions that disrupt the normal recycling or degradation of the GCSFR to mute the signal.
Overall, a variety of CSF3R mutations have been identified from patient derived samples. These various mutations may provide the key to understand the divergence of disease phenotypes and can be divided into three groups of mutations: extracellular domain, transmembrane proximal and intracellular truncation mutations (figure 1 and 2).
Extracellular domain mutations
This class of mutations in the CSF3R gene affects the extracellular domain of the G-CSFR and are commonly associated with SCN and chronic idiopathic neutropenic (CIN) patient groups (figure 2 B) (30, 31). These mutants also act in a dominant negative manner to deteriorate co-expressed wild-type receptors leading to a degenerate state of neutrophil production (30). An extracellular mutant P229H was the first reported extracellular CSF3R mutation (30, 31). It changes the conserved di-proline hinge motif on the receptor thus disrupting the ligand binding cytokine receptor homology (CRH) domain and overall architecture of the ligand/receptor complex (31). The change in the receptor architecture limits G-CSF binding and dimerization which leads to an abnormal G-CSF mediated downstream signaling of G-CSFR.
Another series of extracellular domain mutations in CSF3R cause truncations at the WSXWS motif of the receptor (30, 31). These mutation results in the amino acid substitutions at S296G, S319G or S299G causing truncations of the extracellular regions of the receptor and affecting the ligand binding efficiency (29–31). Additional extracellular domain mutation resulting in S624R, causes a frameshift truncation after the fibronectin domain of the receptor and has been reported in CIN patients (31). These extracellular CSF3R mutations do not typically lead to myeloid malignancy, but result in a neutropenic condition; however, extended treatments with G-CSF to boost neutrophils make the neutropenic patient group more susceptible to a leukemic progression due to secondary mutations.
Transmembrane proximal mutations
Transmembrane proximal point mutations in CSF3R affect the transmembrane domain and adjacent residues of the G-CSFR. These mutations are believed to act by stabilizing the receptor transmembrane helix-helix interactions even in the absence of the G-CSF ligand which leads to constitutively active signaling (32). The most prevalent proximal mutation is T618I (32). This class of mutation does not change the receptor expression compared to wild type so it is not clear how in maintains it continuous activity. One possibility may be that the mutated receptor facilitates spontaneous dimerization, constitutive activation then and get endocytosed and rapidly recycled back to the plasma membrane in a manner similar to a mutated receptor tyrosine kinase, MET or hepatocyte growth factor receptor (HGFR) (33-36). Strikingly, the point mutations M1268T and D1246N of MET receptors change its endocytic routing leading to impaired degradation and increased receptor internalization and recycling back to the plasma membrane (33-36). Similar to G-CSFR-T618I, the MET point mutations also result in a constitutively active receptor (33-36). Mutated MET receptor showed an enhanced signaling and cellular transformation in both in vitro and in vivo studies (33–35). Therefore, it is possible that the CNL phenotype exhibited by G-CSFR-T618I is also due to aberrant receptor recycling mechanisms, as this is consistent with higher receptor expression on the cell surface by leukemic cells and MET receptor in human papillary renal carcinoma (36, 53). However, this possibility requires an experimental validation.
Initially, T618I was identified in a SCN/AML patient already carrying other CSF3R mutations (32, 37). In recent studies around 83% of CNL patients show this mutation (38). This mutation has also been shown to have a predisposition towards myeloid malignancies in the neutropenic patient (39, 40). Furthermore, T618I is observed in aCML, chronic myelomonocytic leukemia (CMML), de novo AML, and in early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) but with lower frequencies (41–43). Proximal mutations also exhibit ligand (G-CSF) independent growth of BaF3 cells (41). Specifically, T618I transduced BaF3 showed activation of JAK2, SRC, TNK, STAT3 and STAT5 consistent with the constitutive activation (39–41). The major pathway activated by this class of mutation is JAK/STAT (figure 2). Consistent with this observation, ruxolitinib, a JAK kinase inhibitor, has been shown to inhibit the downstream JAK/STAT signaling in an in vitro system carrying a proximally mutated G-CSFR (41).
Additional proximal mutations have been reported with various phenotypes. For example, T617A which is found adjacent to T618I has been detected with rare occurrence in CNL and AML cases (41). One patient with this mutation had higher CD34+ cells indicating that this mutation may play a role in the proliferation of the myeloid progenitor cells, while a mouse model of G-CSFR-T617I showed a myeloproliferative neoplasm like disease phenotypes (42, 43). Other less frequent mutations of this class are N630K, R631del, leading to a prolonged activation of STATs similar to G-CSFR-T618I after stimulation with G-CSF (42, 43).
Intracellular truncation mutations
These mutations consist of nonsense or frameshift somatic mutations in CSF3R which cause a truncation between 82 and 98 amino acids from the carboxyl-terminus of the G-CSFR receptor (31, 44-46). The mutants of this class show a normal affinity for G-CSF, however mediate a higher proliferation and lower differentiation in response to G-CSF (45, 47). Intracellular truncation mutations have not been demonstrated to cause SCN; however, SCN patients carrying this class of mutations have a strong predisposition to MDS or AML (46). Intracellular truncation mutations are the most common type of mutation in the SCN/AML patient (~82%) (47, 48). Mild neutropenia with a higher number of immature myeloid cells has been shown in a mouse model of truncated G-CSFR (49). Based on other mouse model studies involving the Csf3r mutations, an increased number of neutrophils are recorded due to the higher myeloid progenitor proliferation (49, 50). A possible reason why truncation mutations are predisposed to MDS/AML, might be due to a strong clonal HSC advantage depending on exogenous G-CSF supply in SCN patients (51). G-CSFR truncated receptors also show an impaired internalization of the receptor after activation due the loss of a conserved di-leucine containing motif in the cytoplasmic tail leading to a higher and more sustained receptor signaling with hypersensitivity to G-CSF compared to the wild type G-CSFR (figure 2 (D) and 3 (C)) (51–53). The loss of the di-leucine motif might be related to higher expression of the receptor on the cell surface as well (53). Unlike transmembrane proximal mutation with constitutive activation primarily through JAK/STAT (based on findings of the studies involving JAK2 inhibition) (41), downstream signaling of the G-CSFR truncated receptor is reported to be primarily mediated by SRC kinases and inhibited by dasatinib treatment, a SRC kinase inhibitor (41). The truncation also causes a loss of direct negative regulation due to removal of the binding sites for tyrosine phosphatases SHP-1, SHP-2, and members of the SOCS family, leading to a decreased trafficking of the receptor to lysosomes (figure 3) (54). A reduced STAT3:STAT5 activation ratio has been observed with the truncated receptor describing its hypersensitivity to G-CSF stimulation (21). STAT5 activation downstream of the truncated receptor corroborates the hyper proliferative response and the clonal HSC advantage of the activated receptor (55). Additionally, the other pathways including PI3K, MAPK and STAT3 support overall proliferation and survival (16, 20, 56). Elevated ROS production is also one of the features of the truncated G-CSFR signaling (16, 20).
Figure 3.

G-CSFR routing and recycling. (A) In WT receptor, G-CSF induced activation leads to receptor internalization and endosome formation. While the receptor gets internalized, downstream signaling continues. After this point, the receptor has two modes to move forward: it either gets dephosphorylated and deubiquitinated, further processed in endoplasmic reticulum and Golgi and is recycled to plasma membrane, or get transitioned into the late endosome and degraded by the lysosomal pathway. (B) In membrane proximally mutated receptors, it is postulated that the recycling rate is increased with rapid processing through ER and Golgi. (C) In cases where the mutation results in intracellular domain truncation, the receptor recycling/degradation process is disrupted due to loss of the motif required for the endocytic signaling. This leads to increased receptor expression on the plasma membrane and sustained downstream signaling.
CSF3R mutations in severe congenital neutropenia and acute myeloid leukemia
Treating SCN patients with G-CSF improves peripheral neutrophil counts and decreases the risk of infections. However, over the long term (10–15 years) improvement in neutrophil counts comes at the cost of an increased risk of leukemic progression from SCN to AML. The mechanism underlying the progression of neutropenia to leukemia is not well known. Surprisingly, most of the common mutations (FLT3, KIT, JAK2, NPM1, CEPBA and TP53) observed in de novo AML are not found in SCN/AML cases (11, 12, 13, 33, 38, 40, 57, 71). Loss of chromosome 7 and gain of chromosome 21 are also common features of SCN/AML cases, but are not frequently observed in de novo AML (4, 68). 80% of SCN patients show CSF3R mutations once they progress to AML, suggesting a significant role of CSF3R driving this transition (58). In a study that involved sequential hematopoietic samples from an SCN patient over a period of 17 years, the development of CSF3R mutations were reported as early genetic defects associated with leukemic transformation to AML (37). Specifically, at a single early time point two years after the start of G-CSF therapy, multiple clones showed truncation mutations of CSF3R. One clone also displayed mutation in LLGL2 (human homolog of Drosophila Lgl gene which expresses a cell polarity protein), and at later time points this founder clone developed mutations in the ZC3H18 gene, which encodes an mRNA binding protein with unknown function. Thus, CSF3R truncation mutations arose after diagnosis and G-CSF therapy, and sequential acquisition of mutations likely allowed expansion of a mutant clone. Eventually, AML developed with additional mutations in various key leukemia oncogenes ASXL1, SUZ12, EP300, CREBBP, CCDC155 and FBXO18 (37, 73). ASXL1, SUZ12 and EP300 encoded proteins that play roles in chromatin modification. ASXL1 protein belongs to a polycomb group recessive complex (PRC) which controls the epigenetic regulation of gene expression. Epigenetic marks and transcription is controlled by ASXL1 protein through a direct interaction with PRC proteins and various transcriptional activators/repressors. Mutations in ASXL1 leads to loss of PRC2-mediated methylation of histone protein (H3K27 trimethylation) causing increased transcription of certain genes while repressing the other (81). ASXL1 gene mutations have also been reported in MDS and CML. SUZ12 encodes a protein which is a part of the PRC protein complex. Similar to ASXL1, mutations in SUZ12 gene have also been shown to reduce H3K27 trimethylation and reported in MPN (73, 82). In addition, while SUZ12 mutation have been reported in SCN/AML cases, it has yet to be determined whether these mutations play a role in leukemic transition of neutropenia over a prolonged G-CSF therapy by reducing the methylation status of H3K27. EP300 codes for p300 protein which directs the proliferation and differentiation signaling. Whereas, CREBBP is a transcriptional coactivator protein coding gene and FBXO18 encodes a DNA helicase involved in DNA repair. Mutations in CREBBP and FBXO18 disrupt the normal DNA repair mechanism resulting in leukemic conditions. The function of CCDC155 protein is not resolved yet.
Surprisingly, the CSF3R truncation mutant clones remained indolent over a long period of G-CSF treatment and disappeared during the leukemic progression of neutropenia. Although, most of the truncated clones were not detected in the progressed disease stage, one specific clone Δ715-CSF3R was prominent (37). Later in the analyses, this clone was observed in the evolution of SCN to AML (37). In a separate study, Δ715-CSF3R was showed to have a strong clonal advantage at the HSC level in a truncated receptor knock-in mouse model (51). However, this clonal advantage was dependent on high level exogenous G-CSF administration. Sustained activation of STAT5 was linked to the strong HSC clonal advantage in Δ715-CSF3R expressing bone marrow (51). Due to increased HSC clonal dominance of Δ715-CSF3R, it might contribute to the leukemic transformation of SCN. Interestingly, the AML phase also contained a proximal CSF3R gene mutation. As previously described, this mutation causes ligand independent dimerization that leads to constitutively active receptor. Appearance of the proximal mutation with the hyper-responsive truncated receptor expressing clone strongly supports the concept that perturbed G-CSF mediated signaling of G-CSFR plays a seminal role in the leukemic transformation of SCN (37).
The role of prolonged G-CSF treatment in the acquisition and/or selection of cells bearing these mutants remains to be established. A possible connection between SCN and the leukemic transformation is that the patient group undergoing a long term G-CSF therapy face chronic genotoxic stress in the HSC compartment leading to the acquisition of additional oncogenic mutations. CSF3R mutations might play a role in the clonal expansion of pre-leukemic or leukemic cells in the HSC compartment. Interestingly, leukemic progression from neutropenia appears to be limited to SCN patients. One possibility is that the mutated G-CSFR provides an activated tyrosine kinase signal and assists in leukemogenesis. For example, in a recent study by Maxson et al 2013, a differential tyrosine kinase activation pattern has been reported for separate classes of the CSF3R mutations (41). In this study, proximal mutations have been shown to activate JAK2 while the truncation mutations activate SRC kinase. However, the study did not explore how these differences in kinase activation ultimately lead to diverse disease phenotypes. This highlights a major gap in our knowledge of this complex signaling process in that we have little understanding regarding the interplay among all of the signaling events. Further understanding the phosphorylation networks associated with normal G-CSFR signaling and those associated with dysregulation due to mutations is now becoming feasible with the advances in quantitative global phosphorylation profiling (74). Adapting similar phospho-profiling techniques to normal and mutated receptors may provide the activation pattern of the downstream kinases/phosphatases and various transcription factors (e.g. GATA, RUNX, STATs etc.) that differentiate the disease phenotypes in mutated receptors after G-CSF treatment.
Based on murine cell line studies involving the Csf3r truncation mutations, it was shown that the truncation mutations have negative effects on neutrophil differentiation signaling (7, 44–46). This finding further suggests that truncated G-CSFR might play a precursor role in the leukemic progression of SCN to AML/MDS as previously described. Additionally, truncation of G-CSFR eliminates SOCS3 (a negative regulator of G-CSFR signaling) binding sites, and could lead to defective ligand-induced receptor internalization and lysosomal routing (78). However, whether the aberrant receptor recycling is directly connected to the leukemic transition of neutropenia remains to be established. Furthermore, STAT3 mediates SOCS3 induction, which controls the overall G-CSFR signaling. Further support for the sustained activation seen for the truncation mutations is due to loss of the C-terminal domain of G-CSFR which abolishes its interaction with Prdx4/Ptp1b, WD repeat and SOCS binding proteins (WSB) leading to elevated expression of the mutated receptor on the plasma membrane (9). The increased expression of truncated receptor due to its abnormal recycling might be behind the uncontrolled activation of STATs downstream of the activated receptor. The perturbed intracellular trafficking of the truncated receptor may also facilitate leukemic progression of hematopoietic stem and progenitor cells. Therefore, an additional approach to better understand the normal and truncated G-CSFR recycling mechanism are required to assess the SCN/AML transition during long term G-CSF treatment. This could also include phosphoproteomics studies as discussed above or could utilize a Mass Spectrometry interactome-based study to investigate the possibility of any interacting partners of the receptor during its recycling and their effect on receptor concentrations at the plasma membrane in normal and mutated receptors.
A mouse model of Csf3r truncation mutation has shown a sustained G-CSFR induced STAT5 activation and an increased level of ROS production (20, 55). The combination of uncontrolled activation of STAT5 leading to sustained proliferative signals and the increased oxidative stress caused by elevated ROS have been reported for both AML and CML (75). Furthermore, sustained activation of STAT5 by FLT3-ITD has already been reported to play a role in the leukemic transformation of the hematopoietic stem cells (59). STAT5 programs transformation via direct transcriptional activation and chromatin remodeling by performing post-translation modification e.g. acetylation on histone proteins (60, 61, 62). It might be possible that uncontrolled STAT5 and ROS level in truncated G-CSFR result in leukemic phenotype by enhancing proliferation of hematopoietic stem cells and increasing oxidative stress in the system leading to a higher secondary mutation rates (75). PTEN inactivation due to elevated ROS has also been reported in the HSC compartment (4, 9, 37, 75) which is consistent with increase proliferation as seen in truncation mutations. Similarly, sustained STAT5 activation by truncated G-CSFR may have a role in leukemic progression of SCN patients as well, but this needs to be experimentally validated. Previous studies have observed an elevated level of STAT5 and STAT3 activation in the G-CSFR mutants compared to WT receptor (8, 56–60, 63, 71). However, STAT3 has been demonstrated to induce differentiation of myeloid progenitor (37–39) while STAT5 as a pro-survival by enhancing the proliferation. It is not yet clear how an equilibrium is maintained between STAT3:STAT5 activation downstream of activated G-CSFR. To begin to address this interplay one might investigate conditional ablation strategies to inhibit STAT3 or STAT5 one at a time in normal and truncated CSF3R expression background and study the myeloproliferative effects after G-CSF treatment. Using such approaches, one may begin to differentiate how these two transcription activators both provide the normal balance of myeloid proliferation versus differentiation and how this process becomes misbalanced leading to the various disease phenotypes.
A study by Kawakami et al 2010, has demonstrated that protein tyrosine phosphatase SHP-1 can control the activation of STAT5 in myelomonocytic leukemia like diseases (figure 4). It might be possible that SHP-1 control is lost downstream of mutated G-CSFR leading to uncontrolled activation of STAT5. Therefore, it will be helpful to use genetic knock-out and knock-in mouse models of tyrosine phosphatases Shp-1 and Shp-2 in the background of the mutated Csf3r to study the role played by phosphatases (figure 4). The potential role played by phosphatases in G-CSFR signaling has not been fully explored and therefore it might provide a fertile field to solve the intricacies of the truncated receptor signaling in the leukemic progression of SCN. Although it has been challenging to study phosphatases due to their low abundance, deciphering the potential role played by phosphatases in G-CSFR signaling might also provide answers to the differential disease outcome resulting from the mutated receptors (proximal as well as truncations).
Figure 4.
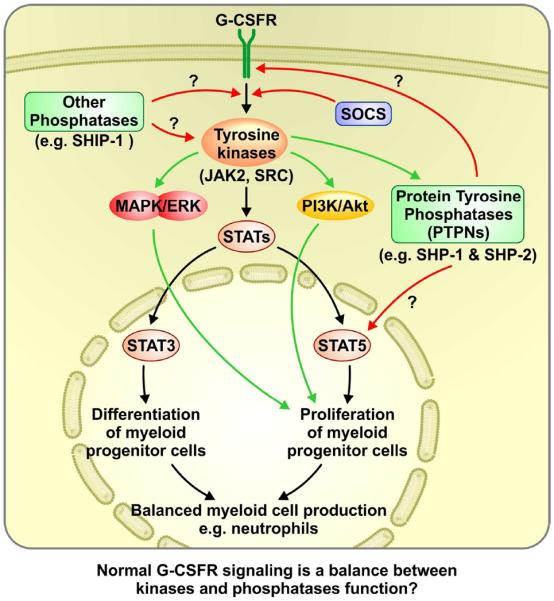
Possible roles played by kinases and phosphatases in the overall G-CSFR signaling and their decisive effects on myeloid cell production, neutrophils.
Chronic neutrophilic leukemia (CNL) and mutations in CSF3R
CNL is a rare but fatal disease with pathological features of refractory neutrophilia, worsening organomegaly and blastic transformation (64). Maxson et al 2013 first described the connection between CSF3R mutation and CNL (41). A number of studies have shown that T618I is the primary mutation in neutrophilic leukemia patients (28, 64–68).
For example, in 12 clinical cases studied by Pardanani et al. 2013, 10 out 12 mutations were the T618I (57) which is consistent with other studies that have shown the frequency of T618I to be >80% (28). Other distal truncation mutations have also been reported in CNL patients but with a much lower frequency (6 of 27) (59). In one study ~40 % of neutrophilic leukemia patients were recorded to have additional SETBP1D868N and ASXL1 gene mutations with proximal mutated CSF3R (38). SETBP1 is gene which encodes SET1 domain binding proteins whereas ASXL1 encodes a chromatin binding protein. The presence of mutations (CSF3R and SETBP/ASXL1) were recorded in granulocytes, mononuclear cells and CD34+ myeloid progenitors, however not in T cells in CNL patients indicating the hematopoietic lineage specific effects (38).
As discussed previously, T618I is an auto-activating transmembrane proximal CSF3R mutation (28, 64–68). In one report, the auto-activation of the receptor was thought to be caused by the loss of an O-glycosylation site on T618I (69). The loss of glycosylation affects the tertiary structure and stability of overall G-CSFR-T618I and thereby making it more stable (69). Furthermore, O-linked glycosylation provides a bulky group to the receptor which sterically hinders the dimer formation and activation. As previously described an increased receptor dimer formation and activation was recorded with T618I due to loss of the glycosylation site (69). Implications on receptor recycling mechanism of the proximally mutated G-CSFR, (as presented in details in the section on transmembrane proximal mutations) will require further study because it is not clear how T618I affects the overall receptor recycling.
It has also been shown that T618I mutated receptor leads to rapid cellular transformation of the myeloid progenitors causing lethal myeloproliferative disorder in a murine bone marrow transplant model (70–72). According to the study done by Maxson et al 2013, this group of mutation activates JAK2 kinase. It might be possible that the constitutively active receptor leads to STATs - mediated activation pathway downstream of JAK2 causing abnormally high level of neutrophils in CNL patients. Conversely, STAT3 is a positive regulator of SOCS3 which mediates the shutdown of G-CSFR signaling (80). Ideally, STAT3 expression should be acting as a limiting factor for G-CSFR signaling through SOCS3, however this does not appear to be the case with the proximal mutation. Unlike in the case of the truncation mutations where the binding site for SOCS3 is eliminated and thus prohibits shutdown of the downstream signaling, the proximal mutation associated with CNL, there is no evidence of aberrant SOCS3 binding to the receptor. Therefore, targeting the signaling mechanism of G-CSFR-T618I with respect to SOCS3 and STAT3 is one avenue to address this question. Similar to the discussion above for SCN and AML, the role of dysregulation of phosphatases in disease initiation and progression as well as receptor recycling mechanism is currently unknown for proximal mutation in CNL and warrant further investigation.
Clinical implications
Mobilization of stem cells for transplantation in healthy donors to neutropenic patients and to maintain normal level of neutrophils in cancer patients undergoing myelotoxic chemotherapy, are a few of the major therapeutic applications of G-CSFR (9). Recombinant G-CSF (filgrastim, filgrastim-sndz) has been widely used in the clinical setting to accomplish the therapeutic benefits of G-CSFR. However, we still do not know the complete signaling biology of G-CSF activated receptor. Therefore, understanding the signaling mechanism of G-CSFR will help us to improve the efficacy of the clinical usage of G-CSF which is required as lifesaving drug for SCN and SCN/AML patients. Furthermore, mutations in CSF3R lead to a variety of diseases (SCN, CNL, CIN, aCML, SCN/AML, SCN/MDS etc.) and most of which ultimately lead to the poor clinical outcomes. It is currently unclear whether different classes of mutant G-CSFR affect different downstream regulators or simply their amplitude or duration. Therefore, understanding the underlying signaling mechanisms is necessary to find targets to treat this group of diseases.
The current therapy for CIN and SCN derived AML/MDS is very limited. Use of JAK2 inhibitor, ruxolitinib has been used as second line of therapy after hydroxyurea, and has been shown to reduce the proliferative signaling from G-CSFR-T618I as well as the level of neutrophils in a CNL patient (66). Dasatinib, a SRC kinase inhibitor selectively inhibits the clones expressing truncated G-CSFR mutants in SCN/AML cases (41). As a combined therapy, ruxolitinib and dasatinib have also been used to treat CNL however with limited success (28, 41, 72). This combined kinase inhibitor therapy has not been tested yet against diseases which carry either/or both the proximal and/or truncation mutations. One concern about kinase therapies involving inhibitors is that they decrease proliferation of progenitor cells while showing an improvement in blood count; however, they do not fully offer a sustainable disease related benefits. Given all these challenges, the best possible treatment available for CNL patients is allogenic stem cell transplant.
For SCN patients, the primary treatment is G-CSF induction therapy. However, many SCN patients develop leukemia over the period of G-CSF therapy. Since CSF3R mutations are present in most SCN-AML, and CSF3R-mutant clones appear to precede leukemic transformation, it will be clinically relevant to monitor the CSF3R gene to evaluate disease progression in SCN patients.
Conclusion and perspective
Although granulocyte-colony stimulating factor has been widely used in clinics to successfully treat a wide range of life-threatening conditions associated with low neutrophil numbers, its prolonged use is related to CSF3R mutations and leukemia in SCN patients. Therefore, the research community and ultimately the SCN patient could greatly benefit from a clear understanding of the molecular mechanism underlying G-CSF therapy and disease progression influenced by its prolong use. One major issue of prolong use of G-CSF is the development of hyporesponse or nonresponse to the treatment in SCN patients. Understanding the signaling mechanism downstream of activated G-CSFR may help us to overcome G-CSF hyporesponse/nonresponse issue in current SCN patients. Similarly, more than 80% of CNL patients carry a CSF3R mutations (specifically T618I) but show a completely different disease characterized by an uncontrolled numbers of neutrophils. Given the high frequency of proximal mutation, it is likely that T618I works as a driver mutation in CNL; however, the mechanism responsible for the T618I mutation leading to CNL remains largely unresolved.
Overall, it is clear that G-CSFR signaling is essential for neutrophil development. However, how the receptor signaling gets differentially perturbed in proximal and truncation mutations leading to different disease conditions, remains to be explored. In the preceding sections of this review, the current knowledge base for our understanding of G-CSFR signaling has been presented along with what is known about the relationship and mechanisms of mutations to the receptor from the various disease manifestations. While considerable progress has been made, there are still major gaps in our understanding of the underlying mechanisms of disease development and progression. These gaps are in part due to both the complexity of the signaling systems and perhaps some of the models systems and technologies that have been applied to study them. For example, while JAK/STAT, PI3K/pAKT, and MAPK/ERK signaling have been established as central players in G-CSFR activation (figures 2), there are clearly other signaling changes that are emerging that are not directly related to these signaling pathway (e.g. redox controlled signaling mechanism involving Prdx1 and Prdx2 etc.) (76, 77). In addition, the role that phosphatases play in controlling these events is just beginning to be explored (SHIPs, SHP1 and SHP2). Additional understanding of the broad phosphorylation network associated with both normal G-CSFR activation and activation in disease-specific mutation will likely provide further insight into the regulation or dysregulation of this complex signaling network. Fortunately, both targeted phosphorylation array technologies and global phosphoproteomics methods have now become more commonplace and thus could represent new areas to develop a deeper understanding of G-CSFR signaling. Another underexplored area is the interplay between STAT3 and STAT5 signaling pathways from normal and disease-specific G-CSFR (truncation versus proximal mutations). How this interplay works between these branches of JAK2 signaling resulting in healthy versus abnormal neutrophil productions requires further investigation possibly utilizing conditional knockdown/knockout of STAT 3/5 in mouse models. These models have been effectively utilized to study JAK/STAT signaling in normal granulopoiesis previously (78–80) but have not yet been integrated into CSF3R mutation models and thus could have an impact on distinguishing their roles in the disease phenotypes.
In conclusion, while the research community continues to gain insight into what regulates and controls the normal process of neutrophil proliferation and differentiation, and how mutations in CSF3R lead to the various clinical phenotypes, the current G-CSFR research still requires investigation of many unexplored avenues to fully understand the signaling biology in normal and diseased/mutated conditions. These new insights could then lead to new avenues for targeted treatment or the prevention of CNL and the transition of a disease like SCN to AML after prolonged induction therapy with G-CSF.
Highlights.
Current mechanistic understanding of G-CSFR signaling is presented
Clinical consequences of G-CSFR mutations and therapeutic options are discussed
Gaps in the current knowledge of G-CSFR biology are reviewed
Discussion of alternative approaches to further understand G-CSFR related diseases
Acknowledgements
The authors thank H. Leighton Grimes for critical reading, edits, and insightful comments. The authors also thank to members of the Greis and Grimes research groups for the valuable suggestions throughout the development of this manuscript and Mr. Glenn Doermann for his expertise in graphic design for the figures. This manuscript and our work associated with G-CSFR is supported by several sources including: NIH 1S10 RR027015-01 (Greis), the University of Cincinnati Millennium Scholars Fund (Greis) and support from Cincinnati Children's Hospital Research Foundation (Greis).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest There are no conflicts of interest.
Human and animal rights and informed consent The proposed article does not contain unpublished studies with human or animal subjects.
References
- 1.Manz MG, Boettcher S. Emergency granulopoiesis. Nature Reviews. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Neutrophils and immunity: challenges and opportunities. Nature Reviews. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 3.Touw IP, Palande K, Beekman R. Granulocyte colony-stimulating factor receptor signaling- implications for G-CSF responses and leukemic progression in severe congenital neutropenia. Hematol Oncol Clin N Am. 2013;27:61–73. doi: 10.1016/j.hoc.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Beekman R, Touw IP. G-CSF and its receptor in myeloid malignancy. Blood. 2010;115:5131–5136. doi: 10.1182/blood-2010-01-234120. [DOI] [PubMed] [Google Scholar]
- 5.Fukunaga R, I.-I E, Pan CX, Seto Y, Nagata S. Functional domains of the granulocyte colony stimulating factor receptor. EMBO Journal. 1991;10:2855–65. doi: 10.1002/j.1460-2075.1991.tb07835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Touw IP, van de Geijn GM. Granulocyte colony stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Frontiers in Bioscience. 2007;12:800–815. doi: 10.2741/2103. [DOI] [PubMed] [Google Scholar]
- 7.Fukunaga R, Ishizaka-Ikeda E, Nagata S. Growth and differentiation signals mediated by different regions in the cytoplasmic domain of granulocyte colony-stimulating factor receptor. Cell. 1993;74:1079–1087. doi: 10.1016/0092-8674(93)90729-a. [DOI] [PubMed] [Google Scholar]
- 8.Santini V, Indik ZK, Gozzini A, Ferrini PR, Schreiber AD. The carboxy-terminal region of the granulocyte colony-stimulating factor receptor transduces a phagocytic signal. Blood. 2003;101(11):4615–4622. doi: 10.1182/blood-2002-07-2271. [DOI] [PubMed] [Google Scholar]
- 9.Palande K, Meenhuis A, Jevdjovic T, Touw IP. Scratching the surface: signaling and routing dynamics of the CSF3 receptor. Front Biosci (Landmark Ed) 2013;18:91–105. doi: 10.2741/4089. [DOI] [PubMed] [Google Scholar]
- 10.Zeidler C, Schwinzer B, Welte K. Congenital neutropenias. Rev Clin Exp Hematol. 2003;7:72–83. [PubMed] [Google Scholar]
- 11.Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109(5):1817–1824. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nature Genetics. 2007;39(1):86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 13.Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannaourakis G, Kinsey SE, Davis R, Scarlata D, Schwinzer B, Zeidler C, Welte K. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol. 2003;72(2):82–93. doi: 10.1002/ajh.10255. [DOI] [PubMed] [Google Scholar]
- 14.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78(11):2791–2808. [PubMed] [Google Scholar]
- 15.Layton JE, Hall NE. The interaction of G-CSF with its receptor. Front Biosci. 2006;11:3181–3189. doi: 10.2741/2041. [DOI] [PubMed] [Google Scholar]
- 16.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with severe congenital neutropenia/ acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189(4):683–692. doi: 10.1084/jem.189.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corey SJ, Burkhardt AL, Bolen JB, Geahlen RL, Tkatch LS, Tweardy DJ. Granulocyte colony-stimulating factor receptor signaling involves the formation of a three-component complex with Lyn and Syk protein-tyrosine kinases. Proc Natl Acad Sci USA. 1994;91:4683–4687. doi: 10.1073/pnas.91.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barge RM, de Koning JP, Pouwels K, Dong F, Lowenberg B, Touw IP. Tryptophan 650 of human granulocyte colony-stimulating factor (G-CSF) receptor, implicated in the activation of JAK2, is also required for G-CSF-mediated activation of signaling complexes of the p21ras route. Blood. 1996;87(6):2148–2153. [PubMed] [Google Scholar]
- 19.Bartalucci N, Tozzi L, Bogani C, Martinelli S, Rotunno G, Villeval JL, Vannucchi A. Co-targeting the PI3K/mTOR and JAK2 signaling pathways produces synergistic activity against myeloproliferative neoplasms. J Cell Mol Med. 2013;17(11):1385–1396. doi: 10.1111/jcmm.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu QS, Xia L, Mills GB, Lowell CA, Touw IP, Corey SJ. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood. 2005;107:1847–1856. doi: 10.1182/blood-2005-04-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward AC, Hermans MH, Smith L, van Aesch YM, Schelen AM, Antonissen C, Touw IP. Tyrosine-dependent and independent mechanism of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood. 1999;93:113–124. [PubMed] [Google Scholar]
- 22.Ward AC, Smith L, de Koning JP, van Aesch Y, Touw IP. Multiple signaling mediate proliferation, differentiation and survival from the granulocyte colony-stimulating factor receptor in myeloid 32D cells. J Biol Chem. 1999;274:14956–14962. doi: 10.1074/jbc.274.21.14956. [DOI] [PubMed] [Google Scholar]
- 23.Lee CK, Raz R, Gimeno R. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17(1):63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 24.Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 (SOCS3) is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol. 2002;169:1219–1227. doi: 10.4049/jimmunol.169.3.1219. [DOI] [PubMed] [Google Scholar]
- 25.Ward AC, Oomen SPMA, Smith L, Gits J, van Leeuwen D, Soede-Bobok AA, Erpelinck-Verschueren CA, Yi T, Touw IP. The SH2- containing protein tyrosine phosphatase SHP-1 is induced by granulocyte colony-stimulating factor (G-CSF) and modulates signaling from the G-CSF receptor. Leukemia. 2000;14:1284–1291. doi: 10.1038/sj.leu.2401822. [DOI] [PubMed] [Google Scholar]
- 26.Hunter MG, Jacob A, O'Donnell LC, Agler A, Druhan LJ, Coggeshall KM, Avalos BR. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J Immunol. 2004;174:5036–5045. doi: 10.4049/jimmunol.173.8.5036. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S, Schwinzer B, Zeidler C, Welte K, Dale DC. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107(12):4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. doi: 10.1182/blood-2013-05-500959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos NP, Eliopoulos GD. Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood. 2005;105:584–591. doi: 10.1182/blood-2004-07-2613. [DOI] [PubMed] [Google Scholar]
- 30.Papadaki HA, Kosteas T, Genetzi C, Damianaki A, Anagnou NP, Eliopoulos GD. Acute myeloid/NK precursor cell leukemia with trisomy 4 and a novel point mutation in the extracellular domain of the G-CSF receptor in a patient with chronic idiopathic neutropenia. Ann Hematol. 2004;83:345–348. doi: 10.1007/s00277-004-0862-y. [DOI] [PubMed] [Google Scholar]
- 31.Liongue C, Ward AC. Granulocyte colony-stimulating factor receptor mutations in myeloid malignancy. Front Oncology. 2014;93(4):1–7. doi: 10.3389/fonc.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Touw IP, Beekman R. Severe congenital neutropenia and chronic neutrophilic leukemia: an intriguing molecular connection unveiled by oncogenic mutations in CSF3R. Haematologica. 2013;98:1490–1492. doi: 10.3324/haematol.2013.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeffers M, Schmidt l, Nakaigawa N, Webb CP, Weirich G, Kishida T, Zbar B, Vande Woude GF. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci USA. 1997;94:11445–11450. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 35.Joffre C, Barrow R, Menard L, Calleja V, Hart IP, Kermorgant S. A direct role for Met endocytosis in tumorigenesis. Nat Cell Biol. 2011;13:827–837. doi: 10.1038/ncb2257. [DOI] [PubMed] [Google Scholar]
- 36.Miaczynska M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2013;5:a009035. doi: 10.1101/cshperspect.a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, Geertsma-Kleinekoort WM, Veerman AJ, Valk PJ, Verhaak RG, Lowenberg B, Touw IP. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119:5071–5077. doi: 10.1182/blood-2012-01-406116. [DOI] [PubMed] [Google Scholar]
- 38.Elliott MA, Tefferi A. Chronic neutrophilic leukemia 2014: Update on diagnosis, molecular genetics, and management. American Journal of Hematology. 2014;89(6):652–658. doi: 10.1002/ajh.23667. [DOI] [PubMed] [Google Scholar]
- 39.Mehta HM, Glaubach T, Long A, Lu H, Przychodzen B, Makishima H, McDevitt MA, Cross NC, Maciejewski J, Corey SJ. Granulocyte colony-stimulating factor receptor T595I (T618I) mutation confers ligand independence and enhanced signaling. Leukemia. 2013;27:2407–2410. doi: 10.1038/leu.2013.164. [DOI] [PubMed] [Google Scholar]
- 40.Beekman R, Valkhof M, van Strien P, Valk PJ, Touw IP. Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica. 2013;98:62–63. doi: 10.3324/haematol.2013.085050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, Bottomly D, Wilmot B, McWeeney SK, Tognon CE, Pond JB, Collins RH, Goueli B, Oh ST, Deininger MW, Chang BH, Loriaux MM, Druker BJ, Tyner JW. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368:1781–1790. doi: 10.1056/NEJMoa1214514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forbes LV, Gale RE, Pizzey A, Pouwels K, Nathwani A, Linch DC. An activating mutation in the transmembrane domain of the granulocyte colony-stimulating factor receptor in patients with acute myeloid leukemia. Oncogene. 2002;21:5981–5989. doi: 10.1038/sj.onc.1205767. [DOI] [PubMed] [Google Scholar]
- 43.Awaya N, Uchida H, Miyakawa Y, Kinjo K, Matsushita H, Nakajima H, Ikeda Y, Kizaki M. Novel variation isoform of G-CSF receptor involved in induction of proliferation of FDCP-2 cells: relevance to the pathogenesis of myelodysplastic syndrome. J Cell Physiol. 2002;191:327–335. doi: 10.1002/jcp.10102. [DOI] [PubMed] [Google Scholar]
- 44.Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333:487–493. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- 45.Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, Palmblad J, Briars GL, Carlsson G, Veerman AJ, Welte K, Lowenberg B, Touw IP. Mutations in the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Leukemia. 1997;11:120–125. doi: 10.1038/sj.leu.2400537. [DOI] [PubMed] [Google Scholar]
- 46.Dong F, van Buitenen C, Pouwels K, Hoefsloot LH, Lowenberg B, Touw IP. Distinct cytoplasmic regions of the human granulocyte colony-stimulating factor receptor involved in induction of proliferation and maturation. Mol Cell Biol. 1993;13:7774–7781. doi: 10.1128/mcb.13.12.7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Germeshausen M, Ballmaier M, Welte K. Implications of mutations in hematopoietic growth factor genes in congenital cytopenias. Ann N Y Acad Sci. 2001;938:305–320. doi: 10.1111/j.1749-6632.2001.tb03599.x. [DOI] [PubMed] [Google Scholar]
- 48.Freedman MH, Alter BP. Risk of myelodysplastic syndrome and acute myeloid leukemia in congenital neutropenias. Semin Hematol. 2002;39:128–133. doi: 10.1053/shem.2002.31912. [DOI] [PubMed] [Google Scholar]
- 49.Mitsui T, Watanabe S, Taniguchi Y, Hanada S, Ebihara Y, Sato T, Heike T, Mitsuyama M, Nakahata T, Tsuji K. Impaired neutrophil maturation in truncated murine G-CSF receptor transgenic mice. Blood. 2003;101:2990–2995. doi: 10.1182/blood.V101.8.2990. [DOI] [PubMed] [Google Scholar]
- 50.McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest. 1998;102:483–492. doi: 10.1172/JCI3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F, Kunter G, Krem MM, Eades WC, Cain JC, Tomasson MH, Hennighausen L, Link DC. Csf3r mutations in mice confer a strong clonal HSC advantage via activation of Stat5. J Clin Invest. 2008;118:946–955. doi: 10.1172/JCI32704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunter G, Woloszynek JR, Link DC. A truncation mutant of Csf3r cooperates with PML-RARalpha to induce acute myeloid leukemia in mice. Exp Hematol. 2011;39:1136–1143. doi: 10.1016/j.exphem.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward AC, van Aesch YM, Schelen AM, Touw IP. Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood. 1999;93:447–458. [PubMed] [Google Scholar]
- 54.Dong F, Qiu Y, Yi T, Touw IP, Larner AC. The carboxyl terminus of the granulocyte colony-stimulating factor receptor, truncated in patients with severe congenital neutropenia/acute myeloid leukemia, is required for SH2-containing phosphatase-1 suppression of Stat activation. I Immunol. 2001;167:6447–6452. doi: 10.4049/jimmunol.167.11.6447. [DOI] [PubMed] [Google Scholar]
- 55.Ilaria RL, Jr, Hawley RG, Van Etten RA. Dominant negative mutants implicate STAT5 in myeloid cell proliferation and neutrophil differentiation. Blood. 1999;93:4154–4166. [PubMed] [Google Scholar]
- 56.Gits J, van Leeuwen D, Carroll HP, Touw IP, Ward AC. Multiple pathways contribute to the hyperproliferative responses from truncated granulocyte colony-stimulating factor receptors. Leukemia. 2006;20:2111–2118. doi: 10.1038/sj.leu.2404448. [DOI] [PubMed] [Google Scholar]
- 57.Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, Ketterling RP, Maxson JE, Tyner JW, Tefferi A. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27(9):1870–1873. doi: 10.1038/leu.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandenberghe P, Beel K. Severe congenital neutropenia, a genetically heterogeneous disease group with an increased risk of AML/MDS. Pediatr Rep. 2011;3(Suppl 2):e9. doi: 10.4081/pr.2011.s2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tefferi A, Elliott M, Pardanani A. Chronic neutrophilic leukemia: novel mutations and their impact on clinical practice. Curr Opin Hematol. 2015;22:171–176. doi: 10.1097/MOH.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 60.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sragin B, Ueker A, Bohmer FD, Berdel WE, Muller-Tidow C, Serve H. Activation mechanism of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110(1):370–374. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 61.Kramer OH, Moriggl R. Acetylation and sumoyulation control STAT5 activation antagonistically. JAK-STAT. 2012;1(3):203–207. doi: 10.4161/jkst.21232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rascle A, Lees E. Chromatin acetylation and remodeling at the Cis promoter during STAT5-induced transcription. Nucle Acids Res. 2003;31(23):6882–6890. doi: 10.1093/nar/gkg907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plo I, Zhang Y, Le Couedic JP, Nakatake M, Boulet JM, Itaya M, Smith SO, Debili N, Constantinescu SN, Vainchenker W, Louache F, de Bottom S. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J Exp Med. 2009;206:1701–1707. doi: 10.1084/jem.20090693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uppal G, Gong J. Chronic neutrophilic leukemia. J Clin Pathol. 2015;0:1–5. doi: 10.1136/jclinpath-2015-203060. [DOI] [PubMed] [Google Scholar]
- 65.Bacher U, Schnittger S, Kern W, Weiss T, Haferlach T, Haferlach C. Distribution of cytogenetic abnormalities in myelodysplastic syndromes, Philadelphia negative myeloproliferative neoplasms, and the overlap MDS/MPN category. Ann Hematol. 2009;88(12):1207–1213. doi: 10.1007/s00277-009-0745-3. [DOI] [PubMed] [Google Scholar]
- 66.Dao KH, Solti MB, Maxson JE. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leukemia Res Rep. 2014;3:67–69. doi: 10.1016/j.lrr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Donadieu J, Leblanc T, Bader Meunier B, Barkaoui M, Fenneteau O, Bertrand Y. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica. 2005;90:45–53. [PubMed] [Google Scholar]
- 68.Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, Brown S, Cham B, Kannourakis G, Kinsey SE, Mori PG, Cottle T, Welte K, Dale DC. Myelodysplastic syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96(2):429–436. [PubMed] [Google Scholar]
- 69.Maxson JE, Luty SB, MacManiman JD, Abel ML, Druker BJ, Tyner JW. Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. The Journal of Biological Chemistry. 2014;289(9):5820–5827. doi: 10.1074/jbc.M113.508440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li L, Modi H, McDonald T, Rossi J, Yee JK, Bhatia R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood. 2011;118(6):1504–1515. doi: 10.1182/blood-2010-06-288910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mehta HM, Malandra M, Corey SJ. G-CSF and GM-CSF in Neutropenia. J Immunol. 2015;195(4):1341–1349. doi: 10.4049/jimmunol.1500861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fleischman AG, Maxson JE, Luty SB, Agarwal A, Royer LR, Abel ML, MacManiman JD, Loriaux MM, Drunker BJ, Tyner JW. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 2013;122:3628–3631. doi: 10.1182/blood-2013-06-509976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brecqueville M, Cervera N, Adelaide J, Rey J, Carbuccia N, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D, Gelsi-Boyer V, Murati A. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J. 2011;1(18):e33. doi: 10.1038/bcj.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, In vivo, and site specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 75.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 76.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW. Regulation of PDGF signaling and vascular remodeling by peroxiredoxin II. Nature. 2005;435(7040):347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 77.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140(4):517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116(14):2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Panopoulos AD, Zhang L, Snow JW, Jones DM, Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, Watowich SS. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108(12):3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ, Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 2007;26(7):1782–1793. doi: 10.1038/sj.emboj.7601640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, Beran M, Gilliland DG, Levine RL, Tefferi A. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–1202. doi: 10.1038/leu.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin in Cell Biol. 2015;37:42–48. doi: 10.1016/j.ceb.2015.10.003. [DOI] [PubMed] [Google Scholar]