

Abstract
Background and purpose
Myocarditis is an inflammatory and autoimmune cardiovascular disease that causes dilated myocardiopathy and is responsible for high morbidity and mortality worldwide. Cortistatin is a neuropeptide produced by neurons and cells of the immune and vascular systems. Besides its action in locomotor activity and sleep, cortistatin inhibits inflammation in different experimental models of autoimmune diseases. However, its role in inflammatory cardiovascular disorders is unexplored. Here, we investigated the therapeutic effects of cortistatin in a well‐established preclinical model of experimental autoimmune myocarditis (EAM).
Experimental Approach
We induced EAM by immunization with a fragment of cardiac myosin in susceptible Balb/c mice. Cortistatin was administered i.p. starting 7, 11 or 15 days after EAM induction. At day 21, we evaluated heart hypertrophy, myocardial injury, cardiac inflammatory infiltration and levels of serum and cardiac inflammatory cytokines, cortistatin and autoantibodies. We determined proliferation and cytokine production by heart draining lymph node cells in response to cardiac myosin restimulation.
Key Results
Systemic injection of cortistatin during the effector phase of the disease significantly reduced its prevalence and signs of heart hypertrophy and injury (decreased the levels of brain natriuretic peptide) and impaired myocardial inflammatory cell infiltration. This effect was accompanied by a reduction in self‐antigen‐specific T‐cell responses in lymph nodes and in the levels of cardiomyogenic antibodies and inflammatory cytokines in serum and myocardium. Finally, we found a positive correlation between cardiac and systemic cortistatin levels and EAM severity.
Conclusions and Implications
Cortistatin emerges as a new candidate to treat inflammatory dilated cardiomyopathy.
Abbreviations
- BNP
brain natriuretic peptide
- DC
dendritic cell
- DLN
draining lymph node
- EAM
experimental autoimmune myocarditis
- MyHC‐α
α‐myosin heavy chain
- Th
T helper
- Treg
regulatory T cell
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Cardiovascular disease has become the most frequent cause of death globally. Although many diseases affect the cardiovascular system, myocarditis and subsequent dilated cardiomyopathy are major causes of heart failure in young patients. Myocarditis caused by exposure to toxins or infection with bacteria or viruses is characterized by infiltration of inflammatory cells into the myocardium with consequent loss of myocytes and development of fibrosis (Cooper, 2009). Myocardial injury can also induce post‐infectious autoimmune responses to heart tissue, which are critical in the pathogenesis of dilated cardiomyopathy (Cihakova and Rose, 2008; Cooper, 2009; Mann, 2011). Studies in rodent models have revealed that myocarditis can be induced by coxsakievirus B3 infection, by immunization with cardiac myosin or myosin‐derived peptides in adjuvant, or by adoptive transfer of cardiac myosin‐activated CD4+ T cells (Rose, 2014). Moreover, specific immune responses against cardiac myosin are directed by self‐antibodies and T cells [mainly T helper‐17 (Th17)] targeting the myocardium (Stephenson et al., 2016). These findings raise the possibility that immunomodulatory therapeutic approaches may be successful in this cardiovascular disease.
Cortistatin is a neuropeptide discovered in brain cortex, based on its inhibitory neuronal activities, that shows remarkable sequential/structural resemblances to somatostatin (de Lecea et al., 1996). Through its binding to all the somatostatin‐receptors (sst1–5), cortistatin exerts many somatostatin‐like functions, especially concerning hormonal and neuronal regulation (Gahete et al., 2008; Siehler et al., 2008). However, cortistatin plays unique functions in the brain and immune system that are related to its capacity to activate receptors other than those of somatostatin, mainly ghrelin receptor 1 (Gonzalez‐Rey et al., 2006a; Gahete et al., 2008; Gonzalez‐Rey and Delgado, 2008; Siehler et al., 2008). Although its role in the cardiovascular system is largely unknown, recent evidence induced us to the study of the effect of cortistatin in inflammatory cardiovascular diseases. First, the cardiovascular system produces cortistatin, especially in response to injury. Thus, heart, arterial endothelium and arterial smooth muscle cells express cortistatin and its receptors (Duran‐Prado et al., 2013; Robas et al., 2003; Zhang et al., 2015a). Moreover, cortistatin is elevated in human atherosclerotic plaques, in mouse arteries subjected to blood‐flow alterations and in the plasma of patients with coronary heart disease (Tian et al., 2006; Duran‐Prado et al., 2013; Zhang et al., 2015b). Second, cortistatin improves the vasculopathies associated with vascular remodelling and stenosis and reduces vascular calcification in animals (Liu et al., 2010; Duran‐Prado et al., 2013). Finally, cortistatin has emerged as a potent anti‐inflammatory factor that regulates both innate and adaptive immune responses in many inflammatory and autoimmune disorders (Gonzalez‐Rey et al., 2006a,b, 2007; Souza‐Moreira et al., 2013).
Therefore, here, we investigated the effect of cortistatin in a preclinical model of autoimmune myocarditis induced in a susceptible mouse strain by immunization with a self‐peptide derived from the α‐myosin heavy chain (MyHC‐α).
Methods
Animals and ethical statement
Female and male Balb/c and male C57Bl/6 mice (7–8 weeks old) were obtained from Charles River and were housed in a controlled‐temperature/humidity environment (22 ± 1°C, 60–70% relative humidity) in individual cages (10 mice per cage, with wood shaving bedding and nesting material), with a 12 h light/dark cycle (lights on at 0700 h) and were fed rodent chow (Global Diet 2018, Harlan) and tap water ad libitum. Mice were allowed to acclimatize to their housing environment for at least 5 days prior to experimentation and to the experimental room for 1 h before experiments. The experimental protocols of this study conform to EU Directive 2010/63 and followed the ethical guidelines for investigations with experimental animals approved by the Ethics Review Committee for Animal Experimentation of Spanish Council of Scientific Research. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Induction of experimental autoimmune myocarditis (EAM)
In this study, we used a well‐established murine model of autoimmune myocarditis, which mirrors important aspects of human inflammatory dilated cardiomyopathy and which is widely used to assay novel therapeutic agents against miocarditis (Cihakova and Rose, 2008). We immunized Balb/c mice s.c. with 100 μg of a murine heart muscle‐specific peptide derived from MyHC‐α (MyHC614–629, Ac‐RSLKLMATLFSTYASADR‐OH, in which two arginines were included at both ends to increase its solubility; purity >93%, Anaspec) emulsified 1/1 in complete Freund's adjuvant (Difco) on days 0 and 7. At day 7, animals were randomly distributed in different experimental groups: (1) mice that received i.p. PBS (control group) three times per week starting at day 7; (2) mice that received i.p. cortistatin (1 nmol, which corresponds to 1.7 μg of cortistatin‐14, from Bachem) three times per week starting at day 7; (3) mice that received i.p. cortistatin at 0.1, 0.5 or 1 nmol starting at day 11; or (4) mice that received i.p. cortistatin at 1 nmol starting at day 15 (a total of six injections in all cases, see scheme depicted in Figure S1). Serum levels of cortistatin increased from 32 pg·mL−1 (basal levels before injection) to 62 and 55 pg·mL−1 at 10 and 30 min after i.p. injection of cortistatin, respectively, and returned to basal levels 1 h later. At day 21 after experimental autoimmune myocarditis (EAM) induction, we collected blood by cardiac puncture, perfused the animals with cold‐PBS and removed hearts, spleen and cranial mediastinal lymph nodes. We isolated serum from blood to determine the levels of the cardiac damage biomarker brain natriuretic peptide (BNP), cortistatin, inflammatory cytokines and MyHC‐specific antibodies as described below. Hearts were weighed, macroscopically scored and processed for histopathological analysis and for RNA isolation (see below). Haematoxylin‐eosin‐stained heart sections were scored blindly by two individuals according to a semi‐quantitative scale of inflammatory infiltrates (0, indicated no focal inflammatory infiltrates; 1, up to 5% of the cross‐sectional area of the heart section; 2, 6 to 20%; 3, 21 to 50%; 4, >50% of a cross‐section involved). Images were acquired using an Olympus microscope, and the area of myocardium and surrounding tissue affected by myocarditis (consisting of inflammatory cells and myocardial necrosis) relative to the entire area was determined by a computer‐assisted analyser (Image J software, NIH) as previously described (Suzuki et al., 2006) in a blind fashion by two independent investigators. Values for three ventricular regions were averaged for each heart, and the mean percentage of affected area for each group was calculated. In addition, the presence of inflammatory infiltrates was confirmed by immunofluorescence analysis of CD45+ leukocytes in the heart sections and by flow cytometric analysis of mononuclear cells isolated from heart (Afanasyeva et al., 2004) as described below. The heart sections were also stained with Picric Sirius Red to evaluate collagen deposits and initial signs of fibrosis, and the area ratio (affected/entire area as a percentage) was automatically quantified with the Fibrosis HR software (ImaGesp) as previously described (Masseroli et al., 1998). Draining lymph nodes (DLNs) and spleen cell suspensions were isolated 14 or 21 days after EAM induction and used for flow cytometric analysis and to study self‐reactive responses as described below.
Measurement of levels of BNP, cortistatin, cytokines and anti‐MyHC antibodies in serum
BNP and cortistatin were determined by using a competitive elisa (RayBio and Phoenix Pharmaceuticals, respectively), and the cytokines IL‐1β, IL‐17, IL‐6 and TNFα were measured by using specific sandwich elisas (BD Pharmingen) following the manufacturer's recommendations. To determine anti‐MyHC IgG antibodies, elisa 96‐well plates were coated overnight at 4°C with 2 μg·mL−1 MyHC614–629 in PBS. After washing with PBS containing 0.05% Tween‐20, nonspecific binding was blocked with PBS containing 10% BFS for 2 h at room temperature. After being washed three times, serum samples diluted 1/50 were added and incubated for 2 h at room temperature. After four washes, biotin‐conjugated goat anti‐mouse IgG, IgG1 or IgG2a antibodies (Jackson Immunoresearch Laboratories) were added and incubated at room temperature for 1 h, followed by six washes and incubation with avidin‐peroxidase for 30 min. After eight washes, plates were developed using 2,2′‐azino‐di‐(3‐ethyl benzothiazoline‐6‐sulphonic acid) as a substrate for peroxidase, and the optical density was measured using a microplate reader.
Measurement of cortistatin and cytokine gene expression in myocardial tissue
Total RNA was isolated from hearts following the manufacturer's protocol (Tripure, Roche). Precipitated RNA was treated with DNase 1 (Sigma‐Aldrich) before reverse transcription (RevertAid First Strand cDNA Synthesis Kit, Thermo Fisher Scientific). SYBER green quantitative PCR (SensiFast Sybr No‐Rox mix, Bioline) was performed on the Bio‐Rad CFX using the following conditions: 95°C for 5 min followed by 35 cycles at 95°C for 30 s with annealing (see Table S1 for annealing temperature and time used for each gene) and extension at 72°C for 30 s. Primer sequences are listed in Table S1. The expression of each gene was normalized against the expression of the housekeeping gene GAPDH in every PCR reaction.
Measurement of autoreactive response in EAM
Single‐cell suspensions (106 cells·mL−1) from DLNs and spleen were obtained 21 days post‐immunization and were stimulated in complete RPMI medium (RPMI‐1640 supplemented with 100 U·mL−1 penicillin/streptomycin, 2 mM L‐glutamine, 50 μM 2‐mercaptoethanol and 10% FCS) with MyHC614–629 (10 μM). To evaluate polyclonal stimulation, cells were cultured with 1 μg·mL−1 anti‐CD3 antibody or with 2.5 μg·mL−1 concanavalin A (Sigma). After 72 h of culture, we evaluated cell proliferation by adding 2.5 μCi·mL−1 [3H]‐thymidine during the last 8 h of culture and determining the cpm. After 48 h of culture, we measured the concentrations of cytokines and chemokines in culture supernatants by specific sandwich elisas (BD Pharmingen). To determine whether cortistatin affects the autoreactive response in vitro, DLN cells isolated at day 21 from mice with EAM were stimulated with MyHC614–629 (10 μM) in the absence or presence of cortistatin (10 nM). In some experiments, antagonists for various cortistatin receptors (all obtained from Sigma‐Aldrich), including cyclosomatostatin (an antagonist for all sst1–5 receptors), CYN‐154806 (a selective sst2 receptor antagonist) and GHRP6 (a specific ghrelin receptor antagonist) were added at 1 μM to MyHC‐activated DLN cell cultures in the absence or presence of cortistatin (10 nM). Moreover, to neutralize cortistatin secreted by MyHC‐activated DLN cells, a rabbit anti‐mouse cortistatin antibody (Phoenix Pharmaceuticals) or control IgG antibody (BD Bioscience) were added at 20 μg·mL−1 to cell cultures. The production of cytokines and the proliferative response were determined at 48 and 72 h respectively.
Immunofluorescence
Sections of hearts were fixed in 4% phosphate‐buffered formaldehyde solution (10 min, 20°C), blocked with 10% goat serum (30 min, 20°C) and incubated with phycoerythrin‐anti‐mouse CD45 antibody (1:500, BD Biosciences). Nuclei were Hoechst‐counterstained, and slices were examined in an Olympus fluorescence microscope.
Flow cytometric analysis
For analysis of heart‐infiltrating cells, single cell suspensions were isolated 21 days after EAM induction and were incubated with anti‐2.4G2 antibody (Mouse BD Fc Block™, 1:100, 4°C, 10 min) to avoid non‐specific binding to Fc‐receptors and with 7‐aminoactinomycin D (Calbiochem, 1:100) to exclude dead cells, washed in PBS/0.1% BSA. Cells were surface stained with allophycocyanin‐labelled anti‐CD4, FITC‐labelled anti‐CD11b and phycoerythrin‐labelled anti‐CD45 monoclonal antibodies (each at 4–5 μg·mL−1, BD Bioscience, 30 min, 4°C) and were analysed in a FACScalibur flow cytometer (BD Biosciences). Data were acquired until at least 100.000 events were collected from a live gate using forward/side scatter plots and 7‐aminoactinomycin D staining.
To analyse the number of cells expressing cytokines, we assayed intracellular cytokines by flow cytometry. Infiltrating inflammatory cells isolated from hearts of mice with EAM at day 21 as well as spleen and DLN cells isolated at days 14 and 21 after EAM induction were activated with 25 ng·mL−1 phorbol 12‐myristate 13‐acetate for 14 h in the presence of monensin (1.33 μM) for the last 6 h. Cells were incubated with anti‐2.4G2 antibody plus 7‐aminoactinomycin D, washed in PBS per 0.1% BSA and were then stained with allophycocyanin‐labelled anti‐CD4 monoclonal antibody as described above. After extensive washing, cells were fixed/permeabilized with Cytofix/Cytoperm solutions (BD Biosciences), stained with phycoerythrin‐labelled anti‐IL‐17 and FITC‐labelled anti‐IFNγ monoclonal antibodies (BD Pharmingen, 2 μg·mL−1, 30 min, 4°C) and analysed in a FACScalibur flow cytometer.
For FoxP3 staining, DLN cells were isolated from mice 14 and 21 days after EAM induction and incubated with FITC‐labelled anti‐CD25 and allophycocyanin‐labelled anti‐CD4 monoclonal antibodies (5 μg·mL−1, BD Biosciences) for 1 h at 4°C. After extensive washing, cells were fixed/permeabilized (eBioscience), stained with phycoerythrin‐labelled anti‐FoxP3 antibody (5 μg·mL−1, eBioscience) for 30 min at 4°C and analysed in a FACScalibur flow cytometer. In all cases, we used isotype‐matched antibodies (BD Biosciences) as controls.
Measurement of dendritic cell function
Dendritic cells (DCs) were differentiated from bone marrow cells obtained from femurs and tibiae of male C57Bl/6 mice. Bone marrow cells (2 × 106) were incubated in Petri dishes in complete RPMI medium containing 20 ng·mL−1 of granulocyte macrophage‐colony stimulating factor (PreproTech). At day 6 of culture, non‐adherent cells were collected (routinely containing 80–90% CD11c+ cells) and were incubated with complete medium or stimulated with LPS (1 μg·mL−1, Sigma) in the absence or presence of cortistatin (100 nM). After 48 h of culture, flow cytometric analysis for CD40, CD80 and CD86 was performed as previously described (Chorny et al., 2006), and the content of cytokines in the culture supernatants was determined by elisa. After 24 h of culture, phagocytosis activity of DCs was determined by flow cytometry after incubation with FITC‐labelled dextran (Sigma) during 2 h as previously described (Delgado et al., 2005). In addition, after 24 h of culture, DCs (2 × 105 cells·mL−1) were co‐cultured with allogeneic T cells (106 cells·mL−1) isolated from spleens of male Balb/c mice to determine the proliferative response and production of IFNγ as described above.
Statistical analysis
All data are expressed as mean ± SEM. We analysed data for statistical differences using Student's 2‐tailed unpaired t‐test to compare two groups or one‐way ANOVA followed by Fisher's protected least significant difference test to compare multiple groups. To compare the severity scores of myocarditis between two groups, the Mann–Whitney U‐test was used. Associations between cardiac or serum cortistatin levels and heart‐to‐body‐weight ratios or serum BNP levels were evaluated using Spearman' correlation test. A probability value of P < 0.05 was considered to be statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Treatment with cortistatin reduces disease severity and heart inflammation in animals with EAM
As expected, mice immunized with cardiac myosin peptide and killed at day 21 post‐immunization showed the severe signs of myocarditis, characterized by marked enlargement of heart and sporadic areas of calcification in pericardium (Figure 1A–B). Myocarditis was accompanied by an increase in serum BNP (Figure 1C), a biomarker of cardiac dysfunction (Atisha et al., 2004). The hypertrophy of the heart correlated with the presence of severe and disseminated inflammatory infiltration of leukocytes in myocardium (Figures 1D and S2) and of incipient and focal areas of pericardial myocyte necrosis (Figure 1D) and fibrosis (Figure 1E). Systemic treatment with cortistatin during the progression phase of EAM (from day 7 to day 19 post‐immunization) significantly reduced the disease severity, assessed by improved signs of heart hypertrophy and reduction of serum BNP levels, of myocardial inflammatory infiltration and of epicardial collagen deposits (Figures 1 and S2A). Importantly, injection of cortistatin reduced disease prevalence (percentage of animals with histological scores >0) from 88 to 44% at the end of the study (Figure 1D). A flow cytometric analysis of the heart infiltrates revealed an attenuation of the inflammatory CD45+ leukocytes, CD4+ lymphocytes and CD11b+ cells (mostly monocytes) in cortistatin‐treated mice with EAM (Figure 2). We observed that the effects of cortistatin were dose‐dependent (Figures 3A and S3A) and that delay of the initiation of treatment with cortistatin to day 11, but not to day 15 (a late time point at the progression phase of the disease), still resulted in a protective effect (Figures 3B and S3B).
Figure 1.
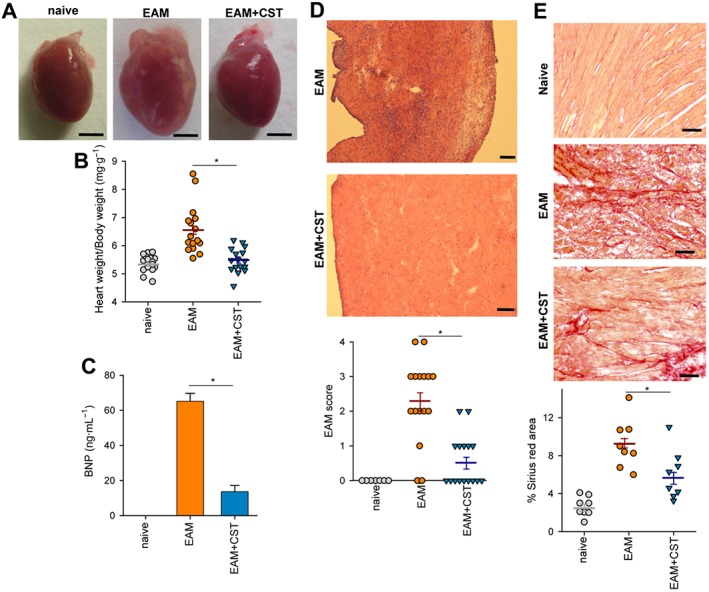
Cortistatin alleviates clinical signs of EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST, 1 nmol per mouse) three times per week for 2 weeks starting at day 7. At day 21, hearts and sera were obtained from each experimental group for analysis. Naïve mice were used as a reference. (A) Macroscopic evaluation of hearts. Scale bars: 2 mm. (B) Ratio between heart weight and body weight. (C) Levels of BNP in serum (n = 16 mice per group). (D) Haematoxylin–eosin staining of heart sections showing areas of myocardial inflammatory infiltration and pericardial myocyte necrosis and histopathological scores measuring extension of myocardial inflammation. Scale bars: 100 μm. The identity of inflammatory infiltrates was confirmed by immunostaining of CD45+ leukocytes in myocardial sections (Figure S2A). (E) Sirius Red staining of heart sections showing areas of incipient collagen deposits in pericardium and the measurement of areas positive for Sirius Red staining. Scale bars: 50 μm. In (B, D and E), each symbol represents one mouse, horizontal lines are the mean and vertical lines represent SEM for each experimental group. Results correspond to three independent experiments. *P < 0.05.
Figure 2.
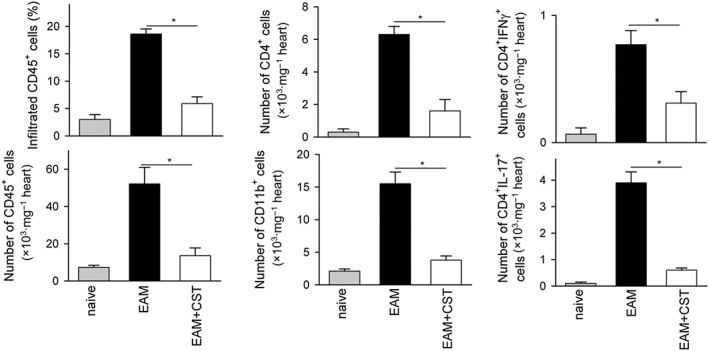
Treatment with cortistatin reduces the number of inflammatory cells infiltrating the heart of mice with EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week for 2 weeks starting at day 7. At day 21, inflammatory cells infiltrating the heart were isolated and analysed by flow cytometry. Frequency and/or absolute number of CD45+, CD4+, CD11b+, IFNγ+ and IL‐17+ cells within live cells were determined. Naïve mice were used as reference. Representative plots are shown in Figure S2B–C. n = 6 mice per group. *P < 0.05.
Figure 3.
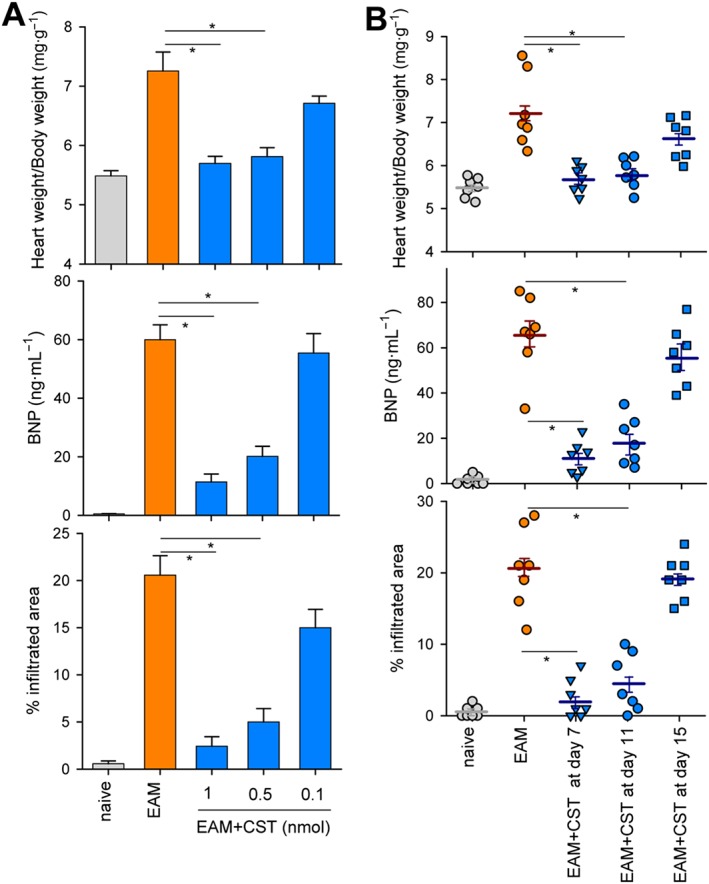
Dose‐ and time‐dependent response to cortistatin in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) at different doses (A) or at 1 nmol per mouse (B) three times per week starting at day 11 (A) or at different time points (B). At day 21, hearts and sera were obtained to determine heart‐to‐body weight ratios, levels of serum BNP and extension of myocardial area with inflammatory infiltration and cardiomyocyte necrosis (representative images of heart sections of each experimental group are shown in Figure S3). Naïve mice were used as reference. n = 7 mice per group. In (B), each symbol represents individual mice, horizontal lines are the mean and vertical lines represent SEM. *P < 0.05.
We next investigated the mechanisms underlying the decrease in severity of EAM following cortistatin treatment. In myocarditis, progression of the autoimmune response involves the development of autoreactive CD4 T cells in peripheral lymphoid organs, their entry into the myocardium and subsequent recruitment of inflammatory cells (Cihakova and Rose, 2008). Moreover, antibodies directed against cardiac myosin are critically involved in the pathogenesis of EAM and dilated cardiomyopathy (Cihakova and Rose, 2008; Mann, 2011). Intracellular cytokine staining of the myocardial infiltrating CD4+ cells revealed that treatment with cortistatin reduced the numbers of IL‐17+ cells (Th17) and IFNγ+ cells (Th1) in mice with EAM (Figure 2). Moreover, cortistatin injection diminished the amounts of inflammatory cytokines related to progression of myocarditis, such as IL‐1β, TNFα, IL‐6, IFNγ and IL‐17, in heart (Figure 4) and/or serum (Figure S4). Importantly, treatment with cortistatin reduced the levels of MyHC‐specific IgGs, particularly IgG2a and IgG1 auto‐antibodies in the sera of mice with EAM (Figure 5).
Figure 4.
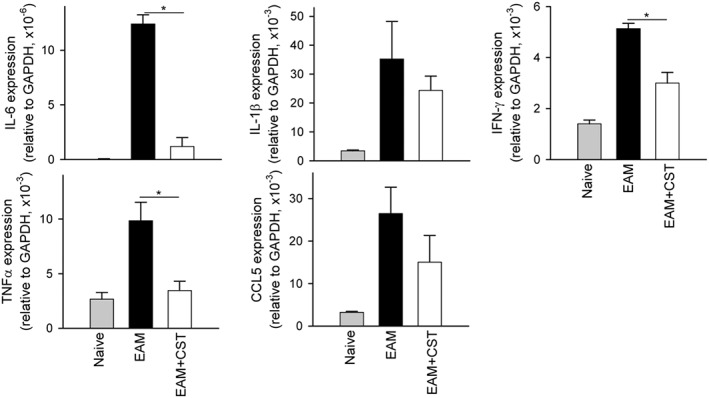
Cortistatin regulates the inflammatory cytokine milieu in the heart during EAM progression. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week for 2 weeks, and the heart was isolated at day 21. Naïve mice were used as reference. RNA expression of cytokines was assayed by quantitative real‐time PCR and normalized with GAPDH gene expression. n = 5 mice per group. *P < 0.05.
Figure 5.
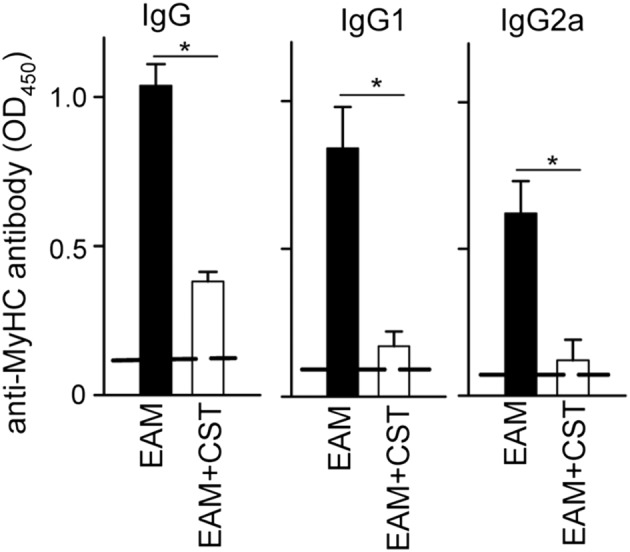
Cortistatin decreases the levels of autoantibodies in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week for 2 weeks. At day 21, sera were obtained and the levels of anti‐MyHC614–629‐specific antibodies were determined by elisa. Naïve mice were used as reference, and their values are shown by dashed lines. n = 8 mice per group, performed in two independent experiments. *P < 0.05.
Cortistatin regulates cardiomyogenic T‐cell responses
We next investigated whether this effect was mediated by regulating cardiomyogenic T cell‐driven responses in peripheral lymphoid organs. Spleen and DLN cells isolated from cortistatin‐treated EAM mice at the peak of the disease (at day 21) showed reduced MyHC‐specific proliferation of effector T cells in comparison with untreated EAM mice (Figure 6A). Moreover, treatment with cortistatin impaired the capacity of DLN T cells to produce IL‐17, IFNγ, IL‐2 and CXCL‐10 in a recall response to the cardiomyogenic antigen (Figure 6A). Interestingly, the suppressive effect of cortistatin was antigen‐specific, because T cells isolated from cortistatin‐treated EAM mice responded adequately to polyclonal stimulation (Figure S5). Intracellular cytokine determination showed that treatment with cortistatin significantly reduced the increase in the number of Th17 cells observed in DLNs of EAM mice, whereas it did not affect the number of IFNγ‐producing T cells (Figure 6B). Moreover, administration of cortistatin significantly increased the proportion of CD25+FoxP3+ regulatory T cells (Treg) in the CD4+ cell population of DLNs isolated during the effector phase of the disease (at day 14), but not isolated at day 21 (Figure 6C). These results suggest that cortistatin could regulate the activation of cardiomyogenic T‐cell responses in peripheral lymphoid organs.
Figure 6.
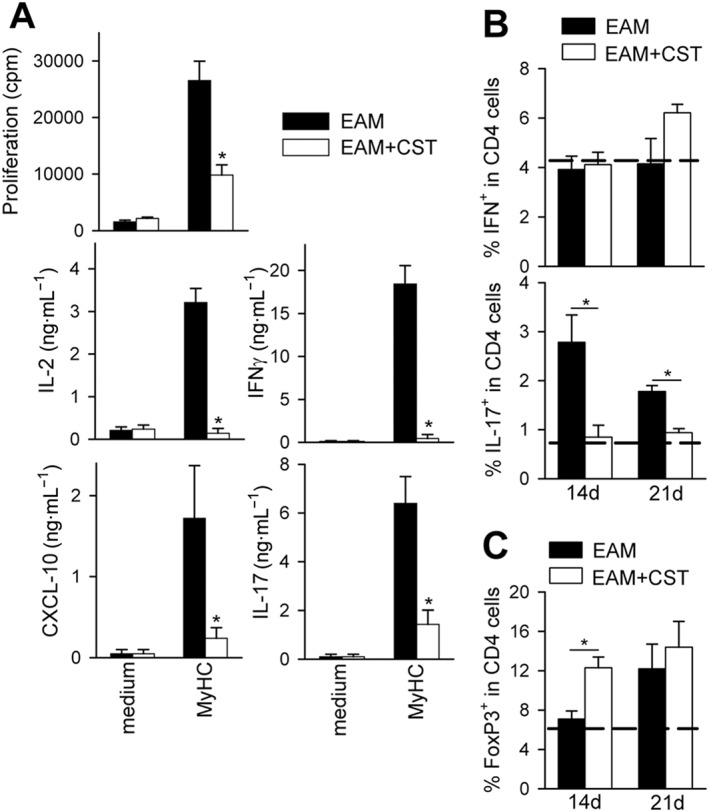
Treatment with cortistatin impairs cardiomyogenic T‐cell responses in vivo. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week for 2 weeks. (A) At day 21, DLN cells were isolated. Proliferation and cytokine production by DLN cells cultured in medium or restimulated with MyHC614–629 was determined. We obtained similar results when splenocytes were analysed. n = 10 mice per group, performed in two independent experiments. *P < 0.05 versus untreated EAM mice. (B) Flow cytometric analysis of intracellular cytokines in the CD4 population of activated DLN cells isolated at days 14 and 21 after EAM induction. n = 8–12 mice per group, performed in three independent experiments. (C) Flow cytometric analysis of CD25+FoxP3+ T cells in gated CD4+ cells of DLN cells isolated 14 and 21 days after EAM induction. Representative dot plots are shown in Figure S6. Dashed lines in (B and C) represent values obtained with DLN cells isolated from naïve mice. n = 8 mice per group, performed in two independent experiments. *P < 0.05.
Therefore, we next evaluated the effect of cortistatin in MyHC‐stimulated T cells isolated from DLNs and spleens of animals with EAM. We found that cortistatin suppressed in vitro cell proliferation and cytokine production by MyHC‐stimulated T cells (Figure 7A), suggesting that cortistatin could exert a direct effect on cardiomyogenic T cells. We further investigated the effect of cortistatin on the activity of DCs. Cortistatin failed to regulate DC functions, including the phagocytosis capacity, the expression of costimulatory molecules, the secretion of inflammatory cytokines and the induction of allogeneic T cell responses (Figure S7).
Figure 7.
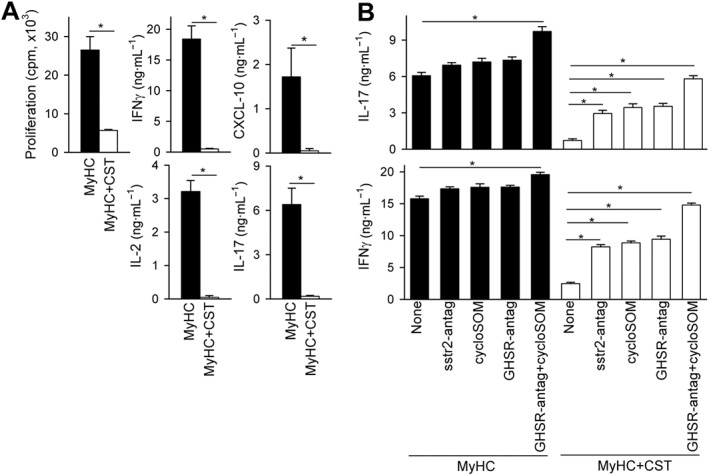
Cortistatin inhibits cardiomyogenic T‐cell responses in vitro. (A) Proliferative response and production of cytokines by DLN cells isolated from mice with EAM at day 21 and restimulated ex vivo with MyHC614–629 in the absence (MyHC) or presence of cortistatin (MyHC + CST). n = 9 mice per group, performed in two independent experiments. (B) Effect of antagonists for somatostatin receptors and the ghrelin receptor on cytokine production by MyHC‐activated DLN cells isolated from mice with EAM. n = 5 mice per group, performed in duplicate. *P < 0.05.
Involvement of cortistatin receptors in the impairment of the cardiomyogenic response
We next investigated the involvement of specific receptors in the actions exerted by cortistatin on cardiomyogenic T cell responses in vitro. Because cortistatin binds to all somatostatin‐receptors (sstr1–5) and ghrelin‐receptor GHSR1, and immune cells express some of these receptors, including sstr2 and GHSR1 (Gonzalez‐Rey and Delgado, 2007), they emerge as potential mediators of the immune‐regulatory effects of cortistatin. Indeed, treatment either with the non‐selective antagonist for sst1–5‐receptors cyclosomatostatin, with the specific antagonist for sst2‐receptor CYN‐154806 or with the GHSR1 antagonist GHRP6 partially reversed the inhibitory effects of cortistatin in the MyHC‐induced production of IL‐17 and IFNγ by DLN cells isolated from mice with EAM (Figure 7B). As expected, co‐treatment with antagonists for both sst1–5‐receptors and GHSR1 showed a significant additive effect and almost completely abrogated the inhibitory activity of cortistatin in cardiomyogenic T cells (Figure 7B). Interestingly, the blockade of both types of cortistatin‐receptors in MyHC‐activated DLN cells, in the absence of exogenous cortistatin, resulted in a significant increase in the production of IL‐17 and IFNγ (Figure 7B), suggesting that cortistatin secreted by immune cells could play a role in the endogenous regulation of the cardiomyogenic response. This hypothesis was confirmed by the fact that the addition of a neutralizing antibody against cortistatin increased the secretion of IL‐17 in MyHC‐activated DLN cultures (Figure S8).
Positive correlation between cortistatin levels and EAM severity
Because cortistatin is produced by heart and inflammatory cells, we finally evaluated whether the amounts of cortistatin changed in hearts of mice with EAM. Cortistatin gene expression significantly increased in myocardial tissue isolated on day 21 after EAM induction compared with that observed in hearts of naïve mice (Figure 8A). Similarly, we observed significantly higher amounts of cortistatin in serum of animals with EAM than in serum of naïve mice (Figure 8B). Moreover, we observed decreased levels of cardiac and serum cortistatin in parallel to reduced disease severities in cortistatin‐treated mice with EAM (Figure 8A). Indeed, we found a positive correlation between cardiac cortistatin levels and EAM severity (Figure 8C) and between levels of cortistatin and BNP in serum of animals with EAM (Figure 8D).
Figure 8.
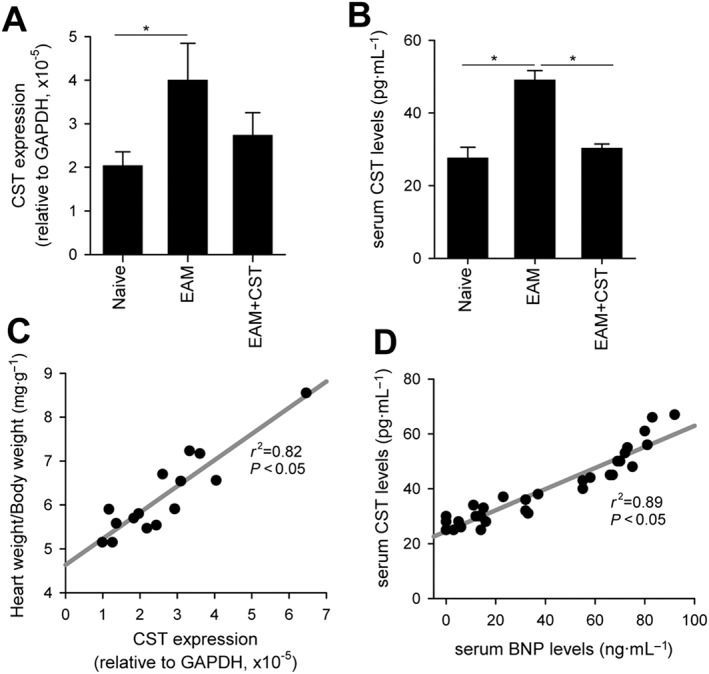
Cortistatin levels in myocardium and serum positively correlate with EAM severity. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week for 2 weeks. At day 21, heart RNA and sera were isolated and subjected to quantitative real‐time PCR to determine cortistatin gene expression and to elisa to determine blood levels of cortistatin and BNP. (A) Results are expressed as cortistatin gene expression relative to GAPDH. n = 5 mice per group. *P < 0.05. (B) Cortistatin levels in sera. n = 16 mice per group. *P < 0.05. (C) Correlation between cortistatin gene expression in the heart and EAM severity (measured as heart‐to‐body weight ratios). (D) Correlation between cortistatin and BNP levels in serum of mice with EAM. Each symbol in (C and D) represents individual mice; r 2 corresponds to the value of correlation obtained with the linear regression analysis.
Discussion and conclusions
Cortistatin is a neuropeptide that has emerged as an anti‐inflammatory factor that regulates self‐reactive responses in various experimental models of autoimmune disorders (Gonzalez‐Rey et al., 2006b, 2007; Souza‐Moreira et al., 2013). In this study, we provide evidence that cortistatin could be considered a protective therapeutic agent for cardiovascular diseases caused by exacerbated inflammatory and autoimmune responses. Using a well‐characterized mouse model of autoimmune myocarditis, which mirrors important aspects of human inflammatory dilated cardiomyopathy, we demonstrate that cortistatin ameliorated heart hypertrophy as well as myocardial inflammation and injury. In the model of EAM used in this study, signs of inflammation in the heart and autoantibodies against cardiac myosin in sera are only evident during the progression phase of the disease, between 10 and 21 days post‐immunization (Cihakova and Rose, 2008). During the late phase, following day 21 post‐immunization, myocardial inflammation declines and fibrosis gradually replaces it (Cihakova and Rose, 2008). This model is useful to study pathogenic mechanisms and to assay novel therapeutic agents against the inflammatory chronic phase of the disease where autoimmunity is the prevailing cause for ongoing disease after clearance of the virus. Data collected from clinical trials showed that immunosuppressive treatments were mainly beneficial for affected patients showing chronic inflammatory heart disease and absence of viral particles in heart, also suggesting a role of the immune system in active myocarditis (Frustaci et al., 2003; Hia et al., 2004; Frustaci et al., 2009; Frustaci and Chimenti, 2015). Our data indicate that the effects of cortistatin on EAM are mainly exerted during the progression phase of the disease, in which innate and adaptive immune responses play pivotal roles.
The beneficial effects of cortistatin on EAM were associated with inhibition of the production of autoantibodies and inflammatory cytokines. This effect is mostly exerted by regulating the cardiomyogenic sensitization in the peripheral immune compartment. Our findings suggest that treatment with cortistatin could impair the activation and/or expansion of tissue‐specific self‐reactive Th17‐ and Th1‐cell clones. Although the role played by Th1 cells in EAM remains controversial, ample evidence indicates that Th17 cells are critically involved in the generation of cardiomyogenic T cell responses and the establishment of myocardial inflammation (Cihakova and Rose, 2008; Stephenson et al., 2016). In fact, mice suffering EAM increased the percentage of Th17 cells in peripheral lymphoid organs and heart, and treatment with cortistatin reduced it. Importantly, this treatment did not result in a general suppression of the immune response, since cortistatin did not affect T cell responses, including Th17‐mediated response, to a polyclonal restimulation. This effect may suppose an advantage versus nonspecific immunosuppressants currently used in clinic to treat myocarditis (Frustaci et al., 2003; Hia et al., 2004; Frustaci et al., 2009; Frustaci and Chimenti, 2015). This finding is also important for designing future clinical trials in human myocarditis, because it has been described that, in patients suffering hyper IgE syndromes with recurrent bacterial and fungal infections, reduced Th17 function has been associated with vascular aneurism development (Chandesris et al., 2012; Freeman et al., 2011). This kind of selectivity in the inhibitory action of cortistatin in T‐cell responses was previously demonstrated in other autoimmune disorders, and it has been shown that the effect is partially associated to the generation of Treg cells by cortistatin (Gonzalez‐Rey et al., 2006b, 2007; Wang et al., 2008; Souza‐Moreira et al., 2013). In EAM, we also observed that cortistatin increased the percentage of CD25+FoxP3+ Treg cells in DLNs during the progression phase of the disease. The effect of cortistatin on Treg cells together with the impairment of Th17 cell expansion implies normalization of the Th17/Treg cell ratio in mice with EAM, which is a challenge recently proposed for the treatment of human myocarditis (Stephenson et al., 2016). Regarding the mechanisms involved in the regulation of cardiomyogenic response by cortistatin, we present data that argue against an indirect effect of this neuropeptide on antigen‐presenting cells and that support a direct action on differentiating effector T cells and/or on generation of antigen‐specific Treg cells.
Although our data indicate that cortistatin could alleviate myocarditis during the effector phase of disease by impairing cardiomyogenic T cell responses in the periphery, we cannot rule‐out local effects of cortistatin in the myocardium. Thus, cortistatin could deactivate the inflammatory response of macrophages once they infiltrated the heart. Evidence indicates that cortistatin is a potent macrophage‐deactivating factor that down‐regulates the secretion of a plethora of inflammatory cytokines and enzymes that are involved in myocardial injury (Gonzalez‐Rey et al., 2006a). We found that delayed administration of cortistatin to animals with established inflammatory cell infiltration in the heart slightly, although not significantly, improved the clinical signs, indicating that cortistatin could also act at the cardiac level. Moreover, it has been reported that, through its binding to ghrelin‐receptor, cortistatin is able to protect myocardial cells from necrosis and apoptosis and to deactivate inflammasome in cardiac fibroblasts in a model of systemic inflammation (Zhang et al., 2015a,b), two processes that are involved in the progression to the late phase of myocarditis and dilated cardiomyopathy. In agreement with this hypothesis, a recent study described the protective effect of ghrelin on a model of dilated cardiomyopathy generated in the absence of myocardial inflammation (Du et al., 2014), supporting the possibility that cortistatin could regulate cardiac remodelling and fibrosis in late phases of the disease by its signalling through ghrelin‐receptors. Finally, since cortistatin has been reported to regulate the secretion and activity of several mediators produced by the peripheral nervous system and endocrine system, which shown prominent immunomodulatory actions, including ghrelin, prolactin, growth hormone, acetylcholine, corticosterone and noradrenergic agonists (Spier and de Lecea, 2000; Córdoba‐Chacón et al., 2011; Souza‐Moreira et al., 2013), it remains to determine their potential involvement in the protective effect observed for cortistatin on EAM.
An important issue that needs to be addressed is related to the role played by endogenous cortistatin in the regulation of cardiovascular disorders. Indirect evidence indicates that cortistatin must exert an important protective role. Previous studies indicated that cortistatin is expressed by the heart and cardiovascular system, mainly following injury (Tian et al., 2006; Duran‐Prado et al., 2013; Zhang et al., 2015b). Similarly, we observed a positive correlation between cardiac and systemic cortistatin levels and the severity of EAM and markers of heart failure, suggesting that cortistatin is locally produced in response to EAM in an attempt to limit the destructive inflammatory response in the heart. Because cortistatin is also produced by monocytes and macrophages, we cannot discard the possibility that the increase in cardiac cortistatin during EAM progression is due to the presence of inflammatory cell infiltrates. Accordingly with its beneficial effect on vascular system, animals lacking cortistatin developed exacerbated restenosis and arterial remodelling after vascular injury (Duran‐Prado et al., 2013). Moreover, macrophages isolated from cortistatin‐deficient mice showed increased inflammatory responses, and T cells lacking cortistatin produce 10 times higher levels of IL‐17 than wild‐type T cells upon stimulation (Souza‐Moreira et al., 2013). Furthermore, spleens of mice deficient in the ghrelin receptor (GHSR1a), a receptor mainly used by cortistatin to regulate immune responses, showed enhanced Th17 cell differentiation (Xu et al., 2015). Here, we found that neutralization of secreted cortistatin with antibodies or the pharmacological blockade of the cortistatin‐receptors sst1–5 and ghrelin receptor in lymph node cells isolated from mice with EAM caused an exacerbated cardiomiogenic T cell response, supporting the role of cortistatin as an endogenous inhibitor of inflammatory and autoimmune responses. A limitation of the present study is that we did not investigate its endogenous role in vivo in inflammatory cardiovascular diseases. However, the complex immune response previously described in cortistatin‐deficient mice at the systemic level would make it difficult to evaluate. Thus, the development of systemic inflammatory and autoimmune disorders was partially inhibited in cortistatin‐deficient mice despite the finding that T cells isolated from these animals showed exacerbated autoimmune recall responses (Souza‐Moreira et al., 2013). This paradoxical effect was attributable to a deregulated hypothalamus‐pituitary–adrenal axis and an increase in circulating glucocorticoids, which confer a certain immunosuppressive state to these mice.
Although further pharmacological studies are needed in order to identify specific receptors and signalling involved in the therapeutic action of cortistatin in myocarditis in vivo, our results indicate that both somatostatin‐receptors (most probably sst2‐receptor) and ghrelin receptors are involved in the action exerted by cortistatin on the cardiomyogenic T cells responses. Previous reports have suggested that the ability of cortistatin to bind to these two different types of receptor gives it an advantage over its related peptides ghrelin and somatostatin (Gahete et al., 2008). Thus, cortistatin is more effective than somatostatin or ghrelin in providing protection from autoimmune and inflammatory disorders, vascular remodelling or pain relief (Gonzalez‐Rey et al., 2006a,b; Duran‐Prado et al., 2013; Morell et al., 2014). However, the effect of ghrelin or somatostatin on myocarditis has not been reported to date, our study being the first to describe the therapeutic potential of any of these three related neuropeptides in EAM.
In conclusion, our findings indicate that cortistatin should be considered an effective therapeutic agent for cardiovascular disorders caused by exacerbated inflammatory and autoimmune responses, like myocarditis. Because the effects observed are based on a mouse model of borderline‐like myocarditis in the absence of viral infection, extrapolations to clinical practice have to be made with caution, and future clinical trials with cortistatin‐based therapies should discriminate between interventions in active and borderline myocarditis. Moreover, because the protective effects of cortistatin in EAM were observed mostly during the effector phase of the disease, this will allow a relatively narrow therapeutic window that should be taken into consideration for future clinical translation. Notably, injection of cortistatin into humans has been proven safe (no side effects were observed) and effective in the treatment of patients with Cushing's disease and with acromegaly or prolactinoma (Grottoli et al., 2006; Giordano et al., 2007). These findings encourage further studies aimed to assess whether cortistatin can be used as a pharmaceutical agent to treat chronic heart inflammation and dilated myocardiopathy.
Author contributions
V.D.‐M. performed most of the experiments and analysed data; M.M., C.P.F., N.A. and F.O. performed most of the histological studies; I.F.‐L. performed experiments of gene expression and of autoreactive responses; G.R. performed experiments with DCs; E.M.‐G. performed experiments of autoreactive responses; E.G.‐R. and A.H.L. designed part of the study and analysed data; M.D. conceived and designed the study and wrote the paper; all authors edited and reviewed the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Sequences of primers and their temperature and time of annealing used for quantitative real‐time PCR analysis.
Figure S1 Scheme illustrating the experimental procedure used to induce and treat mice with experimental autoimmune myocarditis (EAM). Mice were injected s.c. with MyHC614–629 on days 0 and 7 (red arrows) and were then randomly distributed in different experimental groups that were treated i.p. with PBS (controls) or with cortistatin (CST, blue arrows) following three different profiles: treatment 1 consisted in six injections of 1 nmol of cortistatin starting at day 7 (early at the effector phase); treatment 2 consisted in six injections of 1, 0.5 or 0.1 nmol of cortisatin starting at day 11 (during the effector phase); and treatment 3 consisted in six injections of 1 nmol of cortistatin starting at day 15 (late during the effector phase). Samples were collected at day 14 (lymph nodes and spleen) or at day 21 (hearts, sera, lymph nodes and spleen) from each experimental group for analysis.
Figure S2 Cortistatin reduces inflammatory infiltration in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. At day 21, hearts were obtained from each experimental group for analysis. Naïve mice were used as reference. A, Identity of inflammatory infiltrates in myocardium was revealed by immunefluorescence for CD45+ leukocytes in heart sections. Nuclei were Hoechst‐counterstained. Scale bars: 100 μm. B, Inflammatory cells infiltrating the heart were isolated and analysed by flow cytometry. Representative dot plots showing flow cytometric analysis of CD45+ leukocytes in live cells are shown (upper plots) and of CD11b+ monocytes and CD4+ lymphocytes in gated CD45+ cells (lower plots). C, Infiltrating inflammatory cells isolated from hearts were activated with phorbol 12‐myristate 13‐acetate in the presence of monensin and analysed by flow cytometry for the expression of intracellular IFNγ and IL‐17 in gated CD4+ lymphocytes. Numbers in dot plots correspond to the percentage of positive cells in each quadrant and the mean of six experiments is shown in Figure 2.
Figure S3 Cortistatin alleviates clinical signs in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) at different doses (A) or at 1 nmol per mouse (B) starting at day 11 (A) or at the indicated time points (B) as depicted in Figure S1. At day 21, hearts were obtained, sectioned and stained with haematoxylin–eosin to determine the extension of myocardial area with inflammatory infiltration and cardiomyocyte necrosis (see Figure 3 for quantitative results). Images are representative of 7 mice per group. Scale bars: 100 μm.
Figure S4 Cortistatin decreases inflammatory response in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. Sera were isolated at day 21, and the content of cytokines was assayed by elisa. n = 11 mice per group, performed in two independent experiments. *P < 0.05.
Figure S5 Proliferation and cytokine production by draining lymph node cells isolated at day 21 from untreated (EAM) or cortistatin‐treated (EAM + CST) mice with EAM and stimulated ex vivo with Concanavalin A (Con A). We obtained similar results with spleen cells stimulated with ConA and with draining lymph node cells stimulated with an anti‐CD3 antibody. n = 10 mice/group, performed in two independent experiments. *P < 0.05 versus untreated EAM mice.
Figure S6 Effect of cortistatin in the generation of Th1, Th17 and Treg cells in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. At days 14 or 21 after EAM induction, draining lymph node cells were isolated and analysed by flow cytometry for intracellular cytokine expression (A) or for CD25 and FoxP3 (B) expression in gated CD4+ cells. Naïve mice were used as reference. Numbers in plots correspond to the percentage of positive cells in each quadrant and the means of 8–12 mice per group are shown Figure 6B and 6C.
Figure S7 Cortistatin does not affect dendritic cell (DC) function. A and B, Phagocytosis of FIT‐Clabeled dextran (A) and expression of costimulatory molecules and production of cytokines (B) by DCs cultured with medium or matured/activated with LPS in the absence or presence of 100 nM cortistatin. MCF: mean channel fluorescence. n = 6 experiments performed in duplicates. C, DCs isolated form C57Bl/6 mice were cultured with medium or matured with LPS in the absence or presence of cortistatin for 24 h and then co‐incubated with allogeneic T cells isolated from spleens of Balb/c mice. Cell proliferation and the production of IFNγ were determined 72 h and 48 h later respectively. Cultures of T cells and DCs alone were used as basal controls. n = 5 experiments performed in duplicates.
Figure S8 Role of endogenous cortistatin in the control of cardiomyogenic T cell responses in vitro. Production of IL‐17 by DLN cells isolated from mice with EAM at day 21 and restimulated ex vivo with MyHC614–629 in the absence (none) or presence of a neutralizing anti‐cortistatin antibody or a control IgG antibody (control isotype). n = 5 mice per group, performed in duplicate. *P < 0.05.
Acknowledgements
Work supported by grants from Spanish Ministry of Economy and Competitiveness and Excellence Program from Andalusian Government and by JAE‐Predoc fellowship.
Delgado‐Maroto, V. , Falo, C. P. , Forte‐Lago, I. , Adan, N. , Morell, M. , Maganto‐Garcia, E. , Robledo, G. , O'Valle, F. , Lichtman, A. H. , Gonzalez‐Rey, E. , and Delgado, M. (2017) The neuropeptide cortistatin attenuates experimental autoimmune myocarditis via inhibition of cardiomyogenic T cell‐driven inflammatory responses. British Journal of Pharmacology, 174: 267–280. doi: 10.1111/bph.13682.
References
- Afanasyeva M, Georgakopoulos D, Belardi DF, Ramsundar AC, Barin JG, Kass DA et al. (2004). Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am J Pathol 164: 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atisha D, Bhalla MA, Morrison LK, Felicio L, Clopton P, Gardetto N et al. (2004). A prospective study in search of an optimal B‐natriuretic peptide level to screen patients for cardiac dysfunction. Am Heart J 148: 518–523. [DOI] [PubMed] [Google Scholar]
- Chandesris MO, Azarine A, Ong KT, Taleb S, Boutouyrie P, Mousseaux E et al. (2012). Frequent and widespread vascular abnormalities in human signal transducer and activator of transcription 3 deficiency. Circ Cardiovasc Genet 5: 25–34. [DOI] [PubMed] [Google Scholar]
- Chorny A, Gonzalez‐Rey E, Fernandez‐Martin A, Pozo D, Ganea D, Delgado M (2006). Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci U S A 102: 13562–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihakova D, Rose NR (2008). Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol 99: 95–114. [DOI] [PubMed] [Google Scholar]
- Cooper LT (2009). Myocarditis. N Engl J Med 360: 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Córdoba‐Chacón J, Gahete MD, Pozo‐Salas AI, Martínez‐Fuentes AJ, de Lecea L, Gracia‐Navarro F et al. (2011). Cortistatin is not a somatostatin analogue but stimulates prolactin release and inhibits GH and ACTH in a gender‐dependent fashion: potential role of ghrelin. Endocrinology 152: 4800–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Criado JR, Prospero‐Garcia O, Gautvik KM, Schweitzer P, Danielson PE et al. (1996). A cortical neuropeptide with neuronal depressant and sleep‐modulating properties. Nature 381: 242–245. [DOI] [PubMed] [Google Scholar]
- Delgado M, Gonzalez‐Rey E, Ganea D (2005). The neuropeptide vasoactive intestinal peptide generates tolerogenic dendritic cells. J Immunol 175: 7311–7324. [DOI] [PubMed] [Google Scholar]
- Du CK, Zhan DY, Morimoto S, Akiyama T, Schwenke DO, Hosoda H et al. (2014). Survival benefit of ghrelin in the heart failure due to dilated cardiomyopathy. Pharmacol Res Perspect 2: e00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran‐Prado M, Morell M, Delgado‐Maroto V, Castano JP, Aneiros‐Fernandez J, de Lecea L et al. (2013). Cortistatin inhibits migration and proliferation of human vascular smooth muscle cells and decreases neointimal formation on carotid artery ligation. Circ Res 112: 1444–1455. [DOI] [PubMed] [Google Scholar]
- Freeman AF, Avila EM, Shaw PA, Davis J, Hsu AP, Welch P et al. (2011). Coronary artery abnormalities in Hyper‐IgE syndrome. J Clin Immunol 31: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frustaci A, Chimenti C (2015). Immunosuppressive therapy in myocarditis. Circ J 79: 4–7. [DOI] [PubMed] [Google Scholar]
- Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A (2003). Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation 107: 857–863. [DOI] [PubMed] [Google Scholar]
- Frustaci A, Russo MA, Chimenti C (2009). Randomized study on the efficacy of immunosuppressive therapy in patients with virus‐negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J 30: 1995–2002. [DOI] [PubMed] [Google Scholar]
- Gahete MD, Duran‐Prado M, Luque RM, Martinez‐Fuentes AJ, Vazquez‐Martinez R, Malagon MM et al. (2008). Are somatostatin and cortistatin two siblings in regulating endocrine secretions? In vitro work ahead. Mol Cell Endocrinol 286: 128–134. [DOI] [PubMed] [Google Scholar]
- Giordano R, Picu A, Bonelli L, Broglio F, Prodam F, Grottoli S et al. (2007). The activation of somatostatinergic receptors by either somatostatin‐14 or cortistatin‐17 often inhibits ACTH hypersecretion in patients with Cushing's disease. Eur J Endocrinol 157: 393–398. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Delgado M (2007). Anti‐inflammatory neuropeptide receptors: new therapeutic targets for immune disorders? Trends Pharmacol Sci 28: 482–491. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Delgado M (2008). Emergence of cortistatin as a new immunomodulatory factor with therapeutic potential in immune disorders. Mol Cell Endocrinol 286: 135–140. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Chorny A, Robledo G, Delgado M (2006a). Cortistatin, a new antiinflammatory peptide with therapeutic effect on lethal endotoxemia. J Exp Med 203: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Varela N, Sheibanie AF, Chorny A, Ganea D, Delgado M (2006b). Cortistatin, an antiinflammatory peptide with therapeutic action in inflammatory bowel disease. Proc Natl Acad Sci U S A 103: 4228–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Rey E, Chorny A, Del Moral RG, Varela N, Delgado M (2007). Therapeutic effect of cortistatin on experimental arthritis by downregulating inflammatory and Th1 responses. Ann Rheum Dis 66: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grottoli S, Gasco V, Broglio F, Baldelli R, Ragazzoni F, Gallenca F et al. (2006). Cortistatin‐17 and somatostatin‐14 display the same effects on growth hormone, prolactin, and insulin secretion in patients with acromegaly or prolactinoma. J Clin Endocrinol Metab 91: 1595–1599. [DOI] [PubMed] [Google Scholar]
- Hia CP, Yip WC, Tai BC, Quek SC (2004). Immunosuppressive therapy in acute myocarditis: an 18 year systematic review. Arch Dis Child 89: 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou YB, Zhang GG, Cai Y, Duan XH, Teng X et al. (2010). Cortistatin attenuates vascular calcification in rats. Regul Pept 159: 35–43. [DOI] [PubMed] [Google Scholar]
- Mann DL (2011). The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res 108: 1133–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masseroli M, O'Valle F, Andújar M, Ramírez C, Gómez‐Morales M, de Dios Luna J et al. (1998). Design and validation of a new image analysis method for automatic quantication of interstitial fibrosis and glomerular morphometry. Lab Invest 78: 511–522. [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell M, Camprubi‐Robles M, Culler MD, de Lecea L, Delgado M (2014). Cortistatin attenuates inflammatory pain via spinal and peripheral actions. Neurobiol Dis 63: 141–154. [DOI] [PubMed] [Google Scholar]
- Robas N, Mead E, Fidock M (2003). MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. J Biol Chem 278: 44400–44404. [DOI] [PubMed] [Google Scholar]
- Rose NR (2014). Learning from myocarditis: mimicry, chaos and black holes. F1000Prime Rep 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siehler S, Nunn C, Hannon J, Feuerbach D, Hoyer D (2008). Pharmacological profile of somatostatin and cortistatin receptors. Mol Cell Endocrinol 286: 26–34. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza‐Moreira L, Morell M, Delgado‐Maroto V, Pedreno M, Martinez‐Escudero L, Caro M et al. (2013). Paradoxical effect of cortistatin treatment and its deficiency on experimental autoimmune encephalomyelitis. J Immunol 191: 2144–2154. [DOI] [PubMed] [Google Scholar]
- Spier AD, de Lecea L (2000). Cortistatin: a member of the somatostatin neuropeptide family with distinct physiological functions. Brain Res Brain Res Rev 33: 228–241. [DOI] [PubMed] [Google Scholar]
- Stephenson E, Savvatis K, Mohiddin SA, Marelli‐Berg FM (2016). T‐cell immunity in myocardial inflammation: pathogenic role and therapeutic manipulation. Br J Pharmacol. doi:10.1111/bph.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Ogawa M, Futamatsu H, Kosuge H, Tanaka H, Isobe M (2006). A cyclooxygenase‐2 inhibitor alters Th1/Th2 cytokine balance and suppresses autoimmune myocarditis in rats. J Mol Cell Cardiol 40: 688–695. [DOI] [PubMed] [Google Scholar]
- Tian QP, Feng XR, Pang YZ, Tang CS, Liu ML (2006). Relationship between plasma cortistatin and coronary heart disease. Beijing Da Xue Xue Bao 41: 537–540. [PubMed] [Google Scholar]
- Wang J, Zhao R, Zhang F, Li J, Huo B, Cao Y et al. (2008). Control of allograft rejection in mice by applying a novel neuropeptide, cortistatin. Adv Ther 25: 1331–1341. [DOI] [PubMed] [Google Scholar]
- Xu Y, Li Z, Yin Y, Lan H, Wang J, Zhao J et al. (2015). Ghrelin inhibits the differentiation of T helper 17 cells through mTOR/STAT3 signaling pathway. PLoS One 10: e0117081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Liu Y, Sui YB, Cai HQ, Liu WX, Zhu M et al. (2015a). Cortistatin inhibits NLRP3 inflammasome activation of cardiac fibroblasts during sepsis. J Card Fail 21: 426–433. [DOI] [PubMed] [Google Scholar]
- Zhang B, Liu Y, Zhang JS, Zhang XH, Chen WJ, Yin XH et al. (2015b). Cortistatin protects myocardium from endoplasmic reticulum stress induced apoptosis during sepsis. Mol Cell Endocrinol 406: 40–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Sequences of primers and their temperature and time of annealing used for quantitative real‐time PCR analysis.
Figure S1 Scheme illustrating the experimental procedure used to induce and treat mice with experimental autoimmune myocarditis (EAM). Mice were injected s.c. with MyHC614–629 on days 0 and 7 (red arrows) and were then randomly distributed in different experimental groups that were treated i.p. with PBS (controls) or with cortistatin (CST, blue arrows) following three different profiles: treatment 1 consisted in six injections of 1 nmol of cortistatin starting at day 7 (early at the effector phase); treatment 2 consisted in six injections of 1, 0.5 or 0.1 nmol of cortisatin starting at day 11 (during the effector phase); and treatment 3 consisted in six injections of 1 nmol of cortistatin starting at day 15 (late during the effector phase). Samples were collected at day 14 (lymph nodes and spleen) or at day 21 (hearts, sera, lymph nodes and spleen) from each experimental group for analysis.
Figure S2 Cortistatin reduces inflammatory infiltration in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. At day 21, hearts were obtained from each experimental group for analysis. Naïve mice were used as reference. A, Identity of inflammatory infiltrates in myocardium was revealed by immunefluorescence for CD45+ leukocytes in heart sections. Nuclei were Hoechst‐counterstained. Scale bars: 100 μm. B, Inflammatory cells infiltrating the heart were isolated and analysed by flow cytometry. Representative dot plots showing flow cytometric analysis of CD45+ leukocytes in live cells are shown (upper plots) and of CD11b+ monocytes and CD4+ lymphocytes in gated CD45+ cells (lower plots). C, Infiltrating inflammatory cells isolated from hearts were activated with phorbol 12‐myristate 13‐acetate in the presence of monensin and analysed by flow cytometry for the expression of intracellular IFNγ and IL‐17 in gated CD4+ lymphocytes. Numbers in dot plots correspond to the percentage of positive cells in each quadrant and the mean of six experiments is shown in Figure 2.
Figure S3 Cortistatin alleviates clinical signs in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) at different doses (A) or at 1 nmol per mouse (B) starting at day 11 (A) or at the indicated time points (B) as depicted in Figure S1. At day 21, hearts were obtained, sectioned and stained with haematoxylin–eosin to determine the extension of myocardial area with inflammatory infiltration and cardiomyocyte necrosis (see Figure 3 for quantitative results). Images are representative of 7 mice per group. Scale bars: 100 μm.
Figure S4 Cortistatin decreases inflammatory response in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. Sera were isolated at day 21, and the content of cytokines was assayed by elisa. n = 11 mice per group, performed in two independent experiments. *P < 0.05.
Figure S5 Proliferation and cytokine production by draining lymph node cells isolated at day 21 from untreated (EAM) or cortistatin‐treated (EAM + CST) mice with EAM and stimulated ex vivo with Concanavalin A (Con A). We obtained similar results with spleen cells stimulated with ConA and with draining lymph node cells stimulated with an anti‐CD3 antibody. n = 10 mice/group, performed in two independent experiments. *P < 0.05 versus untreated EAM mice.
Figure S6 Effect of cortistatin in the generation of Th1, Th17 and Treg cells in EAM. Mice with MyHC614–629‐induced EAM were treated i.p. with PBS (EAM) or cortistatin (EAM + CST) three times per week during two weeks. At days 14 or 21 after EAM induction, draining lymph node cells were isolated and analysed by flow cytometry for intracellular cytokine expression (A) or for CD25 and FoxP3 (B) expression in gated CD4+ cells. Naïve mice were used as reference. Numbers in plots correspond to the percentage of positive cells in each quadrant and the means of 8–12 mice per group are shown Figure 6B and 6C.
Figure S7 Cortistatin does not affect dendritic cell (DC) function. A and B, Phagocytosis of FIT‐Clabeled dextran (A) and expression of costimulatory molecules and production of cytokines (B) by DCs cultured with medium or matured/activated with LPS in the absence or presence of 100 nM cortistatin. MCF: mean channel fluorescence. n = 6 experiments performed in duplicates. C, DCs isolated form C57Bl/6 mice were cultured with medium or matured with LPS in the absence or presence of cortistatin for 24 h and then co‐incubated with allogeneic T cells isolated from spleens of Balb/c mice. Cell proliferation and the production of IFNγ were determined 72 h and 48 h later respectively. Cultures of T cells and DCs alone were used as basal controls. n = 5 experiments performed in duplicates.
Figure S8 Role of endogenous cortistatin in the control of cardiomyogenic T cell responses in vitro. Production of IL‐17 by DLN cells isolated from mice with EAM at day 21 and restimulated ex vivo with MyHC614–629 in the absence (none) or presence of a neutralizing anti‐cortistatin antibody or a control IgG antibody (control isotype). n = 5 mice per group, performed in duplicate. *P < 0.05.