

Conspectus
Although each type of protein fold and in some cases individual proteins within a fold classification can have very different mechanisms of folding, the underlying biophysical and biochemical principles that operate to cause a linear polypeptide chain to fold into a globular structure must be the same. In an aqueous solution, the protein takes up the thermodynamically most stable structure, but the pathway along which the polypeptide proceeds in order to reach that structure is a function of the amino acid sequence, which must be the final determining factor, not only in shaping the final folded structure, but in dictating the folding pathway. A number of groups have focused on a single protein or group of proteins, to determine in detail the factors that influence the rate and mechanism of folding in a defined system, with the hope that hypothesis-driven experiments can elucidate the underlying principles governing the folding process. Our research group has focused on the folding of the globin family of proteins, and in particular on the monomeric protein apomyoglobin. Apomyoglobin (apoMb) folds relatively slowly (~2 seconds) via an ensemble of obligatory intermediates that form rapidly after the initiation of folding. The folding pathway can be dissected using rapid-mixing techniques, which can probe processes in the millisecond time range. Stopped-flow measurements detected by circular dichroism (CD) or fluorescence spectroscopy give information on the rates of folding events. Quench-flow experiments utilize the differential rates of hydrogen-deuterium exchange of amide protons protected in parts of the structure that are folded early; protection of amides can be detected by mass spectrometry or proton nuclear magnetic resonance spectroscopy (NMR). In addition, apoMb forms an intermediate at equilibrium at pH ~ 4, which is sufficiently stable for it to be structurally characterized by solution methods such as CD, fluorescence and NMR spectroscopies, and the conformational ensembles formed in the presence of denaturing agents and low pH can be characterized as models for the unfolded states of the protein. Newer NMR techniques such as measurement of residual dipolar couplings in the various partly folded states, and relaxation dispersion measurements to probe invisible states present at low concentrations, have contributed to providing a detailed picture of the apomyoglobin folding pathway. The research summarized in this review was aimed at characterizing and comparing the equilibrium and kinetic intermediates both structurally and dynamically, as well as delineating the complete folding pathway at a residue-specific level, in order to answer the question “What is it about the amino acid sequence that causes each molecule in the unfolded protein ensemble to start folding, and, once started, to proceed towards the formation of the correctly folded three-dimensional structure?”
Introduction
Globular proteins must be folded into specific structures in order to perform their biological functions. The fidelity of folding is astonishing: although proteins frequently utilize chaperones and other factors in vivo to reach the correct fold without misfolding or aggregation, in vitro folding experiments with many small proteins commonly show that virtually every molecule folds to the correct structure, in the absence of cellular accessory factors. This argues that the amino acid sequence itself encodes the information for its own folding, and this assumption informs all of the work described in the following pages.
For our laboratory and others, myoglobin (Figure 1) has been the molecule of choice for biophysical studies aimed at elucidation of protein folding. Although our emphasis in the current review is on the story that has emerged from our own laboratory, a number of others (for example, those of Baldwin,1 Roder,2 Gruebele,3 Callender and Dyer4) have contributed important insights. This information has given a new understanding of what features of the amino acid sequence prompt it to fold, and insights into how each molecule in the ensemble can fold to the correct structure despite starting (in the unfolded ensemble) from vastly different structures. We also now understand why the intermediate states in the folding of apomyoglobin cause it to pause in its trajectory to the final folded structure, and why such a pause is advantageous for the formation of correctly-folded molecules. This review summarizes experimental work on apomyoglobin folding, with an emphasis on the understanding that we have generated from our own data and those of other labs.
Figure 1.
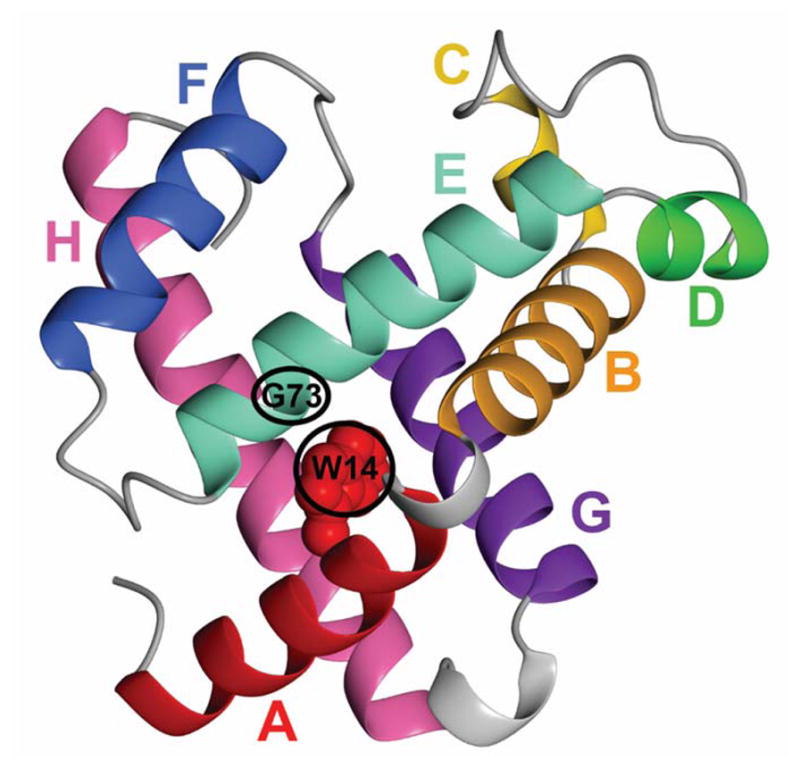
Ribbon diagram of the backbone of sperm whale myoglobin53 showing helices A (red), B (orange), C (yellow), D (green), E (turquoise), F (blue), G (purple) and H (pink). The Trp 14 side chain is shown in its position opposite Gly 73 in the wild-type protein Reproduced with permission from ref.26. Copyright 2005 Elsevier. The figure was prepared using Molmol54.
Unfolding and Refolding of Apomyoglobin
Apomyoglobin unfolds reversibly at acidic pH, and unfolding by the addition of acid occurs in stages that can be monitored by changes in the circular dichroism and fluorescence spectra of the solution.5 ApoMb can readily be refolded from acidic or denaturant solutions. At pH ~ 4, a “molten-globule”5 intermediate, which exists as an equilibrium between two forms (Ia and Ib),6 is populated; at pH ~2, the protein is unfolded but retains a small amount of helical structure, which is eliminated by the addition of denaturants such as urea or guanidine.7 In higher-salt media, the protein populates alternatively-structured forms.8,9
Residue-Level Exploration of Protein Folding
Although numerous experimental techniques have been applied to the exploration of protein folding, the method that can potentially deliver extensive residue- and atomic-level information on the process is solution nuclear magnetic resonance (NMR). Our earliest insights into the folding of apomyoglobin were derived from NMR studies of synthetic peptides with amino acid sequences corresponding to fragments of the apomyoglobin sequence.10 The peptide studies led to hypotheses that were later tested using more advanced NMR and other techniques.
Breakthrough: Hydrogen-Exchange Pulse Labeling by NMR
Atomic-level insights into the folding pathway were not obtained until the introduction of the quench-flow NMR amide exchange technique,11,12 which takes advantage of both the potential for NMR data acquisition on samples obtained by rapid-flow mixing, and the well-known pH dependence of amide proton exchange in proteins, to overcome the limitations imposed by the intrinsically slow acquisition of an NMR spectrum. In a quench-flow experiment, the only amides visible in the NMR spectrum of the folded protein are those that were protected during the refolding process. At the shortest refolding times (approximately 6 ms) only the amides located in the most rapidly folding portions of the protein will remain protonated.
The kinetic folding pathway of apomyoglobin, delineated using 2D 1H NMR spectra,13 showed that folding proceeded via a “burst phase” intermediate that formed within the 6 ms dead time of the quench flow instrument. This kinetic intermediate, as indicated by the pattern of protected amides at the shortest folding times, contained parts of the A, G and H helices. The remaining portions of the protein folded more slowly, over a timescale of seconds. Quench-flow experiments measuring H/D exchange by mass spectrometry14 led to the further conclusion that the kinetic intermediate was obligatory and on-pathway in the folding of all molecules in the ensemble.
Folded and Unfolded States of Apomyoglobin
Apomyoglobin is ideal for folding studies, since it contains no native cysteines or disulfides and exhibits no proline isomerization, and also because it is readily prepared from bacterial expression systems,15 thereby facilitating labeling with 15N and 13C for assignment of all backbone resonances for both the apo- and holoproteins.16,17
At pH 6, apomyoglobin folds into the same topology as the holoprotein, except that the F helix is incompletely folded.18,19 At pH 4, it forms a molten globule, which has been optimized for NMR studies.20 The pH 4 state of apoMb shows amide proton protection21 and native-like helical structure in the A, G and H helices and part of the B helix,20 similar to the results obtained for the burst phase intermediate in the kinetic folding pathway.13 The pH 2 state shows native-like residual helical structure in the H helix, and a small amount of non-native helix between the D and E helices.22
Analysis of relaxation data for the pH 2 state of apoMb suggested the presence of transient long-range contacts between opposite ends of the polypeptide;22 these contacts were verified in a study incorporating spin labels.23 Complete denaturation of apoMb in urea at pH 2 destroys both the residual helical structure and the transient long-range interactions seen in the absence of urea, but subtle sequence-specific variations in the relaxation data were observed.24 These variations likely indicate transient local interactions within clusters containing long hydrophobic side chains, whereas regions containing clusters of small amino acids (glycine and alanine) show significantly greater mobility. The evidence for specific local and long-range interactions in the unfolded states of apoMb led to a hypothesis that clusters of residues with long hydrophobic side chains, including residues such as lysine with its long aliphatic side chain, are instrumental in initiating the folding process (see later section). These clusters can be readily identified as they score high on a scale termed “average area buried upon folding” (AABUF).24–26
Because the unfolded and partially-folded states of apoMb are well-behaved, they provide the means to elucidate the characteristics of unfolded states of proteins in general. Residual dipolar couplings, measured for urea and acid-denatured apoMb transiently aligned in stretched or compressed polyacrylamide gels,27 showed the expected bell-shaped distribution of RDCs in the fully urea-denatured protein. Residual secondary structure propensity in the acid-denatured protein has an effect on the magnitude of the RDC that can be quantified site-specifically to determine the ensemble populations of transiently folded elements of secondary structure.27 Measurements of the proximity of covalently-attached spin labels in the unfolded states of apoMb showed the presence of transient long-range interactions in acid-unfolded apoMb,23 and this technique has been used together with a worm-like chain model to gain new insights into the conformational ensemble of the transient collapsed states present in the unfolded protein at pH 2.3.28
Dissection of the Apomyoglobin Kinetic Folding Pathway
The kinetic folding pathway of apoMb was explored using wild type protein and a series of mutant proteins designed to test hypotheses on the role of factors such as intrinsic secondary structural propensity on folding. Mutation of the H helix sequence to lower its intrinsic helical propensity (N132G/E136G) caused changes in the folding pathway, but these changes were not reflected in the folding rate, which remained the same despite the lowered helical structure in the burst phase intermediate of the mutant protein.29 ApoMb collapses rapidly upon initiation of folding to an intermediate that contains folded helical structure in regions that comprise the A, G, and H helices in the wild-type protein, but the same collapse occurs in the mutant protein, allowing the protein to fold almost normally, despite the absence of the H helix from the intermediate.30 Clearly, although the A(B)GH intermediate is obligatory on the folding pathway,14 the structure of the intermediate itself is not critical and it may be altered quite substantially without changing the kinetics of folding.
Although the conclusions of the early apoMb work13,14 were validated by each new set of experiments, evolution in NMR methodology towards greater precision later showed that sites within individual helices could differ quite substantially in their behavior with slight variations in solution and temperature conditions.31 These studies indicated that the burst phase intermediate identified in the initial quench-flow experiments13 is structurally heterogeneous. The majority of the amides protected in the burst phase of the quench-flow experiment were still observed in the A, G and H helices (and in a small area of the B helix, between Ile28 and Phe33), but partial protection was also observed in the E helix, that is, a percentage of the molecules showed protection of E helix amides in the burst phase, with the same amides in the remainder of the molecules being protected on a slower timescale. This effect was highly dependent on the strength and duration of the high-pH labeling pulse, reflecting different intrinsic stability of local structure at pH 10 that was independent of its rate of folding at pH 7 where the folding process was being examined.
Why Does the Kinetic Folding Intermediate Occur?
A limitation in the experimental protocol for quench-flow experiments led to the development of a robust method to quantitate the proton occupancy of individual amides. All of the early quench flow experiments involved reconstitution of holomyoglobin for NMR analysis.13 Because folding probes consist only of amides that are well-protected in the holoprotein, large portions of the molecule are invisible because they contain no slowly-exchanging amides in the native folded state. Following an idea first suggested in 1995,32 we used an aprotic solvent, DMSO, for NMR analysis of protein solutions generated in the quench-flow experiment.33 With no means of exchange-out of amides during the NMR experiment, folding information could be obtained on all of the amides in the protein except for the F helix, yielding a significantly richer picture of the folding of the wild-type and mutant proteins.
Replacement of hydrophobic side chains in the B helix with alanine results in alteration of the pattern of burst phase proton occupancy.34 Specific long-range interactions were identified between the B helix and other regions of the molecule: in the kinetic intermediate, I30 appears to contact the C helix, and L32 the G helix, both native-like interactions. By contrast, mutation of L29 affects the proton occupancy of residues in the E helix, a contact that does not occur in the native folded state, raising the possibility that local non-native interactions are present in the kinetic intermediate despite its overall native-like topology. Similar long-range contacts were observed when hydrophobic residues in the A, E, G and H helices were substituted by alanine.35 Insights were also obtained into the transition state and the rate-determining step of the visible (~ seconds) folding pathway. Mutations that affect the docking of the E helix onto the A-B-G-H core decrease the folding rate, indicating that correct positioning of the E helix must occur prior to the transition state of the folding reaction. The location of highly protected amides within the individual helices in the burst phase intermediate gave rise to the intriguing hypothesis that the intermediate, despite its obligatory, on-pathway nature, contains a local non-native interaction, specifically that the H helix is translocated by one helical turn from its position in the fully folded apoprotein.35 This non-native interaction must be resolved before E helix docking can occur – this, together with other sites of frustration such as the B/G interface, is the major source of the pause in the folding process where the folding intermediate is observed.
This hypothesis was tested by a two-pronged mutagenesis approach. If the kinetic intermediate indeed contains a translocation of the H helix relative to the G helix, then fixing the relative positions of the G and H helices, for example, by the engineering of a disulfide bond between them, might be expected to have a profound effect on the folding pathway.36 Fluorescence probes were also attached to the G and H helices to measure fluorescence resonance energy transfer (FRET) and fluorescence quenching in various equilibrium states and in the kinetic intermediate.36 The FRET experiments clearly demonstrate that in the equilibrium intermediate state, the H helix slides, in an N-terminal direction, approximately one helical turn towards helix A. Fluorescence quenching experiments confirm this translocation of the H helix, in both the equilibrium intermediate and the Ib kinetic intermediate. Disruption of helix translocation by the formation of a disulfide bond between Cys residues substituted at positions 108 and 135 (to stabilize the native, non-translocated conformation) results in only a slight increase in the refolding rates from urea and acid. There is no change in the structure of the disulfide-bonded protein at neutral pH, but the equilibrium intermediate at pH 4 shows greatly increased helical structure compared to the wild-type protein. These results demonstrate that, although the folding intermediate is locked into the native-like conformation, this is not sufficient to promote correct packing of the E helix, and shows that there must be additional sites of energetic frustration that impede native docking of the E helix and progression to the transition state. In addition to the non-native G-H helix packing, instability of the N-terminal region of helix B37 and burial of the distal His6438 also frustrate the folding process by impeding the native docking interactions of helix E. Translocation of the H helix appears to promote dynamic disorder in the E and F helix regions, preventing excessive stabilization of compact non-native structures and allowing efficient search of conformational space until additional sources of frustration are relieved and folding can progress.36
Sequence Determinants of the Apomyoglobin Folding Pathway
It is clear that for apoMb at least, and likely for most proteins, the amino acid sequence contains complete information not only on the final structure, but determines the pathway of folding. The examination of unfolded and partly-folded forms of apoMb20,22,24 suggested that clusters of side chains that are long and bulky, rather than simply being classified as “hydrophobic”, play a central role in initiation of the folding process. Thus, a lysine side chain, which because of its ζ-amino group is generally defined as “hydrophilic”, is considered to promote folding initiation in this way, while an alanine side chain, although it is hydrophobic, does not, due to its small size. This characteristic was found24,39 to be correlated with the quantity “average area buried upon folding” (AABUF)25 or “hydrophobicity” (as defined by Matheson and Scheraga).40 According to this hypothesis, regions of the amino acid sequence where the AABUF is high should participate in the earliest folding events. To test this hypothesis, we designed mutants that would change the local AABUF but leave the final structure intact.26 Apomyoglobin contains two tryptophan residues, W7 and W14. The structure of the folded protein (Figure 1) shows that the side chain of W14 is located in the hydrophobic core, close to the E helix. Opposite the W14 side chain on the E helix is G73. A double mutant was prepared in which W14 was changed to Gly, and G73 was changed to Trp. Since the bulky tryptophan side chain can be accommodated in the mutant in approximately the position it occupies in the wild-type protein, the structure of the mutant protein at pH 6 is close to that of the wild-type protein. However, the AABUF of the amino acid sequence is drastically changed (Figure 2). Where the wild-type sequence shows a peak in the A helix and a low point in the E helix, consistent with the presence of the A helix in the kinetic and equilibrium molten globule states of the wild-type protein, the mutant protein shows a peak at the E helix and a lowered AABUF at the A helix. The population of protected amides in the burst phase of the kinetic folding of wild-type apoMb includes much of the A, G and H helices and part of the B helix (Figure 3a). This population is changed in the double mutant: the population of protected amides in the A helix is lowered and the population in the E helix is significantly increased (Figure 3b). These results are exactly what would be expected if local regions of the sequence that bury a large surface area on folding, as determined by the AABUF parameter, promote the initial collapse of the polypeptide chain. These conclusions were reinforced by further studies on apoMb mutants where the F helix (normally invisible in NMR experiments) was stabilized.41 These mutant proteins provide excellent examples of role of AABUF in driving the initial collapse and determining the folding pathway.
Figure 2.

Sequence variation of the average area buried upon folding (AABUF)25 for the myoglobin sequence, plotted using a nine-residue moving average. Black curve: wild-type sperm whale sequence. Red dotted curve: sperm whale sequence with the two mutations W14G, G73W Reproduced with permission from ref.26. Copyright 2005 Elsevier.
Figure 3.

Residue-specific plot of proton occupancy in the burst phase intermediate, measured at 6.4 ms folding time in a urea-jump folding experiment, with varying durations of the pH 10.1 labeling pulse. A. wild-type apomyoglobin. B. W14G, G73W mutant apomyoglobin. Red: pulse duration 7 ms; orange, 12 ms; green, 20 ms; blue, 35 ms; purple, 65 ms. The locations of helices A–H are shown Reproduced with permission from ref.26. Copyright 2005 Elsevier.
Measurements of NMR line broadening in paramagnetic spin-labeled apomyoglobin at pH 2.328 show that regions of the unfolded polypeptide with high AABUF values undergo spontaneous local collapse and promote transient, long-range hydrophobic interactions that play a key role in initiating the folding process. The most compact species observed in the acid unfolded protein appear to involve native-like contacts between the A/B and G/H helix regions that bury a large amount of hydrophobic surface and facilitate progression towards the molten globular folding intermediates (Figure 4).
Figure 4.

Schematic diagram of the apomyoglobin folding pathway. The AABUF profile (top of figure) shows peaks where the amino acid sequence indicates local clusters of hydrophobic residues and side chains such as Lys and Glu that contain long aliphatic regions in their side chains. Local interactions between the G and H helices and transient long-range interactions between the A and G/H regions are observed in the acid-unfolded state. The molten globule intermediate Ia is the first observable state, with stabilized helical structure and patterns of protected amide protons that indicate that the H helix is translocated (indicated by arrow and dotted ellipse). The H helix is also translocated in intermediate Ib, which contains more protected amides in the D and E helices, indicating that these helices are partly folded. After a few seconds, the final folded state forms, with all of the helices except F correctly folded and packed.
Apomyoglobin Folding Efficiency is Subordinated to Function
A mutant protein of a completely different type, where the functional distal histidine H64 is changed to a hydrophobic residue, increases the stability of the apoprotein and the equilibrium folding intermediate42 and increases the folding rate.43 This histidine is required for reasons of function, despite its effect in slowing the folding of the protein. Other mutants also demonstrate this subordination of folding efficiency to function. In apoMb at pH 6, the NH resonances of the F helix are invisible,18 probably due to conformational exchange. Mutants with increased helical structure in the F helix (P88K/S92K (F2) and P88K/A90L/S92K/A94L (F4)) were designed to alter the AABUF of the F helix to increase the likelihood that this region of the protein would participate in the earliest steps of folding.41 NH resonances were visible in the HSQC spectrum for the F helix residues of both mutant proteins at neutral pH, indicating that the F helix has been stabilized, but only the F4 mutant protein had a high enough AABUF to stabilize the F helix in the burst phase intermediate. Interestingly, the overall rate of folding is slowed in the F4 mutant, because the folded F helix stabilizes the intermediate and inhibits translocation of the H helix to its native position.41 The structural heterogeneity observed for the F helix in the native wild-type protein at pH 6 may be related to the in vivo insertion of the heme after the protein is folded. Flexibility in the F helix would allow insertion of the bulky heme, after which the heme pocket closes and the F helix becomes stably folded in the holoprotein.18
Extension of Rate Measurements
Conventional stopped-flow and quench flow apparatus is limited by their flow characteristics to the delineation of rates slower than a few milliseconds. Hence, these experiments provide little direct information on processes that occur faster than instrument dead times. Continuous-flow methods have recently been improved to the point where microsecond apomyoglobin refolding processes can be monitored.2,44,45 Quench-flow hydrogen exchange experiments performed using a continuous-flow mixer show that apomyoglobin folds by a hierarchical mechanism. The A, G, and H helix regions undergo rapid collapse within 0.4 ms of initiating refolding, leading to stabilization of helical structure and protection of amides from H/D exchange.45 This state, the Ia intermediate,6,44 corresponds to the burst phase intermediate observed in conventional stopped-flow experiments. After 6 ms, additional helical structure becomes stabilized by docking of the B helix region onto the AGH core, followed by progressive stabilization of structure in the C-terminal region of the E helix, the N-terminal region of E and the C helix to form the Ib intermediate. These intermediates are separated by an energy barrier and differ in the content of helical structure.6,36,44 Ib is a kinetically trapped state in which energetic frustration in the B-G helix packing and translocation of the H helix relative to G impedes progression to the folding transition state.36 Using a fluorescence-detected continuous-flow technique with a dead time as short as 40 μs, an additional intermediate between the unfolded state and Ia has been detected.2
Equilibrium Methods to Probe Alternatively Folded States
Proteins can be unfolded from their native states by altering the solution environment, by lowering or raising the pH, by changing salt concentrations, by the addition of denaturants or by raising or lowering the temperature. Another perturbation that has been applied to apomyoglobin is increase of pressure, which can unfold proteins by causing changes in solution volume. Pressure changes have been applied to myoglobin and changes in the folded state detected by a number of spectroscopic techniques,46–48 including NMR.49,50 The NMR detection of pressure-induced changes in the folded states of proteins has the advantage that site-specific information can be obtained throughout the protein, and the higher-energy conformers induced in the conformational ensemble can readily be detected. For apomyoglobin, the pressure experiments indicated that the protein forms an equilibrium mixture throughout the unfolding transition, populating two defined intermediate conformations as well as fully native and fully unfolded states.50
Another powerful NMR method to explore partly folded states that would be otherwise undetectable is the analysis of relaxation dispersion data.51 This method has been applied to the delineation of states of apomyoglobin that are populated as the pH is slightly decreased from 6.0 to 5.0.52 The F helix itself is invisible in the NMR spectrum, but relaxation dispersion in regions that contact the F helix indicate that these changes are associated with local unfolding and undocking of the F helix. At lower pHs, relaxation dispersion in more widespread regions of the protein indicated a three-state unfolding process with an intermediate state leading to a transiently populated molten globule state that is structurally similar to the equilibrium molten globule state observed at lower pHs.
Conclusions
Intensive research over the past 20 years has provided a coherent picture of the molecular events that accompany folding and unfolding processes in apomyoglobin (Figure 4). The unfolded protein consists of an ensemble of structures, largely unfolded but with local preferences for collapsed states. These preferences are directly related to the local amino acid sequence, in that local collapse initiates in regions with a high AABUF, where a large surface area is buried upon folding. These local hydrophobic clusters coalesce through transient long-range interactions involving widely-separated regions of the polypeptide chain, facilitating formation of compact molten globule intermediates that have native-like topology of the polypeptide chain.28 When the solution conditions are changed from those favoring the unfolded state (high urea, low pH) to those favoring the folded state (low urea, neutral pH), the polypeptide chain undergoes rapid and highly specific collapse, on a timescale faster than ~400 μs,44,45 to form a compact intermediate (Ia) with hydrogen-bonded helical structure in parts of the A, G, and H helix regions. Secondary structure becomes further stabilized as Ia progresses to Ib, with additional transient helical structure forming in local regions of the B, D, and E helices. The apomyoglobin folding landscape is highly rugged and folding is frustrated by energetic bottlenecks at the A-G-H, E-G-H, and B-E-G helix interfaces that impede progression to the transition state.36 Energetic frustration appears to play an important role by preventing excessive stabilization of non-native contacts in compact, partly folded states of correct chain topology while at the same time facilitating dynamic conformational fluctuations in frustrated regions that speed up the search for the native structure.
Biographical Information
H. Jane Dyson received B.Sc.(hons) and Ph.D. degrees from the University of Sydney and a Damon Runyon-Walter Winchell postdoctoral fellowship at Massachusetts Institute of Technology under the direction of Paul Schimmel (1977–78). She was a faculty member in Chemistry at the University of New South Wales (1979–1984), before joining Scripps Research Institute in 1984, where she is currently a Professor. She has been appointed as Editor-In-Chief of Biophysical Journal (2017–2021). Her research interests are in protein folding and dynamics, and structure and functional studies of proteins, both folded and intrinsically disordered, using NMR and other spectroscopic techniques.
Peter E. Wright received B.Sc., M.Sc. and Ph.D. degrees in Chemistry from the University of Auckland and did postdoctoral studies at the University of Oxford, England under the direction of R.J.P. Williams (1972–76). He was a faculty member in Inorganic Chemistry at the University of Sydney (1976–1983) and joined the faculty at The Scripps Research Institute in 1984, serving as Chairman of the Department of Molecular Biology from 1987–2012. He holds the Cecil and Ida Green Chair in Biomedical Research, and has been Editor-in-Chief of J. Mol. Biol. since 1990. He has honorary degrees from the Karolinska Institute (M.D., 1995) and University of Sydney (D.Sc., 2003) and is a member of the American Academy of Arts and Sciences (1995) and the National Academy of Sciences (2008), and a Fellow of the American Association for the Advancement of Science (1998), the International Society for Magnetic Resonance (2006) and the Japanese NMR Society (2006). His research interests are in applications of NMR to elucidate mechanisms of protein folding and unfolding, the function and interactions of intrinsically disordered proteins, the structural basis of protein-protein and protein-nucleic acid interactions in the regulation of gene expression, and the role of dynamics in protein function.
References
- 1.Jamin M, Yeh SR, Rousseau DL, Baldwin RL. Submillisecond unfolding kinetics of apomyoglobin and its pH 4 intermediate. J Mol Biol. 1999;292:731–740. doi: 10.1006/jmbi.1999.3074. [DOI] [PubMed] [Google Scholar]
- 2.Xu M, Beresneva O, Rosario R, Roder H. Microsecond folding dynamics of apomyoglobin at acidic pH. J Phys Chem B. 2012;116:7014–7025. doi: 10.1021/jp3012365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballew RM, Sabelko J, Gruebele M. Direct observation of fast protein folding: The initial collapse of apomyoglobin. Proc Natl Acad Sci US A. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulotta M, Gilmanshin R, Buscher TC, Callender RH, Dyer RB. Core formation in apomyoglobin: probing the upper reaches of the folding energy landscape. Biochemistry. 2001;40:5137–5143. doi: 10.1021/bi002256n. [DOI] [PubMed] [Google Scholar]
- 5.Griko YV, Privalov PL, Venyaminov SY, Kutyshenko VP. Thermodynamic study of the apomyoglobin structure. J Mol Biol. 1988;202:127–138. doi: 10.1016/0022-2836(88)90525-6. [DOI] [PubMed] [Google Scholar]
- 6.Jamin M, Baldwin RL. Two forms of the pH 4 folding intermediate of apomyoglobin. J Mol Biol. 1998;276:491–504. doi: 10.1006/jmbi.1997.1543. [DOI] [PubMed] [Google Scholar]
- 7.Privalov PL. Intermediate states in protein folding. J Mol Biol. 1996;258:707–725. doi: 10.1006/jmbi.1996.0280. [DOI] [PubMed] [Google Scholar]
- 8.Bismuto E, Sirangelo I, Irace G. Salt-induced refolding of myoglobin at acidic pH: molecular properties of a partly folded intermediate. Arch Biochem Biophys. 1992;298:624–629. doi: 10.1016/0003-9861(92)90458-9. [DOI] [PubMed] [Google Scholar]
- 9.Hamada D, Fukada H, Takahashi K, Goto Y. Salt-induced formation of the molten globule state of apomyoglobin studied by isothermal titration calorimetry. Thermochim Acta. 1995;266:385–400. doi: 10.1073/pnas.91.22.10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waltho JP, Feher VA, Merutka G, Dyson HJ, Wright PE. Peptide models of protein folding initiation sites. 1. Secondary structure formation by peptides corresponding to the G- and H-helices of myoglobin. Biochemistry. 1993;32:6337–6347. doi: 10.1021/bi00076a006. [DOI] [PubMed] [Google Scholar]
- 11.Udgaonkar JB, Baldwin RL. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature. 1988;335:694–699. doi: 10.1038/335694a0. [DOI] [PubMed] [Google Scholar]
- 12.Roder H, Elöve GA, Englander SW. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature. 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings PA, Wright PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 14.Tsui V, Garcia C, Cavagnero S, Siuzdak G, Dyson HJ, Wright PE. Quench-flow experiments combined with mass spectrometry show apomyoglobin folds through an obligatory intermediate. Protein Sci. 1999;8:45–49. doi: 10.1110/ps.8.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Springer BA, Sligar SG. High-level expression of sperm whale myoglobin in Escherichia coli. Proc Natl Acad Sci US A. 1987;84:8961–8965. doi: 10.1073/pnas.84.24.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thériault Y, Pochapsky TC, Dalvit C, Chiu ML, Sligar SG, Wright PE. 1H and 15N resonance assignments and secondary structure of the carbon monoxide complex of sperm whale myoglobin. J Biomol NMR. 1994;4:491–504. doi: 10.1007/BF00156616. [DOI] [PubMed] [Google Scholar]
- 17.Jennings PA, Stone MJ, Wright PE. Overexpression of myoglobin and assignment of the amide, Ca and Cb resonances. J Biomol NMR. 1995;6:271–276. doi: 10.1007/BF00197808. [DOI] [PubMed] [Google Scholar]
- 18.Eliezer D, Wright PE. Is apomyoglobin a molten globule? Structural characterization by NMR. J Mol Biol. 1996;263:531–538. doi: 10.1006/jmbi.1996.0596. [DOI] [PubMed] [Google Scholar]
- 19.Lecomte JT, Sukits SF, Bhattacharjya S, Falzone CJ. Conformational properties of native sperm whale apomyoglobin in solution. Protein Sci. 1999;8:1484–1491. doi: 10.1110/ps.8.7.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eliezer D, Chung J, Dyson HJ, Wright PE. Native and non-native secondary structure and dynamics in the pH 4 intermediate of apomyoglobin. Biochemistry. 2000;39:2894–2901. doi: 10.1021/bi992545f. [DOI] [PubMed] [Google Scholar]
- 21.Hughson FM, Wright PE, Baldwin RL. Structural characterization of a partly folded apomyoglobin intermediate. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 22.Yao J, Chung J, Eliezer D, Wright PE, Dyson HJ. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry. 2001;40:3561–3571. doi: 10.1021/bi002776i. [DOI] [PubMed] [Google Scholar]
- 23.Lietzow MA, Jamin M, Dyson HJ, Wright PE. Mapping long-range contacts in a highly unfolded protein. J Mol Biol. 2002;322:655–662. doi: 10.1016/s0022-2836(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 24.Schwarzinger S, Wright PE, Dyson HJ. Molecular hinges in protein folding: the urea-denatured state of apomyoglobin. Biochemistry. 2002;41:12681–12686. doi: 10.1021/bi020381o. [DOI] [PubMed] [Google Scholar]
- 25.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura C, Lietzow MA, Dyson HJ, Wright PE. Sequence determinants of a protein folding pathway. J Mol Biol. 2005;351:383–392. doi: 10.1016/j.jmb.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 27.Mohana-Borges R, Goto NK, Kroon GJA, Dyson HJ, Wright PE. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J Mol Biol. 2004;340:1131–1142. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Felitsky DJ, Lietzow MA, Dyson HJ, Wright PE. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc Natl Acad Sci US A. 2008;105:6278–6283. doi: 10.1073/pnas.0710641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavagnero S, Dyson HJ, Wright PE. Effect of H helix destabilizing mutations on the kinetic and equilibrium folding of apomyoglobin. J Mol Biol. 1999;285:269–282. doi: 10.1006/jmbi.1998.2273. [DOI] [PubMed] [Google Scholar]
- 30.Cavagnero S, Nishimura C, Schwarzinger S, Dyson HJ, Wright PE. Conformational and dynamic characterization of the molten globule state of an apomyoglobin mutant with an altered folding pathway. Biochemistry. 2001;40:14459–14467. doi: 10.1021/bi011500n. [DOI] [PubMed] [Google Scholar]
- 31.Nishimura C, Dyson HJ, Wright PE. The apomyoglobin folding pathway revisited: structural heterogeneity in the kinetic burst phase intermediate. J Mol Biol. 2002;322:483–489. doi: 10.1016/s0022-2836(02)00810-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang YZ, Paterson Y, Roder H. Rapid amide proton exchange rates in peptides and proteins measured by solvent quenching and two-dimensional NMR. Protein Sci. 1995;4:804–814. doi: 10.1002/pro.5560040420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishimura C, Dyson HJ, Wright PE. Enhanced picture of protein-folding intermediates using organic solvents in H/D exchange and quench-flow experiments. Proc Natl Acad Sci US A. 2005;102:4765–4770. doi: 10.1073/pnas.0409538102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura C, Wright PE, Dyson HJ. Role of the B helix in early folding events in apomyoglobin: evidence from site-directed mutagenesis for native-like long range interactions. J Mol Biol. 2003;334:293–307. doi: 10.1016/j.jmb.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 35.Nishimura C, Dyson HJ, Wright PE. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J Mol Biol. 2006;355:139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 36.Aoto PC, Nishimura C, Dyson HJ, Wright PE. Probing the non-native H helix translocation in apomyoglobin folding intermediates. Biochemistry. 2014;53:3767–3780. doi: 10.1021/bi500478m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishimura C, Dyson HJ, Wright PE. Energetic frustration of apomyoglobin folding: role of the B helix. J Mol Biol. 2010;396:1319–1328. doi: 10.1016/j.jmb.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwarzinger S, Mohana-Borges R, Kroon GJA, Dyson HJ, Wright PE. Structural characterization of partially folded intermediates of apomyoglobin H64F. Protein Sci. 2008;17:313–321. doi: 10.1110/ps.073187208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dyson HJ, Wright PE, Scheraga HA. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc Natl Acad Sci US A. 2006;103:13057–13061. doi: 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matheson RR, Jr, Scheraga HA. A method for predicting nucleation sites for protein folding based on hydrophobic contacts. Macromolecules. 1978;11:819–829. [Google Scholar]
- 41.Nishimura C, Dyson HJ, Wright PE. Consequences of stabilizing the natively disordered F helix for the folding pathway of apomyoglobin. J Mol Biol. 2011;411:248–263. doi: 10.1016/j.jmb.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hargrove MS, Singleton EW, Quillin ML, Ortiz LA, Phillips GN, Jr, Olson JS, Mathews AJ. His64(E7)-->Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J Biol Chem. 1994;269:4207–4214. doi: 10.2210/pdb1mgn/pdb. [DOI] [PubMed] [Google Scholar]
- 43.Garcia C, Nishimura C, Cavagnero S, Dyson HJ, Wright PE. Changes in the apomyoglobin folding pathway caused by mutation of the distal histidine residue. Biochemistry. 2000;39:11227–11237. doi: 10.1021/bi0010266. [DOI] [PubMed] [Google Scholar]
- 44.Uzawa T, Akiyama S, Kimura T, Takahashi S, Ishimori K, Morishima I, Fujisawa T. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of a-helical content and compactness. Proc Natl Acad Sci US A. 2004;101:1171–1176. doi: 10.1073/pnas.0305376101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uzawa T, Nishimura C, Akiyama S, Ishimori K, Takahashi S, Dyson HJ, Wright PE. Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR. Proc Natl Acad Sci US A. 2008;105:13859–13864. doi: 10.1073/pnas.0804033105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bismuto E, Sirangelo I, Irace G, Gratton E. Pressure-induced perturbation of apomyoglobin structure: Fluorescence studies on native and acidic compact forms. Biochemistry. 1996;35:1173–1178. doi: 10.1021/bi951163g. [DOI] [PubMed] [Google Scholar]
- 47.Vidugiris GJ, Royer CA. Determination of the volume changes for pressure-induced transitions of apomyoglobin between the native, molten globule, and unfolded states. Biophys J. 1998;75:463–470. doi: 10.1016/S0006-3495(98)77534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bondos SE, Sligar S, Jonas J. High-pressure denaturation of apomyoglobin. Biochim Biophys Acta (Prot Struct Mol Enzymol) 2000;1480:353–364. doi: 10.1016/s0167-4838(00)00088-1. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka N, Ikeda C, Kanaori K, Hiraga K, Konno T, Kunugi S. Pressure effect on the conformational fluctuation of apomyoglobin in the native state. Biochemistry. 2000;39:12063–12068. doi: 10.1021/bi001009g. [DOI] [PubMed] [Google Scholar]
- 50.Kitahara R, Yamada H, Akasaka K, Wright PE. High pressure NMR reveals that apomyoglobin is an equilibrium mixture from the native to the unfolded. J Mol Biol. 2002;320:311–319. doi: 10.1016/S0022-2836(02)00449-7. [DOI] [PubMed] [Google Scholar]
- 51.Loria JP, Rance M, Palmer AG., III A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- 52.Meinhold DW, Wright PE. Measurement of protein unfolding/refolding kinetics and structural characterization of hidden intermediates by NMR relaxation dispersion. Proc Natl Acad Sci US A. 2011;108:9078–9083. doi: 10.1073/pnas.1105682108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuriyan J, Wilz S, Karplus M, Petsko GA. X-ray structure and refinement of carbon-monoxy (Fe II)-myoglobin at 1.5 Å resolution. J Mol Biol. 1986;192:133–154. doi: 10.1016/0022-2836(86)90470-5. [DOI] [PubMed] [Google Scholar]
- 54.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]