

Abstract
Purpose: Depression is a public disorder worldwide. Despite the widespread use of venlafaxine in the treatment of depression, it has been associated with the incidence of toxicities. Hence, the goal of the current investigation was to evaluate the mechanisms of venlafaxine–induced cell death in the model of the freshly isolated rat hepatocytes.
Methods: Collagenase-perfused rat hepatocytes were treated with venlafaxine and other agents. Cell damage, reactive oxygen species (ROS) formation, lipid peroxidation, mitochondrial membrane potential decline, lysosomal damage, glutathione (GSH) level were analyzed. Moreover, rat liver mitochondria were isolated through differential centrifugation to assess respiratory chain functionality.
Results: Our results demonstrated that venlafaxine could induce ROS formation followed by lipid peroxidation, cellular GSH content depletion, elevated GSSG level, loss of lysosmal membrane integrity, MMP collapse and finally cell death in a concentration-dependent manner. N-acetyl cysteine, taurine and quercetine significantly decreased the aforementioned venlafaxine-induced cellular events. Also, radical scavenger (butylatedhydroxytoluene and α-tocopherol), CYP2E1 inhibitor (4-methylpyrazole), lysosomotropic agents (methylamine and chloroquine), ATP generators (L-gluthamine and fructose) and mitochondrial pore sealing agents (trifluoperazine and L-carnitine) considerably reduced cytotoxicity, ROS generation and lysosomal leakage following venlafaxine treatment. Mitochondrion dysfunction was concomitant with the blockade of the electron transfer complexes II and IV of the mitochondrial respiratory system.
Conclusion: Therefore, our data indicate that venlafaxine induces oxidative stress towards hepatocytes and our findings provide evidence to propose that mitochondria and lysosomes are of the primary targets in venlafaxine-mediated cell damage.
Keywords: Venlafaxine, Cytotoxicity, Mitochondria, Lysosome, Oxidative stress, Antioxidants
Introduction
Venlafaxine (VEN) is a well-established antidepressant agent extensively used to combat major depressive disorders, anxiety and social phobia.1,2 Also, VEN is an effective pharmacological agent to ameliorate vasomotor symptoms (VMS) in postmenopausal women.3 However, various data pinpoint VEN might not be as safe as other serotonergic agents, particularly in overdose, leading to severe nervous system depression, seizure and arrhythmias.4,5 Within the blockade of Na channels in cardiac myocytes, VEN exhibits cardiotoxicity.6 Furthermore, VEN in a concentration-dependent manner inhibits cell growth via induction of cytotoxicity in human metastatic breast cancer cell line, MCF-7.7
Considering liver as a dominant site for detoxification of drugs and other xenobiotics, this organ is understandably prone to deleterious drug effects. Post-marketing surveillance of VEN has revealed hepatic events including gamma-glutamyl transpeptidase (GGT) increment, abnormalities of the liver function tests, hepatocytes damages, necrosis, liver failure, and fatty liver disease.8 Some case reports provide the evidence of VEN-induced hepatotoxicity. Hepatocellular injuries concomitant with elevated aspartate transaminase (AST) and alanine transaminase (ALT) enzyme level have been reported even in patients treated with low doses of VEN.9,10 VEN has been associated with cholestatic as well as hepatocellular liver damage in patients without a preceding history of liver dysfunction or marked alcohol use.11 Detry et al., reported a case of a 48-year-old depressed patient who manifested fulminant hepatic failure with a further liver transplantation after developing toxic liver injury, jaundice and encephalopathy.12
Various environmental compound, drugs and xenobiotics induce their deleterious side effects thorough increased reactive oxygen species (ROS) production.13 Besides, ROS formation initiates the activation of several downstream signaling cascades associated with cellular key components damages such as regulatory proteins, DNA and subcellular organelles.14,15 Mitochondrion is an intracellular source of free radicals that is able to potentiate the drug-induced oxidative hazard through increased membrane permeability.16 Lysosomes are the other major intracellular components that compromise hydrolytic enzymes such as glucosidases, lipases and proteases which on release, could degrade intracellular contents.17 ROS formation has a pivotal impact to enhance lysosomal membrane permeability contributed to apoptosis or necrosis eventually.18 The protective effects of many ROS scavenger agents against drug/xenobitic-induced liver injuries has been presented in numerous research papers.19,20
Despite the widespread therapeutic application, the plausible hepatotoxic effects of VEN and the causative mechanisms of cytotoxicity have not been well inspected yet. Therefore, the accelerated cytotoxic mechanism screening (ACMS) was used to examine the potential toxicity of VEN in rat hepatocytes,21 delineation of the molecular aspects involved in the VEN-associated liver toxicity, the protective role of N-acetyl cysteine, taurine and/or quercetine and also the implication of mitochondria and lysosomes and their contribution to damages towards hepatocytes.
Materials and Methods
Chemicals
Thiobarbituric acid (TBA) was obtained from Serva (Heidelberg, Germany). Albumin bovine serum was purchased from the Roche diagnostic corporation (IN). Type II collagenase, taurine, quercetin, N-acetyl cysteine, VEN, 4-methylpyrazole, acridine orange, 1-bromoheptane (BHP), Rhodamine 123, 2’,7’- dichlorofluorescin diacetate (DCFDA), trichloroacetic acid (TCA), ethylenediaminetetraacetic acid (EDTA), butylated hydroxytoluene (BHT), sucrose, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Tris–HCl and all other reagents were obtained from Sigma-Aldrich Chemical Co. (St. Louis, USA).
Animal Care, Isolation and Incubation of Liver Cells
Male Sprague-Dawley rats weighting 250-300 g were kept in standard plastic boxes under normal laboratorial conditions with the temperature set to 21-23 °C and obtained from the animal research center of Tabriz University of Medical Science, Tabriz, Iran. The animals received humane care according to the “European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes” Acts of 1986, and the “Guiding Principles in the Use of Animals in Toxicology”, adopted by the Society of Toxicology in 1989, for the acceptable use of experimental animals. The ethical standards were based on ethical rules of the National Institute of Health (NIH publication No. 85-23, revised 1985). All investigations were fulfilled based on the standard guidance accepted by the Committee of Animal Experimentation of Tabriz University of Medical Sciences, Tabriz, Iran.
Isolation of rat hepatocytes was performed with collagenase-perfusion technique as defined before.22 Hepatocytes with the density of 106 cells/mL were suspended in Krebs–Henseleit buffer (pH 7.4), comprising 12.5 mM HEPES in continuously spinning round bottomed flasks at 37°C water bath under an atmosphere of carbogen gas (5% CO2 and 95% O2). Over 80% cell viability was accepted to continue other procedures.23 The optimal concentration of quercetine, taurine and NAC that engendered appropriate protection was found to be 500 µM, 200 µM and 200 µM respectively. The utilized doses for ROS scavengers (butylated hydroxytoluen (BHT) and α-tocopherol succinate), MPT pore sealing agents (trifluoperazine and carnitine), ATP generators (l-glutamine and fructose), lysosomotropic agents (methylamine and chloroquine) and CYP450 inhibitors (cimetidine and 4-methylpyrazole) were chosen from similar experiments that were done on rat hepatocytes.24-26 We also investigated the aforementioned doses in our isolation system and perceived that they have maximal protective impacts on the cytotoxicity indicators (data not shown).
The Viability Assay
Plasma membrane intactness as a marker of cell viability of isolated hepatocytes was determined via trypan blue (0.2% w/v) exclusion assay microscopically.27 Sampling was done at every 60 min during the 3 hour following hepatocyte incubation.
Reactive oxygen species (ROS) formation
The extent of hepatocyte ROS production by VEN was assessed using dichlorofluorescin-diacetate (DCFH-DA) dye which is hydrolyzed by an intracellular esterase to produce non-fluorescent DCFH in hepatocytes. This agent then reacts with intracellular ROS and becomes a highly fluorescent compound, which emanates the cell. Isolated hepatocytes were incubated with VEN in preloaded cell suspension with DCFH-DA (1.6 μM) and the aliquots of 1ml sampling was performed in 15, 30 and 60 min subsequently at the end of which centrifugation was performed at 3000g for 1 min. Finally the fluorescence of supernatant was measured fluorimetrically at excitation and emission wavelength of 490nm and 520nm respectively with a Jasco FP-750 fluorescence spectrophotometer (Jasco Company, Tokyo, Japan).28
Lipid peroxidation assay
Thiobarbituric acid reactive substances (TBARS) are the markers of membrane lipid peroxidation, which are formed as a result of disintegration of lipid hydroperoxides. Briefly, 1ml of hepatocyte suspension was treated with 250 μl of trichloroacetic acid (TCA, 70%w/v) and centrifuged at 3000g for 15 min. Then 1ml of thiobarbituric acid (0.8%w/v) was added to the supernatant and boiled for 20 min. The absorbance was detected at 532nm with a Ultrospec 2000 UV spectrophotometer.29
Measurement of intracellular GSH and GSSG
The level of intracellular glutathione (GSH) and glutathione disulfide (GSSG) in deproteinized hepatocytes via the addition of 5% metaphosphoric acid were evaluated after derivatization with iodoacetic acid and 1-fluoro-2,4-dinitrobenzene (FDNB), by HPLC, using a µ Bondapak NH2 column (Water Associates, Milford, MA).30,31 External standard solution of GSH and GSSG were prepared prior to hepatocytes isolation. A Waters 6000A solvent delivery system, equipped with a model 660 solvent programmer, aWisp710A automatic injector, and a data module analyzed the data. The process is based upon the primary formation of S-carboxymethyl derivatives of free thiols with iodoacetic acid followed by conversion of free amino groups to 2,4-dinitrophenyl derivatives by reaction with FDNB. Measurement of nanomolar levels of reduced and oxidized glutathione is feasible within this technique. In brief, 1 ml (106) of the cell suspension was spun at 50× g for 40 s before the addition of 1 ml of fresh Krebs-Hensleit medium. The samples were treated with metaphosphoric acid (0.2ml) and centrifuged at 100×g for 5 min after 20min. Afterwards, iodoacetic acid (0.05ml) in the presence of was added to the supernatant (0.5) and left in dark for 1 h. In the second step of derivatization, 0.5 ml of FDNB solution (1.5%, v/v in ethanol) was added to the mixture and left in the dark and room temperature for 6 h before HPLC analysis.
Lysosomal membrane leakiness assay
To determine the integrity of lysosomal membrane the disposition of fluorescent dye, acridine orange was evaluated in isolated rat hepatocytes.32,33 To do the experiment, a volume of 0.5 ml of the pre-stained (acridine orange 5 μM) cell suspension was taken and centrifuged at 800× g for 1min. Thereafter, the cell pellet was resuspended in 2ml of incubation medium. In order to exclude the fluorescent dye the washing process was repeated. The capacity of lysosomes to preserve acridine orange was assessed fluorimetrically using a Jasco FP-750 fluorescence spectrophotometer set at 495nm excitation and 530nm emission wavelengths between control and VEN- damaged cells.
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential (MMP) loss is a prominent index of mitochondrial damage and is measured via the mitochondrion capability to retain Rhodamine 123, as cationic fluorescent prob. When MMP is altered by a drug, toxin or any xenobiotic the facilitated diffusion stops leading to accretion of the level of rhodamine 123 in media. To do the experiment, 1 ml samples of the cell suspension were resuspended in fresh incubation medium encompassing1.5 μM of rhodamine 123 after centrifugation at 1000×g for 1 min. Then, the cell pellet was incubated at 37°C in a water bath with gentle shaking. The centrifugation was repeated and the remained dye in the incubation medium was determined with Jasco FP-750 fluorescence spectrophotometer adjusted to 490 nm excitation and 520 nm emission wavelengths.34,35
Liver Mitochondria Isolation
The continual centrifugation technique was performed to isolate Sprague Dawley rats liver mitochondria.36 The minced rat liver was suspended in an ice cold mannitol solution comprising 75 mM sucrose, 0.225 M D-mannitol and 0.2 mM EDTA and then was centrifuged at 1,000× g for 10 min at 4ºC to sieve the debris. Centrifugation was performed once more to yield a dark layer that was re-suspended in the mannitol solution and re-centrifuged twice at 10,000×g for 10 min. Finally, the produced dens mitochondrial fractions were suspended in Tris solution containing 0.05 M Tris–HCl buffer (pH 7.4), 0.25 M sucrose, 20 mM KCl, 2.0 mM MgCl2, and 1.0 mM Na2HPO4 at 4 °C before the assay. Coomassie blue protein-binding method was utilized to determine mitochondrial protein concentration. Mitochondrial samples of 500 μg protein/mL were used in all experiments.
Mitochondrial Complex II Activity measurement
MTT reduction assay was used to examine the activity of mitochondrial complex II (succinate dehydrogenase).37 Various concentrations of VEN incubated with 100μL of mitochondrial suspensions equal to 500 μg protein/mL at 37 °C for 30 min pursued by the addition of MTT into the medium and on their incubation time for 30 min at 37°C. The absorbance of produced purple formazan was detected with an ELISA reader (Biotek, ELx800, USA).
Measurement of Cytochrome c Oxidase Activity
Complex IV (Cytochrome c oxidase) activity was determined using cytochrome c oxidase assay kit from Sigma-Aldrich (Taufkrichen, Germany). The procedure is based on the oxidation of ferro- cytochrome c to ferri -cytochrome c by cytochrome c oxidase which is concomitant with absorbance reduction. The experiment was followed by the manufacturer's protocol. The obtained data were expressed as units per milligram mitochondrial protein.
Statistical analysis
The normal distribution of data was tested by Kolmogorov-Smirnov test and ANOVA and Tukey's post hoc test were used for analyzing data and multiple mean comparisons, respectively. Statistical significance was considered as a value of P ≤ 0.05 and quantitative data were reported as mean ± SEM. The statistical analysis was performed by SPSS software version 16.0 (SPSS Inc, Chicago, IL, USA).
Results and Discussion
During the incubation period, we observed the minimal viability of 80% in hepatocytes. We utilized EC50 for VEN to avoid either nontoxic or very toxic conditions in the current experiment. Based on the ACMS method, the EC50 of a compound in hepatocyte is described as the concentration, at that cell viability plunges in number to the 50% following the 120 min h of incubation dose–response curves were plotted to achieve EC50 concentration of VEN (Data not shown) which was evaluated to be 2mM. This concentration was utilized during the experiment to assess other biochemical alterations in hepatocytes. As shown in Table 1, VEN (2 mM) markedly enhanced membrane lysis in comparison with control liver cells. Hepatocytes co-treatment with antioxidants (quercetine, NAC and/or taurine was simultaneous with substantial decline of VEN-connected cell death (Table1). In addition, VEN-induced cell death was prohibited by ROS scavengers (α-tocopherol succinate and BHT), MPT pore sealing agents (trifluoperazine and carnitine), ATP generators (l-glutamine and fructose), lysosomotropic agents (methylamine and chloroquine) as well as 4-methylpyrazole (CYP2E1 inhibitor) (Table1).
Table 1. VEN- induced cytotoxicity toward isolated rat hepatocytes .
| Addition | Cytotoxicity% | ||
| Incubation time (min) | 60 | 120 | 180 |
| Control | 18±2 | 22±1 | 24±2 |
| + VEN 2mM | 41±2* | 54±3* | 69±4* |
| +NAC 200 µM | 21±2** | 23±2** | 25±3** |
| + Taurine 200 µM | 24±2** | 27±3** | 30±1** |
| + Quercetine500µM | 18±1** | 24±2** | 26±2** |
| + α-Tocopherol succinate 100μM | 18±1** | 24±2** | 29±2** |
| + BHT50μM | 23±2** | 28±2** | 34±3** |
| + L-Carnitine2mM | 28±1** | 34±2** | 40±2** |
| + Trifluperazine15µM | 25±3** | 29±2** | 33±4** |
| + Fructose1mM | 27±2** | 30±3** | 39±3** |
| + L-Glutamine 1mM | 28±3** | 35±2** | 42±3** |
| +3-Methyladenine 5mM | 25±2** | 29±2** | 34±3** |
| + Chloroquine 100μM | 24±2** | 28±2** | 31±3** |
| + 4-Methylpyrazole 500μM | 30±2** | 38±2** | 42±3** |
| +Cimetidine 1 mM | 44±2 | 68±2** | 88±4** |
| GSH depleted hepatocytes | 20±2 | 24±2 | 26±2 |
| +VEN 2mM | 55±2** | 72±2** | 89±4** |
The Trypan blue exclusion test was used to assess membrane lysis rates induced by VEN. At different time points, the viability of aliquots of 1 ml (106 cells) was evaluated microscopically. Hepatocytes were incubated in Krebs–Henseleit buffer, pH 7.4, at 37 °C. GSH depleted hepatocytes were prepared as described by Abdoli et.al.(Abdoli et al., 2013) Data are given as mean ± SEM for at least three independent experiments.*Significant as compared with the control (Only hepatocytes) group (p < 0.05). **Significant as compared with VEN-treated cells (p < 0.05).
Preincubation of hepatocytes with 1mM cimetidine (non-selective CYP450 inhibitor) caused a significant surge in the rate of VEN-induced cytotoxicity (Table1). Prior depletion of isolated hepatocytes with 1-bromoheptane, elevated VEN-induced cell death (Table 1). All the above mentioned radical scavengers, antioxidants, endocytosis inhibitors, 1-bromoheptan and CYP450 inhibitors did not meaningfully (P < 0.05) alter cell membrane integrity at concentrations used in solitary treating of liver cells (data not shown).
Following the incubation of hepatocytes with VEN at the EC50 for 2h (2mM), ROS formation was significantly increased in comparison with normal hepatocytes. ROS scavengers and/or antioxidants (NAC, taurine, quercetine, α-tocopherol succinate, BHT), CYP2E1 inhibitor (4-methylpyrazole), mitochondrial pore sealing agents (trifluoperazine and L-carnitine), ATP generators (L-gluthamine and fructose) and lysosomotropic agents (methylamine and chloroquine) markedly impeded VEN-induced ROS formation while cimetidine (CYP 2D6 enzyme inhibitor) increased VEN-associated ROS generation. In GSH deprived hepatocytes an augmentation of the VEN-associated ROS generation (p<0.05) was perceived (Table 2).
Table 2. VEN- induced ROS formation toward isolated rat hepatocytes .
| Addition | ROS formation (DCF: fluorescent intensity units) | ||
| Incubation time (min) | 15 | 30 | 60 |
| Control | 95±7 | 109±10 | 138±10 |
| + VEN 2mM | 99±7* | 116±8* | 144±10* |
| +NAC 200 µM | 98±8** | 119±10** | 156±11** |
| + Taurine 200 µM | 98±8** | 118±10** | 145±12** |
| + Quercetine500µM | 102±10** | 110±10** | 138±10** |
| + α-Tocopherol succinate 100μM | 104±6** | 120±8** | 157±10** |
| + BHT50μM | 108±7** | 123±11** | 169±9** |
| + L-Carnitine2mM | 103±7** | 125±10** | 170±10** |
| + Trifluperazine15µM | 108±6** | 123±9** | 172±10** |
| + Fructose1mM | 108±6** | 123±9** | 172±10** |
| + L-Glutamine 1mM | 110±8** | 126±9** | 178±9** |
| +3-Methyladenine 5mM | 110±8** | 126±9** | 178±9** |
| + Chloroquine 100μM | 105±8** | 125±10** | 173±8** |
| + 4-Methylpyrazole 500μM | 107±7** | 132±6** | 174±10** |
| +Cimetidine 1 mM | 130±5** | 189±10** | 268±10** |
| GSH depleted hepatocytes | 92±6 | 118±10 | 142±10 |
| +VEN 2mM | 139±9** | 200±12** | 284±10** |
Hepatocytes were incubated in Krebs–Henseleit buffer, pH 7.4, at 37 °C for 1.0 h after VEN (2mM) treatment. Dichlorofluorescein (DCF) formation was measured as fluorescent intensity units. GSH depleted hepatocytes were prepared as described by Abdoli et.al.(Abdoli et al., 2013) Data are given as mean ± SEM for at least three independent experiments.
*Significant as compared with the control (Only hepatocytes) group (p < 0.05).
**Significant as compared with VEN-treated cells (p < 0.05).
VEN, is a fast becoming suitable drug choice for treatment of clinical manifestations of depression with a dual action of inhibiting serotonin and noradrenaline uptake pumps,38 while literature search has fetched with reported hepatotoxicity.8 However, the precise causative mechanism involved in VEN-induced toxicity has not elucidated so far.39 The latter was investigated through ACMS method in isolated rat hepatocytes. ACMS is a valuable technique to define the cytotoxic impacts of xenobiotics/drugs in rat hepatocytes over a short time period. Determination of hepatotoxic effects of a high dose agent in 2-3 h in vitro, which is in accordance with similar toxic effects the same agent in lower doses over a longer time period in vivo is a prominent property ACMS method.40 Hence, we could express that this technique characterizes and predicts hepatotoxicity in vivo.
The result of this experiment demonstrated that VEN gives rise to liver cell death (cytotoxicity) and oxidative stress in isolated rat hepatocytes. ROS generation is a common primary event in drug/xenobiotic toxicity, mediating signaling pathways, which can result in a plethora of toxic effects and cellular damages.41 Hence, free radicals and overproduction of reactive species might explain a plausible mechanistic hypothesis in VEN-induced cytotoxicity, as DCF fluorescence was significantly increased after administration of this drug in hepatocytes.
Lipid peroxidation (LPO) of cell membrane is often a pursuant event of oxidative stress.42 The amount of TBARS was estimated to inspect the role of VEN in peroxidation of membrane lipids in hepatocytes. It was shown that VEN leads to considerable LPO in isolated rat hepatocytes while taurine, NAC and quercetin diminished the VEN-associated TBARS production significantly (p <0.05) (Figure 1). Oxidative stress is usually concomitant with the propagation of lipid peroxidation, breakage of lipids and formation of reactive compounds that influence the permeability and integrity of plasma and intracellular membranes.43 In this study, induction of LPO in VEN-exposed hepatocytes was also observed. The abovementioned effects were prohibited by antioxidants and/or radical scavengers, which firmly suggest the involvement of reactive species and oxidative damage in the mechanisms involved in of VEN cytotoxicity.
Figure 1.
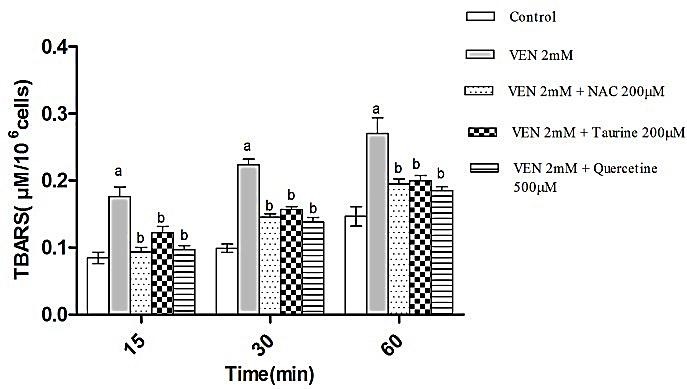
VEN-induced LPO and the protective role of NAC (200μM), Taurine (200μM) and quercetine (500μM) administration. Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37ºC for 1 h following the addition of VEN (2 mM). Data are given as Mean ± SEM for three independent experiments.
aSignificantly higher than control cells (p < 0.05).
bSignificantly lower than VEN-treated cells (p < 0.05).
As demonstrated in Figure 2 and 3, incubation of hepatocytes with VEN (2 mM) induced hepatocyte GSH depletion, which contributed to the production of GSSG. Moreover, treatment of hepatocytes with antioxidants (quercetine, taurine and NAC) significantly prevented both the VEN-linked GSH decrement and GSSG elevation indicating the occurrence of oxidative hazard subsequent to VEN treatment.
Figure 2.
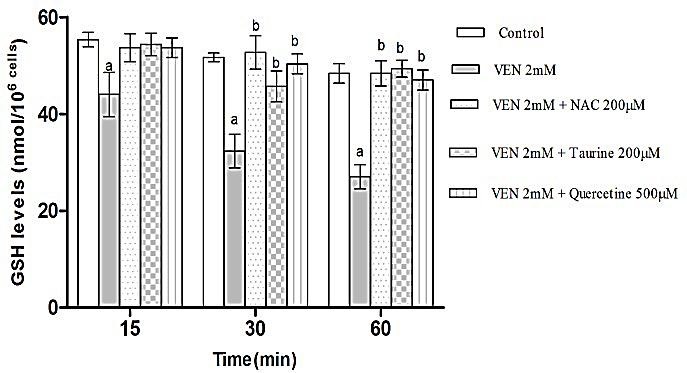
Effects of VEN on the amount of GSH in rat hepatocytes and the protecting role of NAC (200μM), Taurine (200μM) and quercetine (500μM) administration. Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37°C for 1h before the addition of VEN (2 mM). The results are expressed as mean ± SEM of three separate experiments.
aSignificantly different from control (p < 0.05).
bSignificant compared to VEN-treated cells (p < 0.05).
Figure 3.
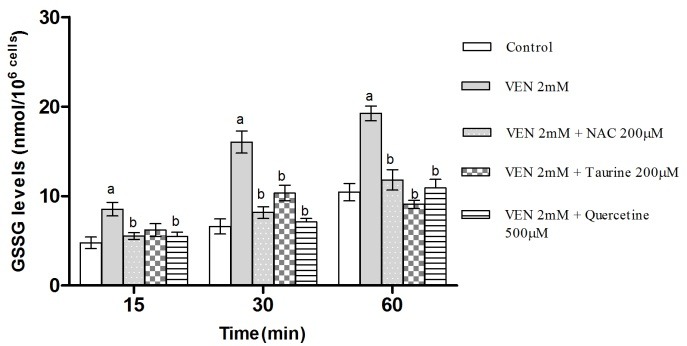
Effects of VEN on the amount of GSSG in rat hepatocytes and the protective role of NAC (200μM), Taurine (200μM) and quercetine (500μM) administration. Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37°C for 1h before the addition of VEN (2 mM).The results are expressed as mean ± SEM of three separate experiments.
aSignificantly different from control (p < 0.05).
bSignificant compared to VEN-treated cells (p < 0.05).
In the current investigation, the role of glutathione in VEN-mediated oxidative hazard in hepatocytes was investigated. GSH is a vital intracellular peptide with divers functions including detoxification, preservation of essential thiol status of proteins and acting as cellular antioxidant reservoir.44 Both the reduced (GSH) and oxidized (GSSG) forms of Glutathione have the ability to convert to each other. Cellular GSH reservoirs are plummeted following oxidative stress which might be a result of their conversion to GSSG.45 VEN-associated oxidative stress led to the GSH diminution in freshly isolated hepatocytes. Therefore, it is justified that oxidative pathway has a leading role in VEN-induced cytotoxicity. Additionally, preceding depletion of hepatocyte GSH content augmented the cytotoxicity and ROS formation proposing the fundamental role of GSH in VEN-induced hepatotoxicity.
As shown in Figure 4, VEN (2mM) caused MMP collapse following treatment of hepatocytes. MMP diminution is a key indicator of toxicity in all mitochondrial damaging cycles. This toxicity was prevented by antioxidants (quercetine, taurine and NAC) (Figure 4) and ATP generators (fructose and L-gluthamine) (Figure 5) (p<0.05). Furthermore, the selected concentration of VEN (100, 200 and 500μM) in our pilot study was investigated in the drug ability to inhibit mitochondrial respiratory chain complexes. As Figure 6 illustrates, VEN (200 and 500μM) dramatically inhibited complexes II and IV while this inhibitory effect was not seen in complexes I and III (data not shown).
Figure 4.
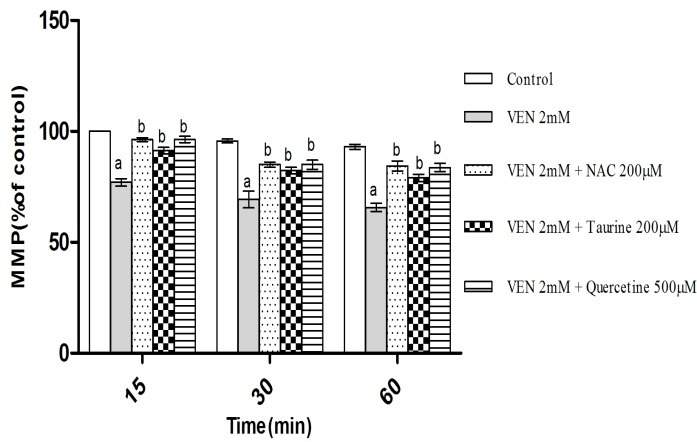
Effect of VEN on mitochondrial membrane potential (MMP) and preventive role of NAC (200μM), Taurine (200μM) and quercetine (500μM) administration. Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37°C for 1 h before the addition of VEN (2 mM). Data are expressed as Mean ± SEM for at least three separate experiments.
aSignificantly lower than control group (p < 0.05).
bSignificantly higher as compared with VEN-treated hepatocytes (p < 0.05).
Figure 5.
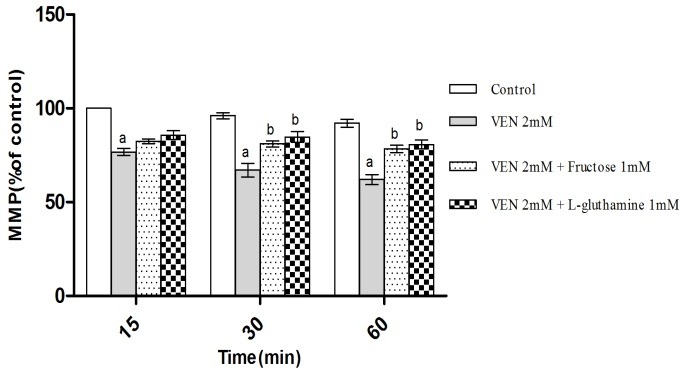
Effect of VEN on mitochondrial membrane potential (MMP) and the protection resulted from Fructos (1mM) and L-Gluthamine (1mM). Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37°C for 1 h following the addition of VEN (2 mM). Data are expressed as Mean ± SEM for at least three separate experiments.
aSignificantly lower than control group (p < 0.05).
bSignificantly higher as compared with VEN-treated hepatocytes (p < 0.05).
Figure 6.
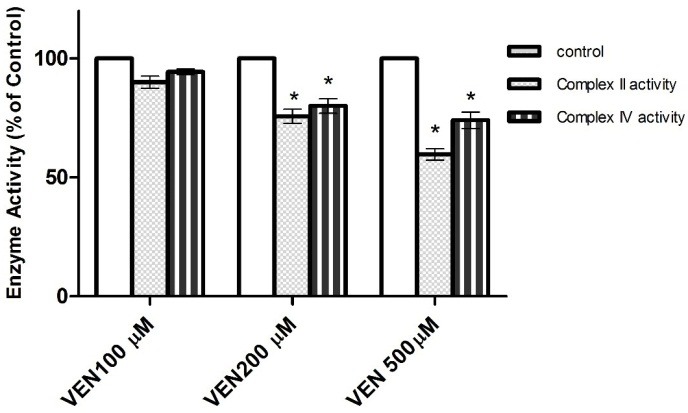
Effect of VEN on respiratory chain complexes II and IV in isolated liver mitochondria. Relative activities are displayed (100% = Control sample without the drug).
* Significant in comparison with Control (p < 0.05).
The activation of NADPH oxidase is the main pathway ROS production in kupffer cells while in hepatocytes, ROS is derived from CYP 2E1 activity.46,47 Attenuated cellular death and also ROS formation with CYP2E1 inhibitor (4-methylpyrazole), mitochondrial pore sealing agents (trifluoperazine and L-carnitine) and lysosomotropic agents (methylamine and chloroquine) provides the evidence of subcellular organelles` participation in VEN-associated oxidative stress and its consequent cytotoxic events.
Mitochondrion is the major cellular source and target of ROS.48 Alterations of MMP, cellular energy production and/or electron transfer chain are among the most important effects of drugs/xenobiotic-induced mitochondrial toxicity.49 It has been shown that antidepressants agents could induce mitochondrial impairment via other pathways including nitric oxide production inhibition.50 Electron transfer from complex I to complexes III and IV comes off in mitochondrial respiratory chains. Our results indicate that, VEN had inhibitory effect in respiratory chain complexes II and IV in isolated liver mitochondria. Our findings are in line with that of Hroudova and Fisar who investigated the effect of antidepressant and mood stabilizer agents on mitochondrial respiratory chain complexes.51 The blocked of respiratory chain complexes has a tight connection with mitochondrial dysfunction which is known to be an important source of reactive species.52 The ROS formed in Mitochondria is a substantial stimulant of many cytotoxic cycles in the cell. Also, mitochondrial membrane and respiratory chains are among the most important targets of ROS.53 In our investigation, the effect of VEN on MMP collapse as a marker of mitochondrial toxicity was measured. Prompt decline of hepatocyte MMP subsequent to VEN administration was postponed by antioxidants pointing out the direct interplay of oxidative stress and mitochondrial toxicity. Moreover, prevention of VEN-associated cytotoxicity and MMP loss by ATP generators indicates that the latter consequences possibly are the result ATP diminution. It has been shown that mitochondrial ATP production decline leads to intracellular acidosis and osmotic injury all of which resulting in cell death.54
Incubation of hepatocytes with VEN considerably injured lysosomes in 1h time period. The aforementioned damage was counteracted by antioxidants (α-tocopherol, taurine, quercetine and NAC), radical scavenger (butylatedhydroxytoluene), CYP2E1 inhibitor (4-methylpyrazole), lysosomotropic agent (methylamine and chloroquine), autophagy inhibitory (3-methyladenine), ATP generators (L-gluthamine and fructose) and mitochondrial pore sealing agents (trifluoperazine and L-carnitine), while prior GSH depletion augmented the release of acridine orange into the cytoplasm (p<0.05) (Table 3).
Table 3. The effect of VEN in lysosomal membrane integrity .
| Treatment | Acridine orange redistribution (intensity unit of diffused cytosolic green fluorescence) | ||
| Incubation time | |||
| 15 min | 30 min | 60 min | |
| Control | 8±2 | 9±2 | 11±3 |
| + VEN 2mM | 35 ± 1* | 52± 2* | 68 ± 2* |
| +NAC 200 µM | 10±2** | 11±3** | 13±2** |
| + Taurine 200 µM | 17±2** | 21±2** | 24±4** |
| + Quercetine500µM | 12±1** | 15±2** | 18±3** |
| + α-Tocopherol succinate 10μM | 13± 1** | 15 ± 1** | 17± 1** |
| +Butylatedhydroxytoluene 50μM | 18 ± 2** | 18 ± 1** | 26 ± 1** |
| +3-Methyladenine 5mM | 15 ± 3** | 17 ± 1** | 19 ± 1** |
| + Chloroquine 100μM | 17 ± 2** | 22 ± 1** | 25 ± 1** |
| + Methylamine 30mM | 17±2** | 23±3** | 27±2** |
| + Fructose 1 mM | 18 ± 1** | 20 ± 1** | 23 ± 2** |
| + L-glutamine 1 mM | 16 ± 0.2** | 18 ± 2** | 21 ± 3** |
| +4-methylpyrazole 500µM | 19 ± 1** | 23± 2** | 24± 2** |
| +Cimetidine 1 mM | 33± 1 | 69 ± 2** | 89 ± 2** |
| +GSH depleted hepatocytes | 37 ± 1 | 70± 2** | 91 ± 1** |
Hepatocytes (106cells/ml) were incubated in Krebs–Henseleit buffer, pH 7.4, at 37 °C for 1.0 h following the addition of VEN (2 mM). Lysosomal membrane damage was determined as difference in redistribution of acridine orange from lysosomes into cytosol between treated cells and control cells. Values are expressed as mean ± SEM of three separate experiments.
* Significant difference in comparison with VPA treated hepatocytes (P < 0.05).
**Significant as compared with VEN-treated hepatocytes (p < 0.05).
Anti-depressant agent have been reported to induce lysosomal membrane damage in lysosomal membrane intactness evaluations.39 The present result also showed that VEN administration decreased lysosomal membrane stability, leading to the membrane leakage. Intracellular oxidative agents, particularly hydrogen peroxide, in combination with lysosomal redox-active iron, generates hydroxyl radicals by the Fenton reaction leading to peroxidation of the lysosomal membrane and subsequent rupture that liberates lysosomal enzymes into the cytoplasm. Released lysosomal enzymes can inflict further damage to lysosomes directly or can in turn trigger mitochondrial apoptotic cascade.55 The deleterious effect of VEN on lysosomal membrane was delayed by antioxidants (α-tocopherol, taurine, quercetine and NAC), radical scavenger (butylatedhydroxytoluene), CYP2E1 inhibitor (4-methylpyrazole), lysosomotropic agents (chloroquine and methylamine), mitochondrial pore sealing agents (trifluoperazine and L-carnitine), ATP generators (L-gluthamine and fructose), lysosomotropic agents (chloroquine and methylamine ) and 3-methyladenine (autophagy inhibitor).
Conclusion
In summary, the present study speculates that VEN caused hepatotoxicity by induction of oxidative stress and subsequent toxic effects including GSH depletion, lipid peroxidation, mitochondrial potential collapse and lysosomal membrane leakiness in isolated rat hepatocyte. Furthermore, mitochondrial functional damage was ensued with inhibition of mitochondrial respiratory complexes II and IV. To the extent of our knowledge this study for the first time represents lysosomes and mitochondria as the earliest targets in VEN-induced hepatotoxicity and characterizes several subcellular associated cascades, mainly as a consequence of oxidative stress initiation. Planning new strategies to protect against cytotoxic effects of VEN in hepatocytes could use the beneficial results of the present study. Hence, further in vivo and clinical evaluations are needed to confirm the protective role of quercetine, NAC and/or taurine in patients receiving VEN.
Acknowledgments
The authors would like to thank Drug Applied Research Center of Tabriz University of Medical Sciences, Tabriz, Iran, for providing technical facilities. The authors are also thankful to the University’s “Students’ Research Committee” for providing technical supports to the study. This is a report of a database from thesis entitled “Evaluation of the mechanisms of hepatic injuries induced by antidepressant drugs” registered and funded by a grant (Grant number: 77/93) from the Drug Applied Research Center of Tabriz University of Medical Sciences, Tabriz, Iran.
Ethical Issues
The ethical issue has been stated on page 522, under the section “Animal Care, Isolation and Incubation of Liver Cells”.
Conflict of Interest
The authors declare no conflict of interests.
References
- 1.Perahia DG, Pritchett YL, Kajdasz DK, Bauer M, Jain R, Russell JM. et al. A randomized, double-blind comparison of duloxetine and venlafaxine in the treatment of patients with major depressive disorder. J Psychiatr Res. 2008;42(1):22–34. doi: 10.1016/j.jpsychires.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Ipser JC, Kariuki CM, Stein DJ. Pharmacotherapy for social anxiety disorder: A systematic review. Expert Rev Neurother. 2008;8(2):235–57. doi: 10.1586/14737175.8.2.235. [DOI] [PubMed] [Google Scholar]
- 3.Caan B, LaCroix AZ, Joffe H, Guthrie KA, Larson JC, Carpenter JS. et al. Effects of estrogen and venlafaxine on menopause-related quality of life in healthy postmenopausal women with hot flashes: A placebo-controlled randomized trial. Menopause. 2015;22(6):607–15. doi: 10.1097/GME.0000000000000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell C, Wilson AD, Waring WS. Cardiovascular toxicity due to venlafaxine poisoning in adults: A review of 235 consecutive cases. Br J Clin Pharmacol. 2007;64(2):192–7. doi: 10.1111/j.1365-2125.2007.02849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckley NA, McManus PR. Fatal toxicity of serotoninergic and other antidepressant drugs: Analysis of united kingdom mortality data. BMJ. 2002;325(7376):1332–3. doi: 10.1136/bmj.325.7376.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalifa M, Daleau P, Turgeon a J. Mechanism of sodium channel block by venlafaxine in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 1999;291(1):280–4. [PubMed] [Google Scholar]
- 7.Bavadekar S, Panchal P, Hanbashi A, Vansal S. Cytotoxic effects of selective serotonin-and serotonin-norepinephrine reuptake inhibitors on human metastatic breast cancer cell line, MCF-7. FASEB J. 2014;28(1):842–3. [Google Scholar]
- 8.Park SH, Ishino R. Liver injury associated with antidepressants. Curr Drug Saf. 2013;8(3):207–23. doi: 10.2174/1574886311308030011. [DOI] [PubMed] [Google Scholar]
- 9.Sencan I, Sahin I, Ozcetin A. Low-dose venlafaxine-associated liver toxicity in chronic hepatitis. Ann Pharmacother. 2004;38(2):352–3. doi: 10.1345/aph.1D205. [DOI] [PubMed] [Google Scholar]
- 10.Phillips BB, Digmann RR, Beck MG. Hepatitis associated with low-dose venlafaxine for postmenopausal vasomotor symptoms. Ann Pharmacother. 2006;40(2):323–7. doi: 10.1345/aph.1G339. [DOI] [PubMed] [Google Scholar]
- 11.Cardona X, Avila A, Castellanos P. Venlafaxine-associated hepatitis. Ann Intern Med. 2000;132(5):417. doi: 10.7326/0003-4819-132-5-200003070-00016. [DOI] [PubMed] [Google Scholar]
- 12.Detry O, Delwaide J, De Roover A, Hans MF, Delbouille MH, Monard J. et al. Fulminant hepatic failure induced by venlafaxine and trazodone therapy: A case report. Transplant Proc. 2009;41(8):3435–6. doi: 10.1016/j.transproceed.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 13.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44(1):88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao C, Ding Y, Zhong L, Jiang L, Geng C, Yao X. et al. Tacrine induces apoptosis through lysosome- and mitochondria-dependent pathway in hepg2 cells. Toxicol In Vitro. 2014;28(4):667–74. doi: 10.1016/j.tiv.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Ahmadian E, Jafari S, Yari Khosroushahi A. Role of angiotensin ii in stem cell therapy of cardiac disease. JRAAS. 2015;16(4):702–11. doi: 10.1177/1470320315621225. [DOI] [PubMed] [Google Scholar]
- 16.Aly HA, Domenech O. Aroclor 1254 induced cytotoxicity and mitochondrial dysfunction in isolated rat hepatocytes. Toxicology. 2009;262(3):175–83. doi: 10.1016/j.tox.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Luzio JP, Pryor PR, Bright NA. Lysosomes: Fusion and function. Nat Rev Mol Cell Biol. 2007;8(8):622–32. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 18.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–51. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Rubio L, Matas P, Miguez MP. Protective effect of melatonin on paraquat-induced cytotoxicity in isolated rat hepatocytes. Hum Exp Toxicol. 2005;24(9):475–80. doi: 10.1191/0960327105ht548oa. [DOI] [PubMed] [Google Scholar]
- 20.Eghbal M, Eftekhari A, Ahmadian E, Azarmi Y, Parvizpur A. A review of biological and pharmacological actions of melatonin: Oxidant and prooxidant properties. J Pharmacol Reports. 2016;1(106):2. doi: 10.4172/jpr.1000106. [DOI] [Google Scholar]
- 21.Pourahmad J. Accelerated cytotoxicity mechanism screening. Asia Pac J Med Toxicol. 2014;3:4–4. doi: 10.22038/apjmt.2014.2811. [DOI] [Google Scholar]
- 22.Eghbal MA, Pennefather PS, O'Brien PJ. H2s cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology. 2004;203(1-3):69–76. doi: 10.1016/j.tox.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 23.Truong DH, Eghbal MA, Hindmarsh W, Roth SH, O'Brien PJ. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab Rev. 2006;38(4):733–44. doi: 10.1080/03602530600959607. [DOI] [PubMed] [Google Scholar]
- 24.Heidari R, Babaei H, Eghbal MA. Cytoprotective effects of taurine against toxicity induced by isoniazid and hydrazine in isolated rat hepatocytes. Arh Hig Rada Toksikol. 2013;64(2):15–24. doi: 10.2478/10004-1254-64-2013-2297. [DOI] [PubMed] [Google Scholar]
- 25.Pourahmad J, Hosseini MJ, Eskandari MR, Shekarabi SM, Daraei B. Mitochondrial/lysosomal toxic cross-talk plays a key role in cisplatin nephrotoxicity. Xenobiotica. 2010;40(11):763–71. doi: 10.3109/00498254.2010.512093. [DOI] [PubMed] [Google Scholar]
- 26.Eftekhari A, Azarmi Y, Parvizpur A, Eghbal MA. Involvement of oxidative stress and mitochondrial/lysosomal cross-talk in olanzapine cytotoxicity in freshly isolated rat hepatocytes. Xenobiotica. 2016;46(4):369–78. doi: 10.3109/00498254.2015.1078522. [DOI] [PubMed] [Google Scholar]
- 27.Moldéus P, Högberg J, Orrenius S. [4] Isolation and use of liver cells. Method Enzymol. 1978;52:60–71. doi: 10.1016/s0076-6879(78)52006-5. [DOI] [PubMed] [Google Scholar]
- 28.Heidari R, Babaei H, Eghbal MA. Cytoprotective effects of organosulfur compounds against methimazole induced toxicity in isolated rat hepatocytes. Adv Pharm Bull. 2013;3(1):135–42. doi: 10.5681/apb.2013.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostafalou S, Abdollahi M, Eghbal MA, Saeedi Kouzehkonani N. Protective effect of nac against malathion-induced oxidative stress in freshly isolated rat hepatocytes. Adv Pharm Bull. 2012;2(1):79–88. doi: 10.5681/apb.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eghbal MA, Tafazoli S, Pennefather P, O'Brien PJ. Peroxidase catalysed formation of cytotoxic prooxidant phenothiazine free radicals at physiological ph. Chem Biol Interact. 2004;151(1):43–51. doi: 10.1016/j.cbi.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Fard JK, Hamzeiy H, Sattari M, Eftekhari A, Ahmadian E, Eghbal MA. Triazole rizatriptan induces liver toxicity through lysosomal/mitochondrial dysfunction. Drug Res (Stuttg) 2016;66(9):470–8. doi: 10.1055/s-0042-110178. [DOI] [PubMed] [Google Scholar]
- 32.Pourahmad J, Hosseini MJ, Eskandari MR, Shekarabi SM, Daraei B. Mitochondrial/lysosomal toxic cross-talk plays a key role in cisplatin nephrotoxicity. Xenobiotica. 2010;40(11):763–71. doi: 10.3109/00498254.2010.512093. [DOI] [PubMed] [Google Scholar]
- 33. Eftekhari A, Ahmadian E, Azarmi Y, Parvizpur A, Hamishehkar H, Eghbal MA. In vitro/vivo studies towards mechanisms of risperidone-induced oxidative stress and the protective role of coenzyme q10 and n-acetylcysteine. Toxicol Mech Methods 2016:1-9. [DOI] [PubMed]
- 34.Abdoli N, Azarmi Y, Eghbal MA. Protective effects of n-acetylcysteine against the statins cytotoxicity in freshly isolated rat hepatocytes. Adv Pharm Bull. 2014;4(3):249–54. doi: 10.5681/apb.2014.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahmadian E, Eftekhari A, Fard JK, Babaei H, Nayebi AM, Mohammadnejad D, et al. In vitro and in vivo evaluation of the mechanisms of citalopram-induced hepatotoxicity. Arch Pharm Res 2016. [DOI] [PubMed]
- 36.Ahadpour M, Eskandari MR, Mashayekhi V, Haj Mohammad Ebrahim Tehrani K , Jafarian I, Naserzadeh P. et al. Mitochondrial oxidative stress and dysfunction induced by isoniazid: Study on isolated rat liver and brain mitochondria. Drug Chem Toxicol. 2016;39(2):224–32. doi: 10.3109/01480545.2015.1092039. [DOI] [PubMed] [Google Scholar]
- 37.Mashayekhi V, Tehrani KH, Hashemzaei M, Tabrizian K, Shahraki J, Hosseini MJ. Mechanistic approach for the toxic effects of perfluorooctanoic acid on isolated rat liver and brain mitochondria. Hum Exp Toxicol. 2015;34(10):985–96. doi: 10.1177/0960327114565492. [DOI] [PubMed] [Google Scholar]
- 38.Rolla R, Gramaglia C, Dalo V, Ressico F, Prosperini P, Vidali M. et al. An observational study of venlafaxine and cyp2d6 in clinical practice. Clin Lab. 2014;60(2):225–31. doi: 10.7754/clin.lab.2013.130141. [DOI] [PubMed] [Google Scholar]
- 39.Minguez L, Farcy E, Ballandonne C, Lepailleur A, Serpentini A, Lebel JM. et al. Acute toxicity of 8 antidepressants: What are their modes of action? Chemosphere. 2014;108:314–9. doi: 10.1016/j.chemosphere.2014.01.057. [DOI] [PubMed] [Google Scholar]
- 40.O'Brien PJ, Siraki AG. Accelerated cytotoxicity mechanism screening using drug metabolising enzyme modulators. Curr Drug Metab. 2005;6(2):101–9. doi: 10.2174/1389200053586082. [DOI] [PubMed] [Google Scholar]
- 41.Deavall DG, Martin EA, Horner JM, Roberts R. Drug-induced oxidative stress and toxicity. J Toxicol. 2012;2012:645460. doi: 10.1155/2012/645460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farmer EE, Mueller MJ. Ros-mediated lipid peroxidation and res-activated signaling. Annu Rev Plant Biol. 2013;64:429–50. doi: 10.1146/annurev-arplant-050312-120132. [DOI] [PubMed] [Google Scholar]
- 43.Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012;2012:137289. doi: 10.5402/2012/137289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1-2):42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J. et al. Redox status expressed as gsh:Gssg ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4(6):1247–53. doi: 10.3892/ol.2012.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegmund SV, Brenner DA. Molecular pathogenesis of alcohol-induced hepatic fibrosis. Alcohol Clin Exp Res. 2005;29(11 Suppl):102S–9S. doi: 10.1097/01.alc.0000189275.97419.58. [DOI] [PubMed] [Google Scholar]
- 47.Ahmadian E, Pennefather PS, Eftekhari A, Heidari R, Eghbal MA. Role of renin-angiotensin system in liver diseases: An outline on the potential therapeutic points of intervention. Expert Rev Gastroenterol Hepatol. 2016;10(11):1279–88. doi: 10.1080/17474124.2016.1207523. [DOI] [PubMed] [Google Scholar]
- 48.Mashayekhi V, Eskandari MR, Kobarfard F, Khajeamiri A, Hosseini MJ. Induction of mitochondrial permeability transition (mpt) pore opening and ros formation as a mechanism for methamphetamine-induced mitochondrial toxicity. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(1):47–58. doi: 10.1007/s00210-013-0919-3. [DOI] [PubMed] [Google Scholar]
- 49.Santos NA, Medina WS, Martins NM, Mingatto FE, Curti C, Santos AC. Aromatic antiepileptic drugs and mitochondrial toxicity: Effects on mitochondria isolated from rat liver. Toxicol In Vitro. 2008;22(5):1143–52. doi: 10.1016/j.tiv.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Hwang J, Zheng LT, Ock J, Lee MG, Kim SH, Lee HW. et al. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology. 2008;55(5):826–34. doi: 10.1016/j.neuropharm.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 51.Hroudova J, Fisar Z. Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuro Endocrinol Lett. 2010;31(3):336–42. [PubMed] [Google Scholar]
- 52.Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T. et al. Evidence of ros generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7(1-2):106–18. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 53.Martins NM, Santos NA, Curti C, Bianchi ML, Santos AC. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J Appl Toxicol. 2008;28(3):337–44. doi: 10.1002/jat.1284. [DOI] [PubMed] [Google Scholar]
- 54.Eskandari MR, Moghaddam F, Shahraki J, Pourahmad J. A comparison of cardiomyocyte cytotoxic mechanisms for 5-fluorouracil and its pro-drug capecitabine. Xenobiotica. 2015;45(1):79–87. doi: 10.3109/00498254.2014.942809. [DOI] [PubMed] [Google Scholar]
- 55.Pourahmad J, Shaki F, Tanbakosazan F, Ghalandari R, Ettehadi HA, Dahaghin E. Protective effects of fungal beta-(1-->3)-d-glucan against oxidative stress cytotoxicity induced by depleted uranium in isolated rat hepatocytes. Hum Exp Toxicol. 2011;30(3):173–81. doi: 10.1177/0960327110372643. [DOI] [PubMed] [Google Scholar]